The FANC/BRCA Pathway Releases Replication Blockades by Eliminating DNA Interstrand Cross-Links


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
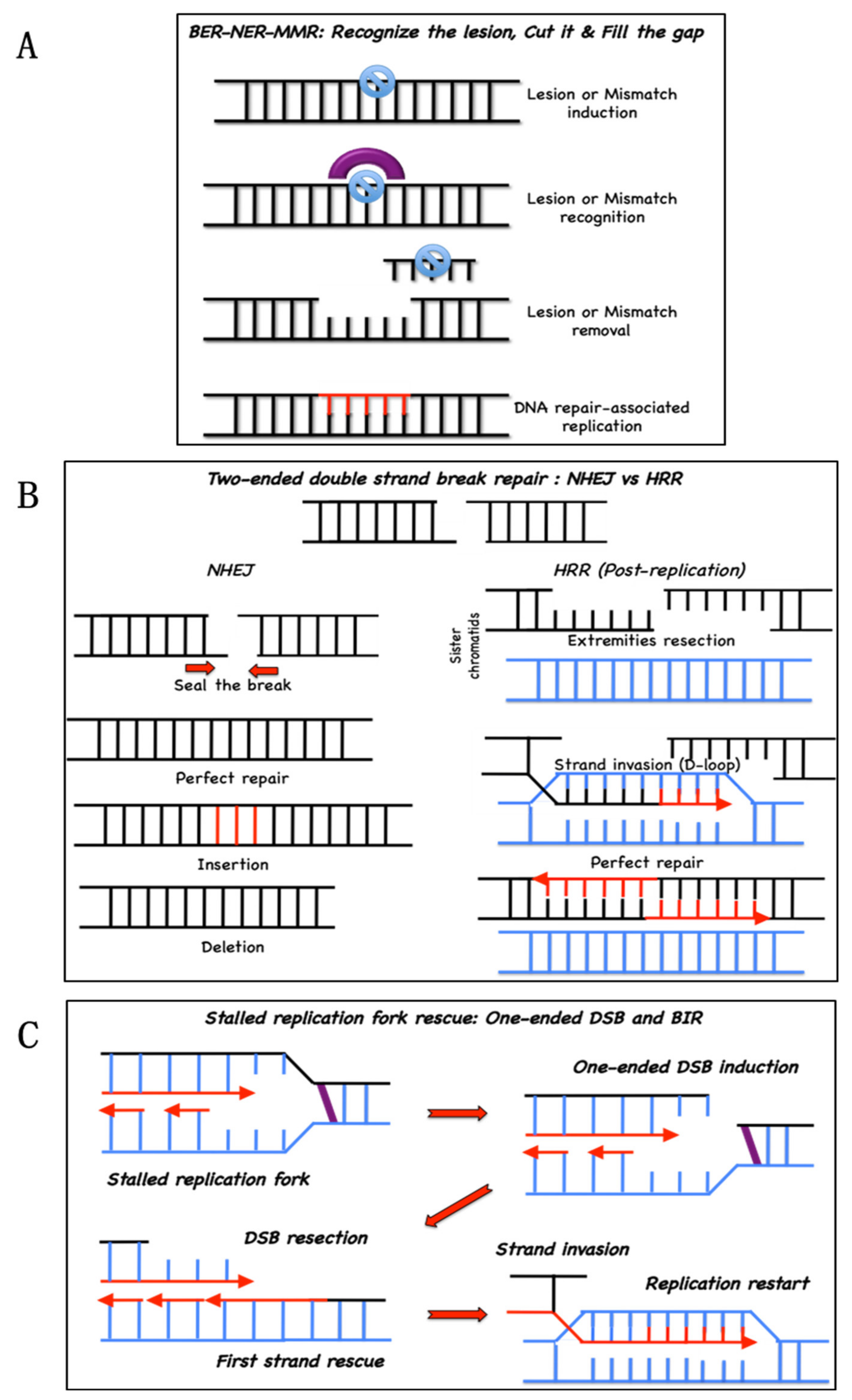
:1. Introduction
2. Origin of ICL in Cells
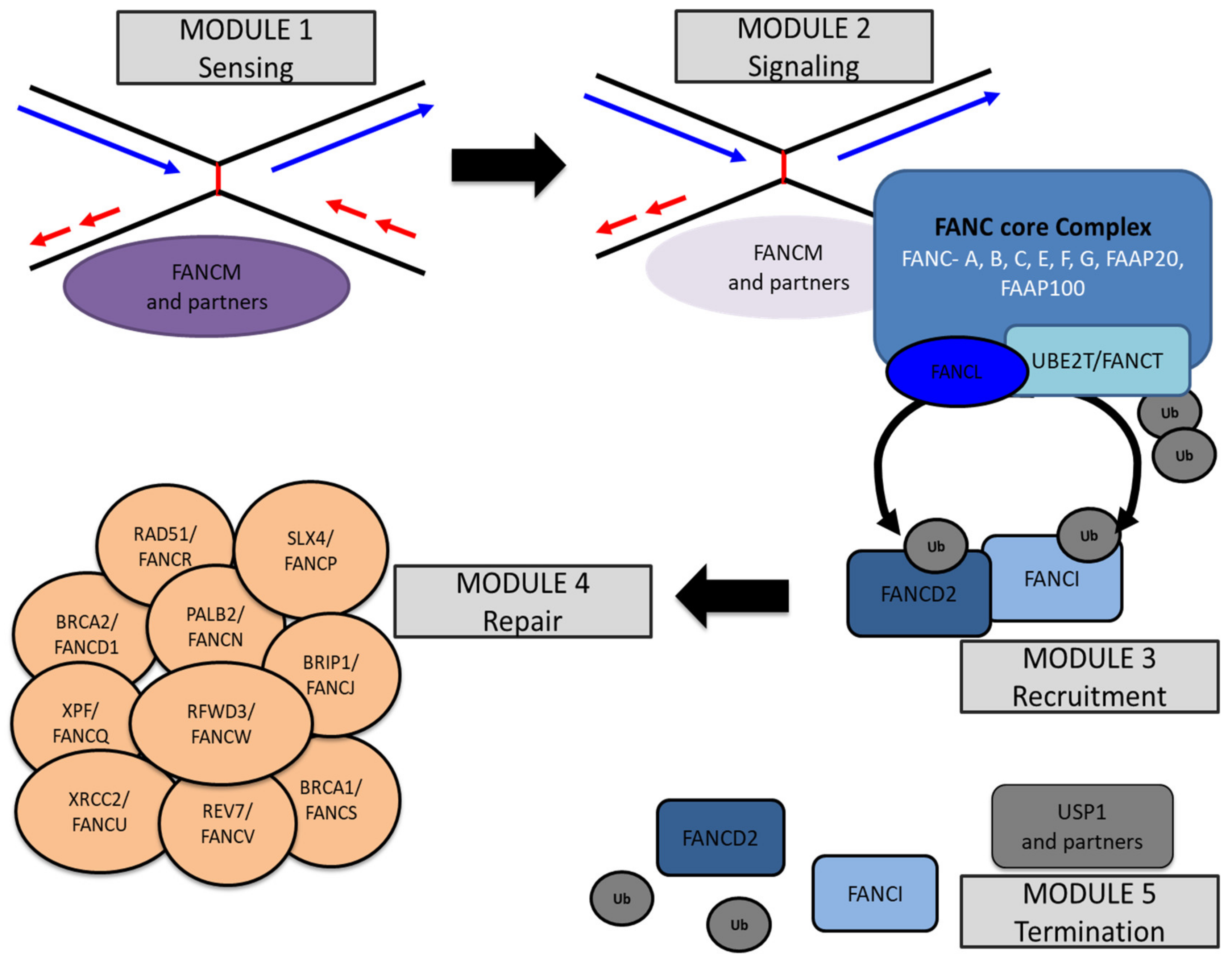
3. Biochemistry of the FANC/BRCA Pathway
3.1. The First Module: FANCM and Its Partners
3.2. The Second Module: The FANC Core Complex
3.3. The Third Module: FANCD2 and FANCI Heterodimer
3.4. The Fourth Module: The HRR at Work
3.5. The Fifth Module: USP1, the Finisher
3.6. Other Partners
4. Evidence for the Essential Role of the FANC/BRCA Pathway in ICL Repair
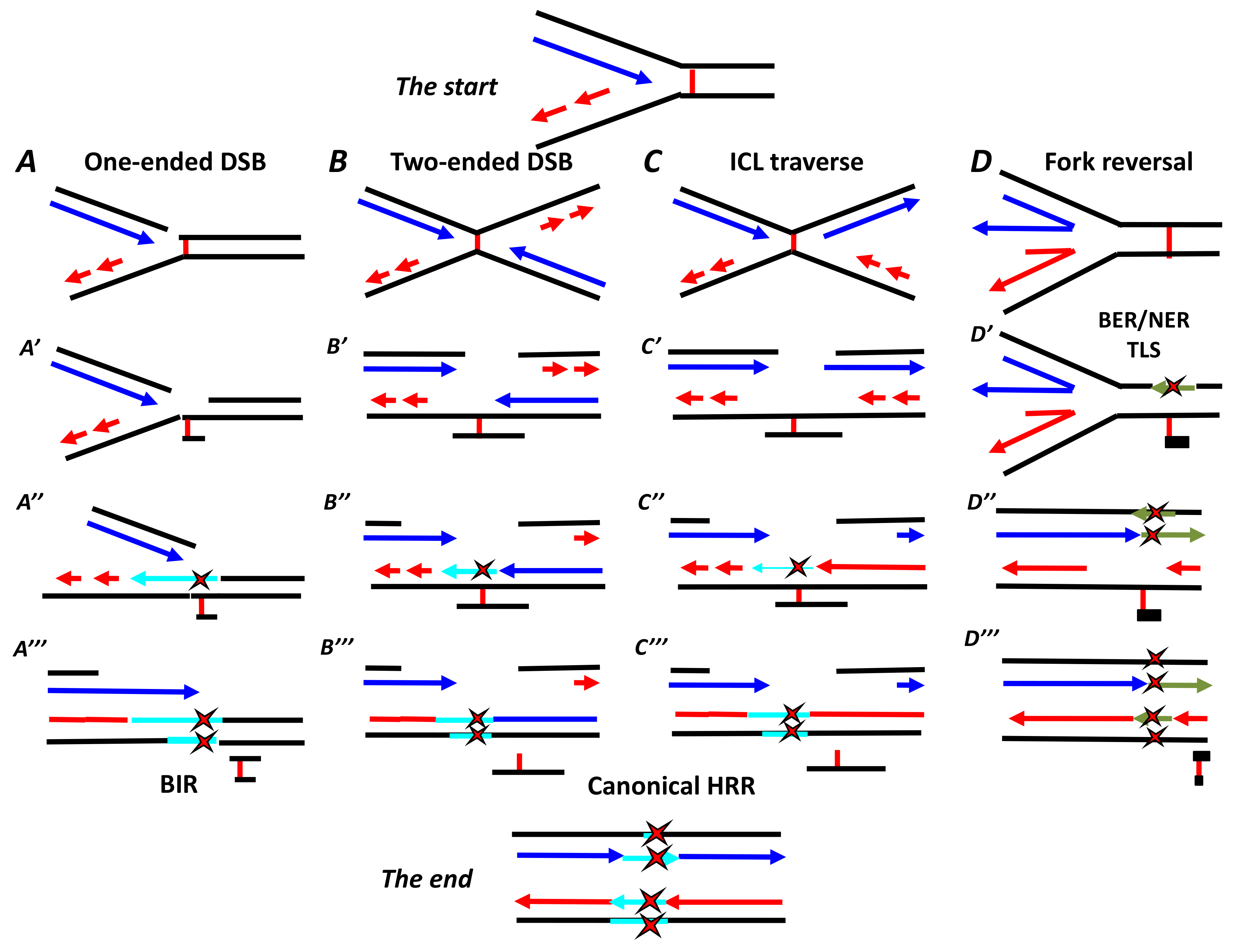
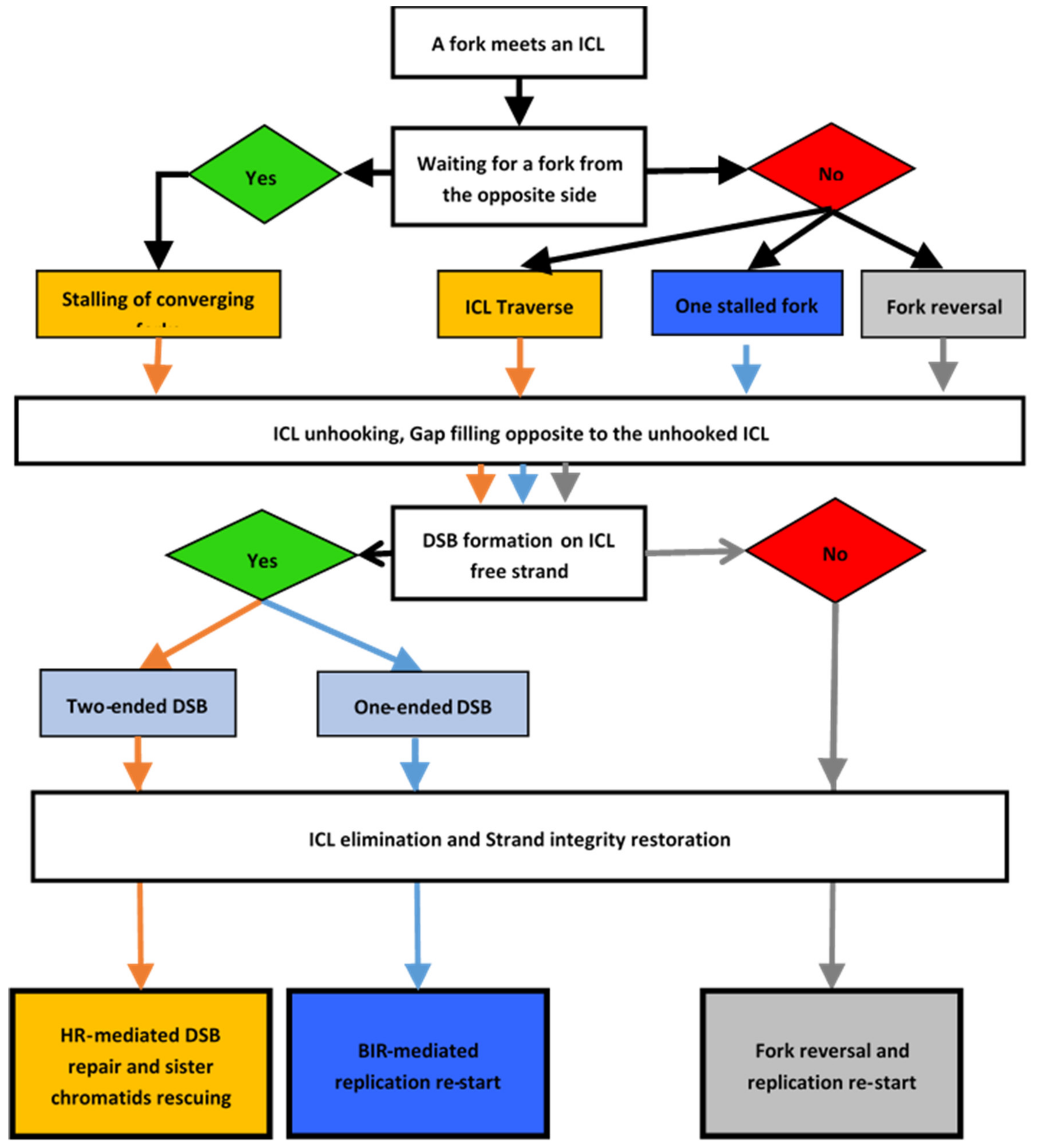
5. First ICL Repair Model: Downstream of a Stalled Single Replication Fork
6. Second ICL Repair Model: Converging Forks at Work
7. Third ICL Repair Model: ICL Traverse
8. Concluding Remarks and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Guainazzi, A.; Schärer, O.D. Using synthetic DNA interstrand crosslinks to elucidate repair pathways and identify new therapeutic targets for cancer chemotherapy. Cell. Mol. Life Sci. 2010, 67, 3683–3697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Legerski, R.J. Repair of DNA interstrand cross-links during S phase of the mammalian cell cycle. Environ. Mol. Mutagen. 2010, 51, 540–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clauson, C.; Schärer, O.D.; Niedernhofer, L. Advances in understanding the complex mechanisms of DNA interstrand cross-link repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012732. [Google Scholar] [CrossRef] [PubMed]
- Hoeijmakers, J.H. Genome maintenance mechanisms for preventing cancer. Nature 2001, 411, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Ait Saada, A.; Lambert, S.A.E.; Carr, A.M. Preserving replication fork integrity and competence via the homologous recombination pathway. DNA Repair 2018, 71, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Carr, A.M.; Lambert, S. Replication stress-induced genome instability: The dark side of replication maintenance by homologous recombination. J. Mol. Biol. 2013, 425, 4733–4744. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Lange, J.; Keeney, S. Genome destabilization by homologous recombination in the germ line. Nat. Rev. Mol. Cell Biol. 2010, 11, 182–195. [Google Scholar] [CrossRef] [Green Version]
- Bétermier, M.; Bertrand, P.; Lopez, B.S. Is non-homologous end-joining really an inherently error-prone process? PLoS Genet. 2014, 10, e1004086. [Google Scholar] [CrossRef] [Green Version]
- Guirouilh-Barbat, J.; Lambert, S.; Bertrand, P.; Lopez, B.S. Is homologous recombination really an error-free process? Front. Genet. 2014, 5, 175. [Google Scholar] [CrossRef] [Green Version]
- Hanlon Newell, A.E.; Hemphill, A.; Akkari, Y.M.N.; Hejna, J.; Moses, R.E.; Olson, S.B. Loss of homologous recombination or non-homologous end-joining leads to radial formation following DNA interstrand crosslink damage. Cytogenet. Genome Res. 2008, 121, 174–180. [Google Scholar] [CrossRef] [Green Version]
- Marini, F.; Rawal, C.C.; Liberi, G.; Pellicioli, A. Regulation of DNA Double Strand Breaks Processing: Focus on Barriers. Front. Mol. Biosci. 2019, 6, 55. [Google Scholar] [CrossRef] [PubMed]
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 2019, 20, 698–714. [Google Scholar] [CrossRef] [PubMed]
- Bogliolo, M.; Surrallés, J. Fanconi anemia: A model disease for studies on human genetics and advanced therapeutics. Curr. Opin. Genet. Dev. 2015, 33, 32–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gueiderikh, A.; Rosselli, F.; Neto, J.B.C. A never-ending story: The steadily growing family of the FA and FA-like genes. Genet. Mol. Biol. 2017, 40, 398–407. [Google Scholar] [CrossRef] [PubMed]
- Fanconi, G. Familiäre infantile perniziosaartige Anämie (perniziöses Blutbild und Konstitution). Jahrb. Für Kinderheilkd. Phys. Erzieh. 5wIEN° 1927, 117, 257–280. [Google Scholar]
- Lobitz, S.; Velleuer, E. Guido Fanconi (1892–1979): A jack of all trades. Nat. Rev. Cancer 2006, 6, 893–898. [Google Scholar] [CrossRef]
- Nalepa, G.; Clapp, D.W. Fanconi anaemia and cancer: An intricate relationship. Nat. Rev. Cancer 2018, 18, 168–185. [Google Scholar] [CrossRef]
- Ceccaldi, R.; Sarangi, P.; D’Andrea, A.D. The Fanconi anaemia pathway: New players and new functions. Nat. Rev. Mol. Cell Biol. 2016, 17, 337–349. [Google Scholar] [CrossRef]
- Geiselhart, A.; Lier, A.; Walter, D.; Milsom, M.D. Disrupted Signaling through the Fanconi Anemia Pathway Leads to Dysfunctional Hematopoietic Stem Cell Biology: Underlying Mechanisms and Potential Therapeutic Strategies. Anemia 2012, 2012, 265790. [Google Scholar] [CrossRef] [Green Version]
- Helbling-Leclerc, A.; Dessarps-Freichey, F.; Evrard, C.; Rosselli, F. Fanconi anemia proteins counteract the implementation of the oncogene-induced senescence program. Sci. Rep. 2019, 9, 17024. [Google Scholar] [CrossRef]
- Lindahl, T.; Barnes, D.E. Repair of endogenous DNA damage. Cold Spring Harb. Symp. Quant. Biol. 2000, 65, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Fink, S.P.; Reddy, G.R.; Marnett, L.J. Mutagenicity in Escherichia coli of the major DNA adduct derived from the endogenous mutagen malondialdehyde. Proc. Natl. Acad. Sci. USA 1997, 94, 8652–8657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukai, F.H.; Goldstein, B.D. Mutagenicity of malonaldehyde, a decomposition product of peroxidized polyunsaturated fatty acids. Science 1976, 191, 868–869. [Google Scholar] [CrossRef] [PubMed]
- Niedernhofer, L.J.; Daniels, J.S.; Rouzer, C.A.; Greene, R.E.; Marnett, L.J. Malondialdehyde, a product of lipid peroxidation, is mutagenic in human cells. J. Biol. Chem. 2003, 278, 31426–31433. [Google Scholar] [CrossRef] [Green Version]
- Langevin, F.; Crossan, G.P.; Rosado, I.V.; Arends, M.J.; Patel, K.J. Fancd2 counteracts the toxic effects of naturally produced aldehydes in mice. Nature 2011, 475, 53–58. [Google Scholar] [CrossRef]
- Rosado, I.V.; Langevin, F.; Crossan, G.P.; Takata, M.; Patel, K.J. Formaldehyde catabolism is essential in cells deficient for the Fanconi anemia DNA-repair pathway. Nat. Struct. Mol. Biol. 2011, 18, 1432–1434. [Google Scholar] [CrossRef]
