Location, Location, Location: The Role of Nuclear Positioning in the Repair of Collapsed Forks and Protection of Genome Stability


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Replication Stress and the Nuclear Pore Complex (NPC)
3. Links between Replication Stress and the NPC: Sumoylation
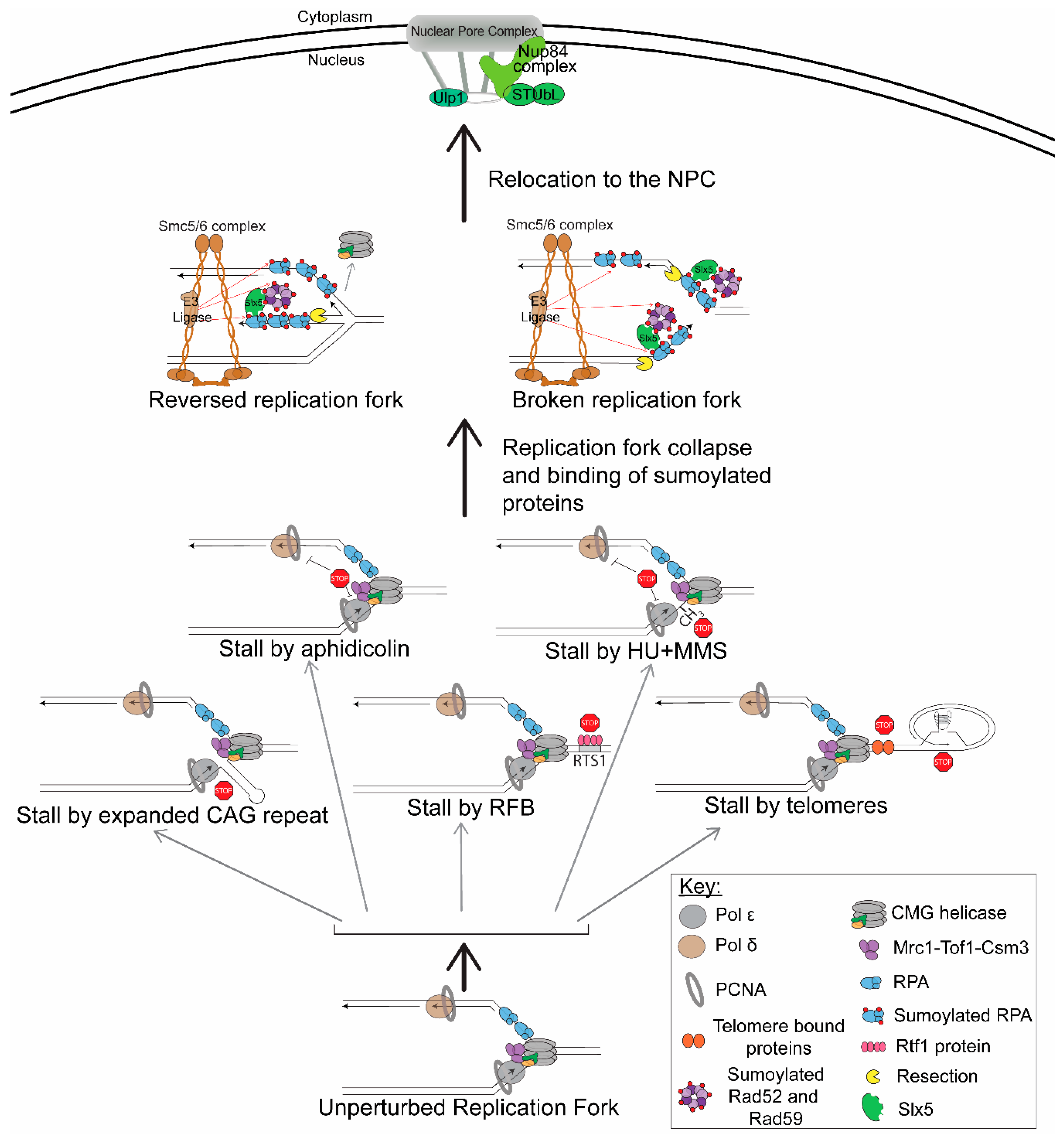
4. Types of Barriers that Inhibit Replication and Relocate to the NPC
5. Sumoylation Requirements for Collapsed Fork Relocation to the NPC
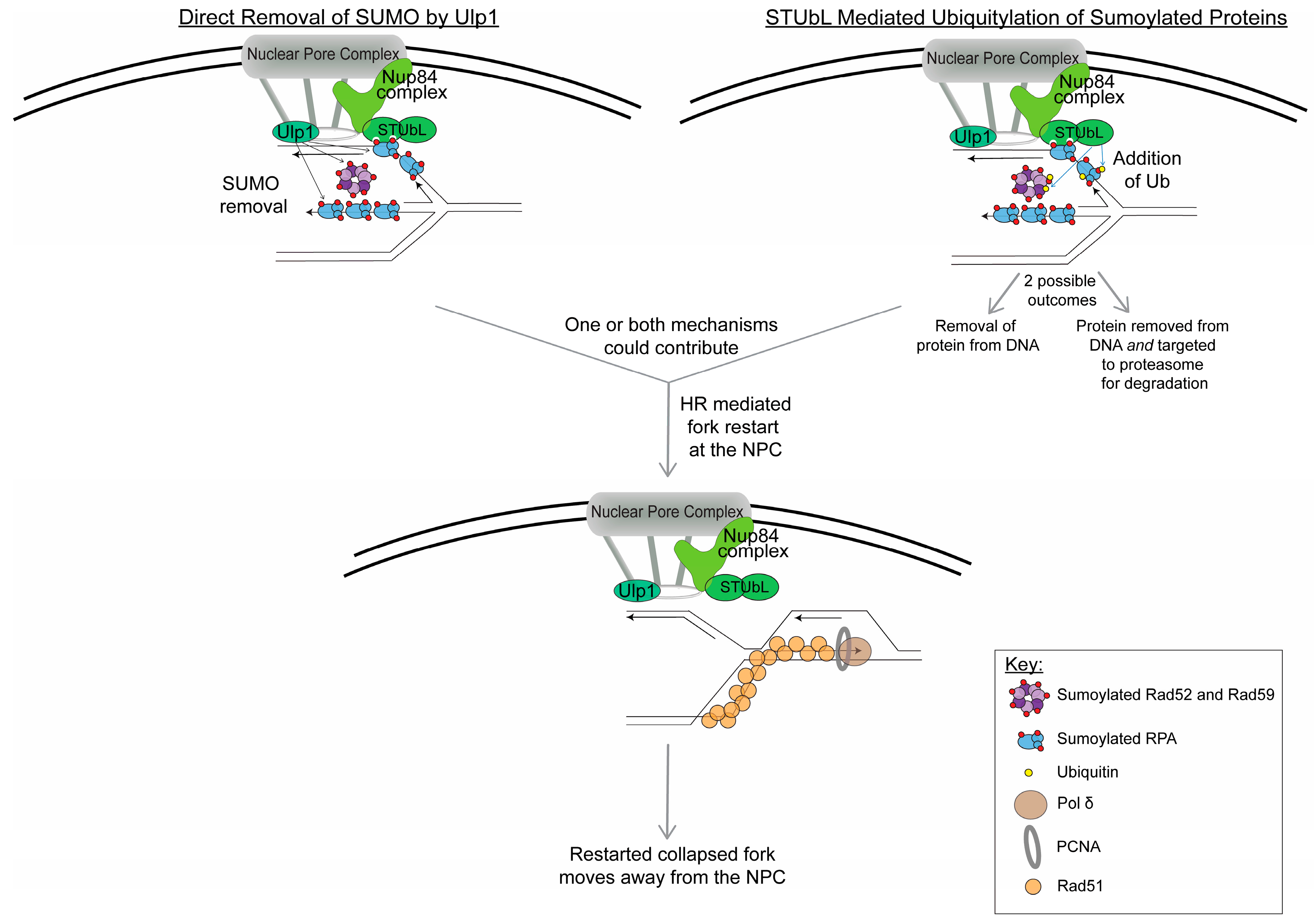
6. How Does Sumoylation Mediate the Movement of Collapsed Forks to the NPC?
7. Additional Requirements for Collapsed Fork Relocation to the NPC
8. Why Do Collapsed Replication Forks Go to the NPC?
9. Gaps in Knowledge
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yeeles, J.T.P.; Janska, A.; Early, A.; Diffley, J.F.X. How the Eukaryotic Replisome Achieves Rapid and Efficient DNA Replication. Mol. Cell 2017, 65, 105–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirkin, E.V.; Mirkin, S.M. Replication Fork Stalling at Natural Impediments. Microbiol. Mol. Biol. Rev. 2007, 71, 13–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, S.; Carr, A.M. Impediments to replication fork movement: Stabilisation, reactivation and genome instability. Chromosoma 2013, 122, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Petermann, E.; Helleday, T. Pathways of mammalian replication fork restart. Nat. Rev. Mol. Cell Biol. 2010, 11, 683–687. [Google Scholar] [CrossRef] [PubMed]