- Garaycoechea, J.I.; Crossan, G.P.; Langevin, F.; Mulderrig, L.; Louzada, S.; Yang, F.; Guilbaud, G.; Park, N.; Roerink, S.; Nik-Zainal, S.; et al. Alcohol and endogenous aldehydes damage chromosomes and mutate stem cells. Nature 2018, 553, 171–177. [Google Scholar] [CrossRef]
- Pontel, L.B.; Rosado, I.V.; Burgos-Barragan, G.; Garaycoechea, J.I.; Yu, R.; Arends, M.J.; Chandrasekaran, G.; Broecker, V.; Wei, W.; Liu, L.; et al. Endogenous Formaldehyde Is a Hematopoietic Stem Cell Genotoxin and Metabolic Carcinogen. Mol. Cell 2015, 60, 177–188. [Google Scholar] [CrossRef] [Green Version]
- Hira, A.; Yabe, H.; Yoshida, K.; Okuno, Y.; Shiraishi, Y.; Chiba, K.; Tanaka, H.; Miyano, S.; Nakamura, J.; Kojima, S.; et al. Variant ALDH2 is associated with accelerated progression of bone marrow failure in Japanese Fanconi anemia patients. Blood 2013, 122, 3206–3209. [Google Scholar] [CrossRef] [Green Version]
- Kirchner, J.J.; Sigurdsson, S.T.; Hopkins, P.B. Interstrand cross-linking of duplex DNA by nitrous acid: Covalent structure of the dG-to-dG cross-link at the sequence 5’-CG. J. Am. Chem. Soc. 1992, 114, 4021–4027. [Google Scholar] [CrossRef]
- Dutta, S.; Chowdhury, G.; Gates, K.S. Interstrand cross-links generated by abasic sites in duplex DNA. J. Am. Chem. Soc. 2007, 129, 1852–1853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Leung, J.W.C.; Lowery, M.; Matsushita, N.; Wang, Y.; Shen, X.; Huong, D.; Takata, M.; Chen, J.; Li, L. Modularized functions of the Fanconi anemia core complex. Cell Rep. 2014, 7, 1849–1857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rickman, K.A.; Lach, F.P.; Abhyankar, A.; Donovan, F.X.; Sanborn, E.M.; Kennedy, J.A.; Sougnez, C.; Gabriel, S.B.; Elemento, O.; Chandrasekharappa, S.C.; et al. Deficiency of UBE2T, the E2 Ubiquitin Ligase Necessary for FANCD2 and FANCI Ubiquitination, Causes FA-T Subtype of Fanconi Anemia. Cell Rep. 2015, 12, 35–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Higuera, I.; Taniguchi, T.; Ganesan, S.; Meyn, M.S.; Timmers, C.; Hejna, J.; Grompe, M.; D’Andrea, A.D. Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol. Cell 2001, 7, 249–262. [Google Scholar] [CrossRef]
- Nijman, S.M.B.; Huang, T.T.; Dirac, A.M.G.; Brummelkamp, T.R.; Kerkhoven, R.M.; D’Andrea, A.D.; Bernards, R. The deubiquitinating enzyme USP1 regulates the Fanconi anemia pathway. Mol. Cell 2005, 17, 331–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meetei, A.R.; Medhurst, A.L.; Ling, C.; Xue, Y.; Singh, T.R.; Bier, P.; Steltenpool, J.; Stone, S.; Dokal, I.; Mathew, C.G.; et al. A human ortholog of archaeal DNA repair protein Hef is defective in Fanconi anemia complementation group M. Nat. Genet. 2005, 37, 958–963. [Google Scholar] [CrossRef] [Green Version]
- Yan, Z.; Delannoy, M.; Ling, C.; Daee, D.; Osman, F.; Muniandy, P.A.; Shen, X.; Oostra, A.B.; Du, H.; Steltenpool, J.; et al. A histone-fold complex and FANCM form a conserved DNA-remodeling complex to maintain genome stability. Mol. Cell 2010, 37, 865–878. [Google Scholar] [CrossRef] [Green Version]
- Ciccia, A.; Ling, C.; Coulthard, R.; Yan, Z.; Xue, Y.; Meetei, A.R.; Laghmani, E.H.; Joenje, H.; McDonald, N.; de Winter, J.P.; et al. Identification of FAAP24, a Fanconi anemia core complex protein that interacts with FANCM. Mol. Cell 2007, 25, 331–343. [Google Scholar] [CrossRef]
- Basbous, J.; Constantinou, A. A tumor suppressive DNA translocase named FANCM. Crit. Rev. Biochem. Mol. Biol. 2019, 54, 27–40. [Google Scholar] [CrossRef]
- Xue, X.; Sung, P.; Zhao, X. Functions and regulation of the multitasking FANCM family of DNA motor proteins. Genes Dev. 2015, 29, 1777–1788. [Google Scholar] [CrossRef] [Green Version]
- Gari, K.; Décaillet, C.; Stasiak, A.Z.; Stasiak, A.; Constantinou, A. The Fanconi anemia protein FANCM can promote branch migration of Holliday junctions and replication forks. Mol. Cell 2008, 29, 141–148. [Google Scholar] [CrossRef]
- Xue, Y.; Li, Y.; Guo, R.; Ling, C.; Wang, W. FANCM of the Fanconi anemia core complex is required for both monoubiquitination and DNA repair. Hum. Mol. Genet. 2008, 17, 1641–1652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deans, A.J.; West, S.C. FANCM connects the genome instability disorders Bloom’s Syndrome and Fanconi Anemia. Mol. Cell 2009, 36, 943–953. [Google Scholar] [CrossRef] [PubMed]
- Hoadley, K.A.; Xue, Y.; Ling, C.; Takata, M.; Wang, W.; Keck, J.L. Defining the molecular interface that connects the Fanconi anemia protein FANCM to the Bloom syndrome dissolvasome. Proc. Natl. Acad. Sci. USA 2012, 109, 4437–4442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karow, J.K.; Constantinou, A.; Li, J.L.; West, S.C.; Hickson, I.D. The Bloom’s syndrome gene product promotes branch migration of holliday junctions. Proc. Natl. Acad. Sci. USA 2000, 97, 6504–6508. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Hickson, I.D. The Bloom’s syndrome helicase suppresses crossing over during homologous recombination. Nature 2003, 426, 870–874. [Google Scholar] [CrossRef]
- Luke-Glaser, S.; Luke, B.; Grossi, S.; Constantinou, A. FANCM regulates DNA chain elongation and is stabilized by S-phase checkpoint signalling. EMBO J. 2010, 29, 795–805. [Google Scholar] [CrossRef] [Green Version]
- Klein Douwel, D.; Boonen, R.A.C.M.; Long, D.T.; Szypowska, A.A.; Räschle, M.; Walter, J.C.; Knipscheer, P. XPF-ERCC1 acts in Unhooking DNA interstrand crosslinks in cooperation with FANCD2 and FANCP/SLX4. Mol. Cell 2014, 54, 460–471. [Google Scholar] [CrossRef] [Green Version]
- Mosedale, G.; Niedzwiedz, W.; Alpi, A.; Perrina, F.; Pereira-Leal, J.B.; Johnson, M.; Langevin, F.; Pace, P.; Patel, K.J. The vertebrate Hef ortholog is a component of the Fanconi anemia tumor-suppressor pathway. Nat. Struct. Mol. Biol. 2005, 12, 763–771. [Google Scholar] [CrossRef]
- Yamamoto, K.N.; Kobayashi, S.; Tsuda, M.; Kurumizaka, H.; Takata, M.; Kono, K.; Jiricny, J.; Takeda, S.; Hirota, K. Involvement of SLX4 in interstrand cross-link repair is regulated by the Fanconi anemia pathway. Proc. Natl. Acad. Sci. USA 2011, 108, 6492–6496. [Google Scholar] [CrossRef] [Green Version]
- Bakker, S.T.; van de Vrugt, H.J.; Rooimans, M.A.; Oostra, A.B.; Steltenpool, J.; Delzenne-Goette, E.; van der Wal, A.; van der Valk, M.; Joenje, H.; te Riele, H.; et al. Fancm-deficient mice reveal unique features of Fanconi anemia complementation group M. Hum. Mol. Genet. 2009, 18, 3484–3495. [Google Scholar] [CrossRef] [PubMed]
- Fouquet, B.; Pawlikowska, P.; Caburet, S.; Guigon, C.; Mäkinen, M.; Tanner, L.; Hietala, M.; Urbanska, K.; Bellutti, L.; Legois, B.; et al. A homozygous FANCM mutation underlies a familial case of non-syndromic primary ovarian insufficiency. eLife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Bogliolo, M.; Bluteau, D.; Lespinasse, J.; Pujol, R.; Vasquez, N.; d’Enghien, C.D.; Stoppa-Lyonnet, D.; Leblanc, T.; Soulier, J.; Surrallés, J. Biallelic truncating FANCM mutations cause early-onset cancer but not Fanconi anemia. Genet. Med. 2018, 20, 458–463. [Google Scholar] [CrossRef] [PubMed]
- Catucci, I.; Osorio, A.; Arver, B.; Neidhardt, G.; Bogliolo, M.; Zanardi, F.; Riboni, M.; Minardi, S.; Pujol, R.; Azzollini, J.; et al. Individuals with FANCM biallelic mutations do not develop Fanconi anemia, but show risk for breast cancer, chemotherapy toxicity and may display chromosome fragility. Genet. Med. 2018, 20, 452–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nurmi, A.; Muranen, T.A.; Pelttari, L.M.; Kiiski, J.I.; Heikkinen, T.; Lehto, S.; Kallioniemi, A.; Schleutker, J.; Bützow, R.; Blomqvist, C.; et al. Recurrent moderate-risk mutations in Finnish breast and ovarian cancer patients. Int. J. Cancer 2019, 145, 2692–2700. [Google Scholar] [CrossRef]
- Schubert, S.; van Luttikhuizen, J.L.; Auber, B.; Schmidt, G.; Hofmann, W.; Penkert, J.; Davenport, C.F.; Hille-Betz, U.; Wendeburg, L.; Bublitz, J.; et al. The identification of pathogenic variants in BRCA1/2 negative, high risk, hereditary breast and/or ovarian cancer patients: High frequency of FANCM pathogenic variants. Int. J. Cancer 2019, 144, 2683–2694. [Google Scholar] [CrossRef]
- Singh, T.R.; Bakker, S.T.; Agarwal, S.; Jansen, M.; Grassman, E.; Godthelp, B.C.; Ali, A.M.; Du, C.; Rooimans, M.A.; Fan, Q.; et al. Impaired FANCD2 monoubiquitination and hypersensitivity to camptothecin uniquely characterize Fanconi anemia complementation group M. Blood 2009, 114, 174–180. [Google Scholar] [CrossRef]
- Shakeel, S.; Rajendra, E.; Alcón, P.; O’Reilly, F.; Chorev, D.S.; Maslen, S.; Degliesposti, G.; Russo, C.J.; He, S.; Hill, C.H.; et al. Structure of the Fanconi anaemia monoubiquitin ligase complex. Nature 2019, 575, 234–237. [Google Scholar] [CrossRef]
- Van Twest, S.; Murphy, V.J.; Hodson, C.; Tan, W.; Swuec, P.; O’Rourke, J.J.; Heierhorst, J.; Crismani, W.; Deans, A.J. Mechanism of Ubiquitination and Deubiquitination in the Fanconi Anemia Pathway. Mol. Cell 2017, 65, 247–259. [Google Scholar] [CrossRef] [Green Version]
- Adachi, D.; Oda, T.; Yagasaki, H.; Nakasato, K.; Taniguchi, T.; D’Andrea, A.D.; Asano, S.; Yamashita, T. Heterogeneous activation of the Fanconi anemia pathway by patient-derived FANCA mutants. Hum. Mol. Genet. 2002, 11, 3125–3134. [Google Scholar] [CrossRef] [Green Version]
- Benitez, A.; Liu, W.; Palovcak, A.; Wang, G.; Moon, J.; An, K.; Kim, A.; Zheng, K.; Zhang, Y.; Bai, F.; et al. FANCA Promotes DNA Double-Strand Break Repair by Catalyzing Single-Strand Annealing and Strand Exchange. Mol. Cell 2018, 71, 621–628. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.V.; Riou, L.; Aoufouchi, S.; Rosselli, F. Fanca deficiency reduces A/T transitions in somatic hypermutation and alters class switch recombination junctions in mouse B cells. J. Exp. Med. 2014, 211, 1011–1018. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.-C.; Li, Z.; Lopez-Martinez, D.; Nicholson, W.V.; Vénien-Bryan, C.; Cohn, M.A. The FANCD2-FANCI complex is recruited to DNA interstrand crosslinks before monoubiquitination of FANCD2. Nat. Commun. 2016, 7, 12124. [Google Scholar] [CrossRef]
- Boisvert, R.A.; Howlett, N.G. The Fanconi anemia ID2 complex: Dueling saxes at the crossroads. Cell Cycle 2014, 13, 2999–3015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renaudin, X.; Koch Lerner, L.; Menck, C.F.M.; Rosselli, F. The ubiquitin family meets the Fanconi anemia proteins. Mutat. Res. Rev. Mutat. Res. 2016, 769, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, N.; Kitao, H.; Ishiai, M.; Nagashima, N.; Hirano, S.; Okawa, K.; Ohta, T.; Yu, D.S.; McHugh, P.J.; Hickson, I.D.; et al. A FancD2-monoubiquitin fusion reveals hidden functions of Fanconi anemia core complex in DNA repair. Mol. Cell 2005, 19, 841–847. [Google Scholar] [CrossRef]
- Tan, W.; van Twest, S.; Murphy, V.J.; Deans, A.J. ATR-Mediated FANCI Phosphorylation Regulates Both Ubiquitination and Deubiquitination of FANCD2. Front. Cell Dev. Biol. 2020, 8, 2. [Google Scholar] [CrossRef] [Green Version]
- Bogliolo, M.; Lyakhovich, A.; Callén, E.; Castellà, M.; Cappelli, E.; Ramírez, M.J.; Creus, A.; Marcos, R.; Kalb, R.; Neveling, K.; et al. Histone H2AX and Fanconi anemia FANCD2 function in the same pathway to maintain chromosome stability. EMBO J. 2007, 26, 1340–1351. [Google Scholar] [CrossRef]
- Kais, Z.; Rondinelli, B.; Holmes, A.; O’Leary, C.; Kozono, D.; D’Andrea, A.D.; Ceccaldi, R. FANCD2 Maintains Fork Stability in BRCA1/2-Deficient Tumors and Promotes Alternative End-Joining DNA Repair. Cell Rep. 2016, 15, 2488–2499. [Google Scholar] [CrossRef] [Green Version]
- Michl, J.; Zimmer, J.; Buffa, F.M.; McDermott, U.; Tarsounas, M. FANCD2 limits replication stress and genome instability in cells lacking BRCA2. Nat. Struct. Mol. Biol. 2016, 23, 755–757. [Google Scholar] [CrossRef]
- Sato, K.; Shimomuki, M.; Katsuki, Y.; Takahashi, D.; Kobayashi, W.; Ishiai, M.; Miyoshi, H.; Takata, M.; Kurumizaka, H. FANCI-FANCD2 stabilizes the RAD51-DNA complex by binding RAD51 and protects the 5’-DNA end. Nucleic Acids Res. 2016, 44, 10758–10771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlacher, K.; Wu, H.; Jasin, M. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 2012, 22, 106–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howlett, N.G.; Taniguchi, T.; Olson, S.; Cox, B.; Waisfisz, Q.; De Die-Smulders, C.; Persky, N.; Grompe, M.; Joenje, H.; Pals, G.; et al. Biallelic inactivation of BRCA2 in Fanconi anemia. Science 2002, 297, 606–609. [Google Scholar] [CrossRef] [PubMed]