- Woodward, A.M.; Göhler, T.; Luciani, M.G.; Oehlmann, M.; Ge, X.; Gartner, A.; Jackson, D.A.; Blow, J.J. Excess Mcm2-7 license dormant origins of replication that can be used under conditions of replicative stress. J. Cell Biol. 2006, 173, 673–683. [Google Scholar] [CrossRef] [Green Version]
- Ge, X.Q.; Jackson, D.A.; Blow, J.J. Dormant origins licensed by excess Mcm2-7 are required for human cells to survive replicative stress. Genes Dev. 2007, 21, 3331–3341. [Google Scholar] [CrossRef] [Green Version]
- McIntosh, D.; Blow, J.J. Dormant origins, the licensing checkpoint, and the response to replicative stresses. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef]
- Lopes, M.; Foiani, M.; Sogo, J.M. Multiple mechanisms control chromosome integrity after replication fork uncoupling and restart at irreparable UV lesions. Mol. Cell 2006, 21, 15–27. [Google Scholar] [CrossRef]
- Elvers, I.; Johansson, F.; Groth, P.; Erixon, K.; Helleday, T. UV stalled replication forks restart by re-priming in human fibroblasts. Nucleic Acids Res. 2011, 39, 7049–7057. [Google Scholar] [CrossRef] [Green Version]
- Ait Saada, A.; Lambert, S.A.E.; Carr, A.M. Preserving replication fork integrity and competence via the homologous recombination pathway. DNA Repair 2018, 71, 135–147. [Google Scholar] [CrossRef]
- Neelsen, K.J.; Lopes, M. Replication fork reversal in eukaryotes: From dead end to dynamic response. Nat. Rev. Mol. Cell Biol. 2015, 16, 207–220. [Google Scholar] [CrossRef]
- Lopes, M.; Cotta-Ramusino, C.; Pellicoli, A.; Liberi, G.; Plevani, P.; Muzi-Falconi, M.; Newton, C.S.; Foiani, M. The DNA replication checkpoint response stabilizes stalled replication forks. Nature 2001, 412, 557–561. [Google Scholar] [CrossRef]