- Levitus, M.; Waisfisz, Q.; Godthelp, B.C.; de Vries, Y.; Hussain, S.; Wiegant, W.W.; Elghalbzouri-Maghrani, E.; Steltenpool, J.; Rooimans, M.A.; Pals, G.; et al. The DNA helicase BRIP1 is defective in Fanconi anemia complementation group J. Nat. Genet. 2005, 37, 934–935. [Google Scholar] [CrossRef]
- Litman, R.; Peng, M.; Jin, Z.; Zhang, F.; Zhang, J.; Powell, S.; Andreassen, P.R.; Cantor, S.B. BACH1 is critical for homologous recombination and appears to be the Fanconi anemia gene product FANCJ. Cancer Cell 2005, 8, 255–265. [Google Scholar] [CrossRef] [Green Version]
- Reid, S.; Schindler, D.; Hanenberg, H.; Barker, K.; Hanks, S.; Kalb, R.; Neveling, K.; Kelly, P.; Seal, S.; Freund, M.; et al. Biallelic mutations in PALB2 cause Fanconi anemia subtype FA-N and predispose to childhood cancer. Nat. Genet. 2007, 39, 162–164. [Google Scholar] [CrossRef]
- Vaz, F.; Hanenberg, H.; Schuster, B.; Barker, K.; Wiek, C.; Erven, V.; Neveling, K.; Endt, D.; Kesterton, I.; Autore, F.; et al. Mutation of the RAD51C gene in a Fanconi anemia-like disorder. Nat. Genet. 2010, 42, 406–409. [Google Scholar] [CrossRef]
- Wang, A.T.; Kim, T.; Wagner, J.E.; Conti, B.A.; Lach, F.P.; Huang, A.L.; Molina, H.; Sanborn, E.M.; Zierhut, H.; Cornes, B.K.; et al. A Dominant Mutation in Human RAD51 Reveals Its Function in DNA Interstrand Crosslink Repair Independent of Homologous Recombination. Mol. Cell 2015, 59, 478–490. [Google Scholar] [CrossRef] [Green Version]
- Sawyer, S.L.; Tian, L.; Kähkönen, M.; Schwartzentruber, J.; Kircher, M.; University of Washington Centre for Mendelian Genomics; FORGE Canada Consortium; Majewski, J.; Dyment, D.A.; Innes, A.M.; et al. Biallelic mutations in BRCA1 cause a new Fanconi anemia subtype. Cancer Discov. 2015, 5, 135–142. [Google Scholar] [CrossRef] [Green Version]
- Park, J.-Y.; Virts, E.L.; Jankowska, A.; Wiek, C.; Othman, M.; Chakraborty, S.C.; Vance, G.H.; Alkuraya, F.S.; Hanenberg, H.; Andreassen, P.R. Complementation of hypersensitivity to DNA interstrand crosslinking agents demonstrates that XRCC2 is a Fanconi anaemia gene. J. Med. Genet. 2016, 53, 672–680. [Google Scholar] [CrossRef] [Green Version]
- Bogliolo, M.; Schuster, B.; Stoepker, C.; Derkunt, B.; Su, Y.; Raams, A.; Trujillo, J.P.; Minguillón, J.; Ramírez, M.J.; Pujol, R.; et al. Mutations in ERCC4, encoding the DNA-repair endonuclease XPF, cause Fanconi anemia. Am. J. Hum. Genet. 2013, 92, 800–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.; Lach, F.P.; Desetty, R.; Hanenberg, H.; Auerbach, A.D.; Smogorzewska, A. Mutations of the SLX4 gene in Fanconi anemia. Nat. Genet. 2011, 43, 142–146. [Google Scholar] [CrossRef] [PubMed]
- Stoepker, C.; Hain, K.; Schuster, B.; Hilhorst-Hofstee, Y.; Rooimans, M.A.; Steltenpool, J.; Oostra, A.B.; Eirich, K.; Korthof, E.T.; Nieuwint, A.W.M.; et al. SLX4, a coordinator of structure-specific endonucleases, is mutated in a new Fanconi anemia subtype. Nat. Genet. 2011, 43, 138–141. [Google Scholar] [CrossRef] [PubMed]
- Bluteau, D.; Masliah-Planchon, J.; Clairmont, C.; Rousseau, A.; Ceccaldi, R.; d’Enghien, C.D.; Bluteau, O.; Cuccuini, W.; Gachet, S.; de Latour, R.P.; et al. Biallelic inactivation of REV7 is associated with Fanconi anemia. J. Clin. Investig. 2017, 127, 1117. [Google Scholar] [CrossRef] [Green Version]
- Knies, K.; Inano, S.; Ramírez, M.J.; Ishiai, M.; Surrallés, J.; Takata, M.; Schindler, D. Biallelic mutations in the ubiquitin ligase RFWD3 cause Fanconi anemia. J. Clin. Investig. 2017, 127, 3013–3027. [Google Scholar] [CrossRef] [Green Version]
- Cohn, M.A.; Kee, Y.; Haas, W.; Gygi, S.P.; D’Andrea, A.D. UAF1 is a subunit of multiple deubiquitinating enzyme complexes. J. Biol. Chem. 2009, 284, 5343–5351. [Google Scholar] [CrossRef] [Green Version]
- Huang, T.T.; Nijman, S.M.B.; Mirchandani, K.D.; Galardy, P.J.; Cohn, M.A.; Haas, W.; Gygi, S.P.; Ploegh, H.L.; Bernards, R.; D’Andrea, A.D. Regulation of monoubiquitinated PCNA by DUB autocleavage. Nat. Cell Biol. 2006, 8, 339–347. [Google Scholar] [CrossRef]
- Kannouche, P.L.; Wing, J.; Lehmann, A.R. Interaction of human DNA polymerase eta with monoubiquitinated PCNA: A possible mechanism for the polymerase switch in response to DNA damage. Mol. Cell 2004, 14, 491–500. [Google Scholar] [CrossRef]
- Kim, J.M.; Parmar, K.; Huang, M.; Weinstock, D.M.; Ruit, C.A.; Kutok, J.L.; D’Andrea, A.D. Inactivation of murine Usp1 results in genomic instability and a Fanconi anemia phenotype. Dev. Cell 2009, 16, 314–320. [Google Scholar] [CrossRef] [Green Version]
- Oestergaard, V.H.; Langevin, F.; Kuiken, H.J.; Pace, P.; Niedzwiedz, W.; Simpson, L.J.; Ohzeki, M.; Takata, M.; Sale, J.E.; Patel, K.J. Deubiquitination of FANCD2 is required for DNA crosslink repair. Mol. Cell 2007, 28, 798–809. [Google Scholar] [CrossRef]
- Savitsky, K.; Bar-Shira, A.; Gilad, S.; Rotman, G.; Ziv, Y.; Vanagaite, L.; Tagle, D.A.; Smith, S.; Uziel, T.; Sfez, S.; et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science 1995, 268, 1749–1753. [Google Scholar] [CrossRef] [PubMed]
- O’Driscoll, M.; Ruiz-Perez, V.L.; Woods, C.G.; Jeggo, P.A.; Goodship, J.A. A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nat. Genet. 2003, 33, 497–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreassen, P.R.; D’Andrea, A.D.; Taniguchi, T. ATR couples FANCD2 monoubiquitination to the DNA-damage response. Genes Dev. 2004, 18, 1958–1963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, M.-Y.; Dunn, C.E.; Chen, W.; Kochupurakkal, B.S.; Nguyen, H.; Moreau, L.A.; Shapiro, G.I.; Parmar, K.; Kozono, D.; D’Andrea, A.D. Cooperation of the ATM and Fanconi Anemia/BRCA Pathways in Double-Strand Break End Resection. Cell Rep. 2020, 30, 2402–2415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guervilly, J.-H.; Macé-Aimé, G.; Rosselli, F. Loss of CHK1 function impedes DNA damage-induced FANCD2 monoubiquitination but normalizes the abnormal G2 arrest in Fanconi anemia. Hum. Mol. Genet. 2008, 17, 679–689. [Google Scholar] [CrossRef]
- Pichierri, P.; Rosselli, F. The DNA crosslink-induced S-phase checkpoint depends on ATR-CHK1 and ATR-NBS1-FANCD2 pathways. EMBO J. 2004, 23, 1178–1187. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, T.; Garcia-Higuera, I.; Xu, B.; Andreassen, P.R.; Gregory, R.C.; Kim, S.-T.; Lane, W.S.; Kastan, M.B.; D’Andrea, A.D. Convergence of the fanconi anemia and ataxia telangiectasia signaling pathways. Cell 2002, 109, 459–472. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Kennedy, R.D.; Ray, K.; Stuckert, P.; Ellenberger, T.; D’Andrea, A.D. Chk1-mediated phosphorylation of FANCE is required for the Fanconi anemia/BRCA pathway. Mol. Cell. Biol. 2007, 27, 3098–3108. [Google Scholar] [CrossRef] [Green Version]
- Zhu, W.; Dutta, A. An ATR- and BRCA1-mediated Fanconi anemia pathway is required for activating the G2/M checkpoint and DNA damage repair upon rereplication. Mol. Cell. Biol. 2006, 26, 4601–4611. [Google Scholar] [CrossRef] [Green Version]
- Gatei, M.; Zhou, B.B.; Hobson, K.; Scott, S.; Young, D.; Khanna, K.K. Ataxia telangiectasia mutated (ATM) kinase and ATM and Rad3 related kinase mediate phosphorylation of Brca1 at distinct and overlapping sites. In vivo assessment using phospho-specific antibodies. J. Biol. Chem. 2001, 276, 17276–17280. [Google Scholar] [CrossRef] [Green Version]
- Stewart, G.S.; Maser, R.S.; Stankovic, T.; Bressan, D.A.; Kaplan, M.I.; Jaspers, N.G.; Raams, A.; Byrd, P.J.; Petrini, J.H.; Taylor, A.M. The DNA double-strand break repair gene hMRE11 is mutated in individuals with an ataxia-telangiectasia-like disorder. Cell 1999, 99, 577–587. [Google Scholar] [CrossRef] [Green Version]
- Varon, R.; Vissinga, C.; Platzer, M.; Cerosaletti, K.M.; Chrzanowska, K.H.; Saar, K.; Beckmann, G.; Seemanová, E.; Cooper, P.R.; Nowak, N.J.; et al. Nibrin, a novel DNA double-strand break repair protein, is mutated in Nijmegen breakage syndrome. Cell 1998, 93, 467–476. [Google Scholar] [CrossRef] [Green Version]
- Carney, J.P.; Maser, R.S.; Olivares, H.; Davis, E.M.; Le Beau, M.; Yates, J.R.; Hays, L.; Morgan, W.F.; Petrini, J.H. The hMre11/hRad50 protein complex and Nijmegen breakage syndrome: Linkage of double-strand break repair to the cellular DNA damage response. Cell 1998, 93, 477–486. [Google Scholar] [CrossRef] [Green Version]
- Matsuura, S.; Tauchi, H.; Nakamura, A.; Kondo, N.; Sakamoto, S.; Endo, S.; Smeets, D.; Solder, B.; Belohradsky, B.H.; Der Kaloustian, V.M.; et al. Positional cloning of the gene for Nijmegen breakage syndrome. Nat. Genet. 1998, 19, 179–181. [Google Scholar] [CrossRef] [PubMed]
- Uziel, T.; Lerenthal, Y.; Moyal, L.; Andegeko, Y.; Mittelman, L.; Shiloh, Y. Requirement of the MRN complex for ATM activation by DNA damage. EMBO J. 2003, 22, 5612–5621. [Google Scholar] [CrossRef] [Green Version]
- Van den Bosch, M.; Bree, R.T.; Lowndes, N.F. The MRN complex: Coordinating and mediating the response to broken chromosomes. EMBO Rep. 2003, 4, 844–849. [Google Scholar] [CrossRef] [Green Version]
- Chang, E.Y.-C.; Tsai, S.; Aristizabal, M.J.; Wells, J.P.; Coulombe, Y.; Busatto, F.F.; Chan, Y.A.; Kumar, A.; Dan Zhu, Y.; Wang, A.Y.-H.; et al. MRE11-RAD50-NBS1 promotes Fanconi Anemia R-loop suppression at transcription-replication conflicts. Nat. Commun. 2019, 10, 4265. [Google Scholar] [CrossRef]
- Donahue, S.L.; Campbell, C. A Rad50-dependent pathway of DNA repair is deficient in Fanconi anemia fibroblasts. Nucleic Acids Res. 2004, 32, 3248–3257. [Google Scholar] [CrossRef] [Green Version]
- Nakanishi, K.; Taniguchi, T.; Ranganathan, V.; New, H.V.; Moreau, L.A.; Stotsky, M.; Mathew, C.G.; Kastan, M.B.; Weaver, D.T.; D’Andrea, A.D. Interaction of FANCD2 and NBS1 in the DNA damage response. Nat. Cell Biol. 2002, 4, 913–920. [Google Scholar] [CrossRef]
- Pichierri, P.; Averbeck, D.; Rosselli, F. DNA cross-link-dependent RAD50/MRE11/NBS1 subnuclear assembly requires the Fanconi anemia C protein. Hum. Mol. Genet. 2002, 11, 2531–2546. [Google Scholar] [CrossRef] [Green Version]
- Hirano, S.; Yamamoto, K.; Ishiai, M.; Yamazoe, M.; Seki, M.; Matsushita, N.; Ohzeki, M.; Yamashita, Y.M.; Arakawa, H.; Buerstedde, J.-M.; et al. Functional relationships of FANCC to homologous recombination, translesion synthesis, and BLM. EMBO J. 2005, 24, 418–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meetei, A.R.; Sechi, S.; Wallisch, M.; Yang, D.; Young, M.K.; Joenje, H.; Hoatlin, M.E.; Wang, W. A multiprotein nuclear complex connects Fanconi anemia and Bloom syndrome. Mol. Cell. Biol. 2003, 23, 3417–3426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ralf, C.; Hickson, I.D.; Wu, L. The Bloom’s syndrome helicase can promote the regression of a model replication fork. J. Biol. Chem. 2006, 281, 22839–22846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weaver, G.M.; Mettrick, K.A.; Corocher, T.-A.; Graham, A.; Grainge, I. Replication fork collapse at a protein-DNA roadblock leads to fork reversal, promoted by the RecQ helicase. Mol. Microbiol. 2019, 111, 455–472. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.-L.; North, P.S.; Hickson, I.D. BLM is required for faithful chromosome segregation and its localization defines a class of ultrafine anaphase bridges. EMBO J. 2007, 26, 3397–3409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naim, V.; Rosselli, F. The FANC pathway and BLM collaborate during mitosis to prevent micro-nucleation and chromosome abnormalities. Nat. Cell Biol. 2009, 11, 761–768. [Google Scholar] [CrossRef]
- Tang, J.; Cho, N.W.; Cui, G.; Manion, E.M.; Shanbhag, N.M.; Botuyan, M.V.; Mer, G.; Greenberg, R.A. Acetylation limits 53BP1 association with damaged chromatin to promote homologous recombination. Nat. Struct. Mol. Biol. 2013, 20, 317–325. [Google Scholar] [CrossRef]
- Chailleux, C.; Tyteca, S.; Papin, C.; Boudsocq, F.; Puget, N.; Courilleau, C.; Grigoriev, M.; Canitrot, Y.; Trouche, D. Physical interaction between the histone acetyl transferase Tip60 and the DNA double-strand breaks sensor MRN complex. Biochem. J. 2010, 426, 365–371. [Google Scholar] [CrossRef] [Green Version]
- Ortega-Atienza, S.; Wong, V.C.; DeLoughery, Z.; Luczak, M.W.; Zhitkovich, A. ATM and KAT5 safeguard replicating chromatin against formaldehyde damage. Nucleic Acids Res. 2016, 44, 198–209. [Google Scholar] [CrossRef] [Green Version]
- Hejna, J.; Holtorf, M.; Hines, J.; Mathewson, L.; Hemphill, A.; Al-Dhalimy, M.; Olson, S.B.; Moses, R.E. Tip60 is required for DNA interstrand cross-link repair in the Fanconi anemia pathway. J. Biol. Chem. 2008, 283, 9844–9851. [Google Scholar] [CrossRef] [Green Version]
- Renaud, E.; Barascu, A.; Rosselli, F. Impaired TIP60-mediated H4K16 acetylation accounts for the aberrant chromatin accumulation of 53BP1 and RAP80 in Fanconi anemia pathway-deficient cells. Nucleic Acids Res. 2016, 44, 648–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couvé, S.; Macé-Aimé, G.; Rosselli, F.; Saparbaev, M.K. The human oxidative DNA glycosylase NEIL1 excises psoralen-induced interstrand DNA cross-links in a three-stranded DNA structure. J. Biol. Chem. 2009, 284, 11963–11970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, N.; Wang, J.; Wallace, S.S.; Chen, J.; Zhou, J.; D’Andrea, A.D. Cooperation of the NEIL3 and Fanconi anemia/BRCA pathways in interstrand crosslink repair. Nucleic Acids Res. 2020, 48, 3014–3028. [Google Scholar] [CrossRef] [PubMed]
- Macé-Aimé, G.; Couvé, S.; Khassenov, B.; Rosselli, F.; Saparbaev, M.K. The Fanconi anemia pathway promotes DNA glycosylase-dependent excision of interstrand DNA crosslinks. Environ. Mol. Mutagen. 2010, 51, 508–519. [Google Scholar] [CrossRef] [PubMed]
- Martin, P.R.; Couvé, S.; Zutterling, C.; Albelazi, M.S.; Groisman, R.; Matkarimov, B.T.; Parsons, J.L.; Elder, R.H.; Saparbaev, M.K. The Human DNA glycosylases NEIL1 and NEIL3 Excise Psoralen-Induced DNA-DNA Cross-Links in a Four-Stranded DNA Structure. Sci. Rep. 2017, 7, 17438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semlow, D.R.; Zhang, J.; Budzowska, M.; Drohat, A.C.; Walter, J.C. Replication-Dependent Unhooking of DNA Interstrand Cross-Links by the NEIL3 Glycosylase. Cell 2016, 167, 498–511. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Long, D.T.; Lee, K.Y.; Abbas, T.; Shibata, E.; Negishi, M.; Luo, Y.; Schimenti, J.C.; Gambus, A.; Walter, J.C.; et al. The MCM8-MCM9 complex promotes RAD51 recruitment at DNA damage sites to facilitate homologous recombination. Mol. Cell. Biol. 2013, 33, 1632–1644. [Google Scholar] [CrossRef]
- Nishimura, K.; Ishiai, M.; Horikawa, K.; Fukagawa, T.; Takata, M.; Takisawa, H.; Kanemaki, M.T. Mcm8 and Mcm9 form a complex that functions in homologous recombination repair induced by DNA interstrand crosslinks. Mol. Cell 2012, 47, 511–522. [Google Scholar] [CrossRef] [Green Version]
- Räschle, M.; Smeenk, G.; Hansen, R.K.; Temu, T.; Oka, Y.; Hein, M.Y.; Nagaraj, N.; Long, D.T.; Walter, J.C.; Hofmann, K.; et al. DNA repair. Proteomics reveals dynamic assembly of repair complexes during bypass of DNA cross-links. Science 2015, 348, 1253671. [Google Scholar] [CrossRef] [Green Version]
- Rossi, F.; Helbling-Leclerc, A.; Kawasumi, R.; Jegadesan, N.K.; Xu, X.; Devulder, P.; Abe, T.; Takata, M.; Xu, D.; Rosselli, F.; et al. SMC5/6 acts jointly with Fanconi anemia factors to support DNA repair and genome stability. EMBO Rep. 2020, 21, e48222. [Google Scholar] [CrossRef]
- Papadopoulo, D.; Guillouf, C.; Mohrenweiser, H.; Moustacchi, E. Hypomutability in Fanconi anemia cells is associated with increased deletion frequency at the HPRT locus. Proc. Natl. Acad. Sci. USA 1990, 87, 8383–8387. [Google Scholar] [CrossRef] [Green Version]
- Mirchandani, K.D.; McCaffrey, R.M.; D’Andrea, A.D. The Fanconi anemia core complex is required for efficient point mutagenesis and Rev1 foci assembly. DNA Repair 2008, 7, 902–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosselli, F.; Duchaud, E.; Averbeck, D.; Moustacchi, E. Comparison of the effects of DNA topoisomerase inhibitors on lymphoblasts from normal and Fanconi anemia donors. Mutat. Res. 1994, 325, 137–144. [Google Scholar] [CrossRef]
- Rothfuss, A.; Grompe, M. Repair kinetics of genomic interstrand DNA cross-links: Evidence for DNA double-strand break-dependent activation of the Fanconi anemia/BRCA pathway. Mol. Cell. Biol. 2004, 24, 123–134. [Google Scholar] [CrossRef] [Green Version]
- Zou, X.; Owusu, M.; Harris, R.; Jackson, S.P.; Loizou, J.I.; Nik-Zainal, S. Validating the concept of mutational signatures with isogenic cell models. Nat. Commun. 2018, 9, 1744. [Google Scholar] [CrossRef] [Green Version]
- Guervilly, J.-H.; Renaud, E.; Takata, M.; Rosselli, F. USP1 deubiquitinase maintains phosphorylated CHK1 by limiting its DDB1-dependent degradation. Hum. Mol. Genet. 2011, 20, 2171–2181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, R.D.; Chen, C.C.; Stuckert, P.; Archila, E.M.; De la Vega, M.A.; Moreau, L.A.; Shimamura, A.; D’Andrea, A.D. Fanconi anemia pathway-deficient tumor cells are hypersensitive to inhibition of ataxia telangiectasia mutated. J. Clin. Investig. 2007, 117, 1440–1449. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, K.; Nihrane, A.; Aglipay, J.; Sironi, J.; Arkin, S.; Lipton, J.M.; Ouchi, T.; Liu, J.M. Upregulated ATM gene expression and activated DNA crosslink-induced damage response checkpoint in Fanconi anemia: Implications for carcinogenesis. Mol. Med. 2008, 14, 167–174. [Google Scholar] [CrossRef]
- Heinrich, M.C.; Hoatlin, M.E.; Zigler, A.J.; Silvey, K.V.; Bakke, A.C.; Keeble, W.W.; Zhi, Y.; Reifsteck, C.A.; Grompe, M.; Brown, M.G.; et al. DNA cross-linker-induced G2/M arrest in group C Fanconi anemia lymphoblasts reflects normal checkpoint function. Blood 1998, 91, 275–287. [Google Scholar] [CrossRef]
- Collins, N.B.; Wilson, J.B.; Bush, T.; Thomashevski, A.; Roberts, K.J.; Jones, N.J.; Kupfer, G.M. ATR-dependent phosphorylation of FANCA on serine 1449 after DNA damage is important for FA pathway function. Blood 2009, 113, 2181–2190. [Google Scholar] [CrossRef] [Green Version]
- Ho, G.P.H.; Margossian, S.; Taniguchi, T.; D’Andrea, A.D. Phosphorylation of FANCD2 on two novel sites is required for mitomycin C resistance. Mol. Cell. Biol. 2006, 26, 7005–7015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, T.R.; Ali, A.M.; Paramasivam, M.; Pradhan, A.; Wahengbam, K.; Seidman, M.M.; Meetei, A.R. ATR-dependent phosphorylation of FANCM at serine 1045 is essential for FANCM functions. Cancer Res. 2013, 73, 4300–4310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niedernhofer, L.J.; Lalai, A.S.; Hoeijmakers, J.H.J. Fanconi anemia (cross)linked to DNA repair. Cell 2005, 123, 1191–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knipscheer, P.; Räschle, M.; Schärer, O.D.; Walter, J.C. Replication-coupled DNA interstrand cross-link repair in Xenopus egg extracts. Methods Mol. Biol. Clifton NJ 2012, 920, 221–243. [Google Scholar]
- Räschle, M.; Knipscheer, P.; Knipsheer, P.; Enoiu, M.; Angelov, T.; Sun, J.; Griffith, J.D.; Ellenberger, T.E.; Schärer, O.D.; Walter, J.C. Mechanism of replication-coupled DNA interstrand crosslink repair. Cell 2008, 134, 969–980. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Liu, S.; Bellani, M.A.; Thazhathveetil, A.K.; Ling, C.; de Winter, J.P.; Wang, Y.; Wang, W.; Seidman, M.M. The DNA translocase FANCM/MHF promotes replication traverse of DNA interstrand crosslinks. Mol. Cell 2013, 52, 434–446. [Google Scholar] [CrossRef] [Green Version]
- Bensimon, A.; Simon, A.; Chiffaudel, A.; Croquette, V.; Heslot, F.; Bensimon, D. Alignment and sensitive detection of DNA by a moving interface. Science 1994, 265, 2096–2098. [Google Scholar] [CrossRef]
- Machida, Y.J.; Machida, Y.; Chen, Y.; Gurtan, A.M.; Kupfer, G.M.; D’Andrea, A.D.; Dutta, A. UBE2T is the E2 in the Fanconi anemia pathway and undergoes negative autoregulation. Mol. Cell 2006, 23, 589–596. [Google Scholar] [CrossRef]
- Huang, M.; Kennedy, R.; Ali, A.M.; Moreau, L.A.; Meetei, A.R.; D’Andrea, A.D.; Chen, C.C. Human MutS and FANCM complexes function as redundant DNA damage sensors in the Fanconi Anemia pathway. DNA Repair 2011, 10, 1203–1212. [Google Scholar] [CrossRef]
- Hanada, K.; Budzowska, M.; Modesti, M.; Maas, A.; Wyman, C.; Essers, J.; Kanaar, R. The structure-specific endonuclease Mus81-Eme1 promotes conversion of interstrand DNA crosslinks into double-strands breaks. EMBO J. 2006, 25, 4921–4932. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Ghosal, G.; Yuan, J.; Chen, J.; Huang, J. FAN1 acts with FANCI-FANCD2 to promote DNA interstrand cross-link repair. Science 2010, 329, 693–696. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, I.M.; Hain, K.; Déclais, A.-C.; Gardiner, M.; Toh, G.W.; Sanchez-Pulido, L.; Heuckmann, J.M.; Toth, R.; Macartney, T.; Eppink, B.; et al. Coordination of structure-specific nucleases by human SLX4/BTBD12 is required for DNA repair. Mol. Cell 2009, 35, 116–127. [Google Scholar] [CrossRef]
- Smogorzewska, A.; Desetty, R.; Saito, T.T.; Schlabach, M.; Lach, F.P.; Sowa, M.E.; Clark, A.B.; Kunkel, T.A.; Harper, J.W.; Colaiácovo, M.P.; et al. A genetic screen identifies FAN1, a Fanconi anemia-associated nuclease necessary for DNA interstrand crosslink repair. Mol. Cell 2010, 39, 36–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Walter, J.C. Mechanism and regulation of incisions during DNA interstrand cross-link repair. DNA Repair 2014, 19, 135–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hicks, J.K.; Chute, C.L.; Paulsen, M.T.; Ragland, R.L.; Howlett, N.G.; Guéranger, Q.; Glover, T.W.; Canman, C.E. Differential roles for DNA polymerases eta, zeta, and REV1 in lesion bypass of intrastrand versus interstrand DNA cross-links. Mol. Cell. Biol. 2010, 30, 1217–1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meetei, A.R.; de Winter, J.P.; Medhurst, A.L.; Wallisch, M.; Waisfisz, Q.; van de Vrugt, H.J.; Oostra, A.B.; Yan, Z.; Ling, C.; Bishop, C.E.; et al. A novel ubiquitin ligase is deficient in Fanconi anemia. Nat. Genet. 2003, 35, 165–170. [Google Scholar] [CrossRef]
- Hoege, C.; Pfander, B.; Moldovan, G.-L.; Pyrowolakis, G.; Jentsch, S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 2002, 419, 135–141. [Google Scholar] [CrossRef]
- Niedernhofer, L.J.; Odijk, H.; Budzowska, M.; van Drunen, E.; Maas, A.; Theil, A.F.; de Wit, J.; Jaspers, N.G.J.; Beverloo, H.B.; Hoeijmakers, J.H.J.; et al. The structure-specific endonuclease Ercc1-Xpf is required to resolve DNA interstrand cross-link-induced double-strand breaks. Mol. Cell. Biol. 2004, 24, 5776–5787. [Google Scholar] [CrossRef] [Green Version]
- Kinoshita, E.; van der Linden, E.; Sanchez, H.; Wyman, C. RAD50, an SMC family member with multiple roles in DNA break repair: How does ATP affect function? Chromosome Res. 2009, 17, 277–288. [Google Scholar] [CrossRef] [Green Version]
- Roques, C.; Coulombe, Y.; Delannoy, M.; Vignard, J.; Grossi, S.; Brodeur, I.; Rodrigue, A.; Gautier, J.; Stasiak, A.Z.; Stasiak, A.; et al. MRE11-RAD50-NBS1 is a critical regulator of FANCD2 stability and function during DNA double-strand break repair. EMBO J. 2009, 28, 2400–2413. [Google Scholar] [CrossRef] [Green Version]
- Bártová, E.; Legartová, S.; Dundr, M.; Suchánková, J. A role of the 53BP1 protein in genome protection: Structural and functional characteristics of 53BP1-dependent DNA repair. Aging 2019, 11, 2488–2511. [Google Scholar] [CrossRef] [PubMed]
- Adamo, A.; Collis, S.J.; Adelman, C.A.; Silva, N.; Horejsi, Z.; Ward, J.D.; Martinez-Perez, E.; Boulton, S.J.; La Volpe, A. Preventing nonhomologous end joining suppresses DNA repair defects of Fanconi anemia. Mol. Cell 2010, 39, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Pace, P.; Mosedale, G.; Hodskinson, M.R.; Rosado, I.V.; Sivasubramaniam, M.; Patel, K.J. Ku70 corrupts DNA repair in the absence of the Fanconi anemia pathway. Science 2010, 329, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.L.; Palmai-Pallag, T.; Ying, S.; Hickson, I.D. Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat. Cell Biol. 2009, 11, 753–760. [Google Scholar] [CrossRef]
- Lordier, L.; Pan, J.; Naim, V.; Jalil, A.; Badirou, I.; Rameau, P.; Larghero, J.; Debili, N.; Rosselli, F.; Vainchenker, W.; et al. Presence of a defect in karyokinesis during megakaryocyte endomitosis. Cell Cycle 2012, 11, 4385–4389. [Google Scholar] [CrossRef]
- Naim, V.; Rosselli, F. The FANC pathway and mitosis: A replication legacy. Cell Cycle 2009, 8, 2907–2911. [Google Scholar] [CrossRef]
- Naim, V.; Wilhelm, T.; Debatisse, M.; Rosselli, F. ERCC1 and MUS81-EME1 promote sister chromatid separation by processing late replication intermediates at common fragile sites during mitosis. Nat. Cell Biol. 2013, 15, 1008–1015. [Google Scholar] [CrossRef]
- Pawlikowska, P.; Fouchet, P.; Vainchenker, W.; Rosselli, F.; Naim, V. Defective endomitosis during megakaryopoiesis leads to thrombocytopenia in Fanca-/- mice. Blood 2014, 124, 3613–3623. [Google Scholar] [CrossRef] [Green Version]
- Knipscheer, P.; Räschle, M.; Smogorzewska, A.; Enoiu, M.; Ho, T.V.; Schärer, O.D.; Elledge, S.J.; Walter, J.C. The Fanconi anemia pathway promotes replication-dependent DNA interstrand cross-link repair. Science 2009, 326, 1698–1701. [Google Scholar] [CrossRef] [Green Version]
- Sparks, J.; Walter, J.C. Extracts for Analysis of DNA Replication in a Nucleus-Free System. Cold Spring Harb. Protoc. 2019, 2019. [Google Scholar] [CrossRef]
- Zhang, J.; Dewar, J.M.; Budzowska, M.; Motnenko, A.; Cohn, M.A.; Walter, J.C. DNA interstrand cross-link repair requires replication-fork convergence. Nat. Struct. Mol. Biol. 2015, 22, 242–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodskinson, M.R.; Bolner, A.; Sato, K.; Kamimae-Lanning, A.N.; Rooijers, K.; Witte, M.; Mahesh, M.; Silhan, J.; Petek, M.; Williams, D.M.; et al. Alcohol-derived DNA crosslinks are repaired by two distinct mechanisms. Nature 2020, 579, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Yang, K.; Dejsuphong, D.; D’Andrea, A.D. Regulation of Rev1 by the Fanconi anemia core complex. Nat. Struct. Mol. Biol. 2012, 19, 164–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renaud, E.; Rosselli, F. FANC pathway promotes UV-induced stalled replication forks recovery by acting both upstream and downstream Polη and Rev1. PLoS ONE 2013, 8, e53693. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, Z.; Wang, F.; Temviriyanukul, P.; Ma, X.; Tu, Y.; Lv, L.; Lin, Y.-F.; Huang, M.; Zhang, T.; et al. FANCD2 and REV1 cooperate in the protection of nascent DNA strands in response to replication stress. Nucleic Acids Res. 2015, 43, 8325–8339. [Google Scholar] [CrossRef] [Green Version]
- Jayabal, P.; Ma, C.; Nepal, M.; Shen, Y.; Che, R.; Turkson, J.; Fei, P. Involvement of FANCD2 in Energy Metabolism via ATP5α. Sci. Rep. 2017, 7, 4921. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.S.; Leung, K.S.; Hicks, M.J.; Hastings, P.J.; Youssoufian, H.; Plon, S.E. Defective mitochondrial peroxiredoxin-3 results in sensitivity to oxidative stress in Fanconi anemia. J. Cell Biol. 2006, 175, 225–235. [Google Scholar] [CrossRef] [Green Version]
- Cappelli, E.; Ravera, S.; Vaccaro, D.; Cuccarolo, P.; Bartolucci, M.; Panfoli, I.; Dufour, C.; Degan, P. Mitochondrial respiratory complex I defects in Fanconi anemia. Trends Mol. Med. 2013, 19, 513–514. [Google Scholar] [CrossRef]
- Kumari, U.; Ya Jun, W.; Huat Bay, B.; Lyakhovich, A. Evidence of mitochondrial dysfunction and impaired ROS detoxifying machinery in Fanconi anemia cells. Oncogene 2014, 33, 165–172. [Google Scholar] [CrossRef] [Green Version]
- Rousset, S.; Nocentini, S.; Rouillard, D.; Baroche, C.; Moustacchi, E. Mitochondrial alterations in fanconi anemia fibroblasts following ultraviolet A or psoralen photoactivation. Photochem. Photobiol. 2002, 75, 159–166. [Google Scholar] [CrossRef]
- Solanki, A.; Rajendran, A.; Mohan, S.; Raj, R.; Vundinti, B.R. Mitochondrial DNA variations and mitochondrial dysfunction in Fanconi anemia. PLoS ONE 2020, 15, e0227603. [Google Scholar] [CrossRef] [PubMed]
- Falkenberg, M. Mitochondrial DNA replication in mammalian cells: Overview of the pathway. Essays Biochem. 2018, 62, 287–296. [Google Scholar] [PubMed]
- Alam, R.; Thazhathveetil, A.K.; Li, H.; Seidman, M.M. Preparation and application of triple helix forming oligonucleotides and single strand oligonucleotide donors for gene correction. Methods Mol. Biol. Clifton NJ 2014, 1114, 103–113. [Google Scholar]
- Huang, J.; Gali, H.; Paramasivam, M.; Muniandy, P.; Gichimu, J.; Bellani, M.A.; Seidman, M.M. Single Molecule Analysis of Laser Localized Interstrand Crosslinks. Front. Genet. 2016, 7, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thazhathveetil, A.K.; Liu, S.-T.; Indig, F.E.; Seidman, M.M. Psoralen conjugates for visualization of genomic interstrand cross-links localized by laser photoactivation. Bioconjug. Chem. 2007, 18, 431–437. [Google Scholar] [CrossRef]
- Ling, C.; Huang, J.; Yan, Z.; Li, Y.; Ohzeki, M.; Ishiai, M.; Xu, D.; Takata, M.; Seidman, M.; Wang, W. Bloom syndrome complex promotes FANCM recruitment to stalled replication forks and facilitates both repair and traverse of DNA interstrand crosslinks. Cell Discov. 2016, 2, 16047. [Google Scholar] [CrossRef]
- Rohleder, F.; Huang, J.; Xue, Y.; Kuper, J.; Round, A.; Seidman, M.; Wang, W.; Kisker, C. FANCM interacts with PCNA to promote replication traverse of DNA interstrand crosslinks. Nucleic Acids Res. 2016, 44, 3219–3232. [Google Scholar] [CrossRef] [Green Version]
- Gari, K.; Décaillet, C.; Delannoy, M.; Wu, L.; Constantinou, A. Remodeling of DNA replication structures by the branch point translocase FANCM. Proc. Natl. Acad. Sci. USA 2008, 105, 16107–16112. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Zhang, J.; Bellani, M.A.; Pokharel, D.; Gichimu, J.; James, R.C.; Gali, H.; Ling, C.; Yan, Z.; Xu, D.; et al. Remodeling of Interstrand Crosslink Proximal Replisomes Is Dependent on ATR, FANCM, and FANCD2. Cell Rep. 2019, 27, 1794–1808. [Google Scholar] [CrossRef] [Green Version]
- Guillouf, C.; Laquerbe, A.; Moustacchi, E.; Papadopoulo, D. Mutagenic processing of psoralen monoadducts differ in normal and Fanconi anemia cells. Mutagenesis 1993, 8, 355–361. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Renaudin, X.; Rosselli, F. The FANC/BRCA Pathway Releases Replication Blockades by Eliminating DNA Interstrand Cross-Links. Genes 2020, 11, 585. https://doi.org/10.3390/genes11050585
Renaudin X, Rosselli F. The FANC/BRCA Pathway Releases Replication Blockades by Eliminating DNA Interstrand Cross-Links. Genes. 2020; 11(5):585. https://doi.org/10.3390/genes11050585
Chicago/Turabian StyleRenaudin, Xavier, and Filippo Rosselli. 2020. "The FANC/BRCA Pathway Releases Replication Blockades by Eliminating DNA Interstrand Cross-Links" Genes 11, no. 5: 585. https://doi.org/10.3390/genes11050585
APA StyleRenaudin, X., & Rosselli, F. (2020). The FANC/BRCA Pathway Releases Replication Blockades by Eliminating DNA Interstrand Cross-Links. Genes, 11(5), 585. https://doi.org/10.3390/genes11050585