- Cobb, J.A.; Bjergbaek, L.; Shimada, K.; Frei, C.; Gasser, S.M. DNA polymerase stabilization at stalled replication forks requires Mec1 and the RecQ helicase Sgs1. EMBO J. 2003, 22, 4325–4336. [Google Scholar] [CrossRef] [Green Version]
- Cobb, J.A.; Schleker, T.; Rojas, V.; Bjergbaek, L.; Tercero, J.A.; Gasser, S.M. Replisome instability, fork collapse, and gross chromosomal rearrangements arise synergistically from Mec1 kinase and RecQ helicase mutations. Genes Dev. 2005, 19, 3055–3069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [Green Version]
- De Piccoli, G.; Katou, Y.; Itoh, T.; Nakato, R.; Shirahige, K.; Labib, K. Replisome Stability at Defective DNA Replication Forks Is Independent of S Phase Checkpoint Kinases. Mol. Cell 2012, 45, 696–704. [Google Scholar] [CrossRef] [Green Version]
- Cortez, D. Preventing replication fork collapse to maintain genome integrity. DNA Repair 2015, 32, 149–157. [Google Scholar] [CrossRef] [Green Version]
- Beck, M.; Hurt, E. The nuclear pore complex: Understanding its function through structural insight. Nat. Rev. Mol. Cell Biol. 2017, 18, 73–89. [Google Scholar] [CrossRef]
- Schwartz, T.U. The Structure Inventory of the Nuclear Pore Complex. J. Mol. Biol. 2016, 428, 1986–2000. [Google Scholar] [CrossRef] [Green Version]
- Bai, S.W.; Rouquette, J.; Umeda, M.; Faigle, W.; Loew, D.; Sazer, S.; Doye, V. The Fission Yeast Nup107-120 Complex Functionally Interacts with the Small GTPase Ran/Spi1 and Is Required for mRNA Export, Nuclear Pore Distribution, and Proper Cell Division. Mol. Cell. Biol. 2004, 24, 6379–6392. [Google Scholar] [CrossRef] [Green Version]
- Chang, M.; Bellaoui, M.; Boone, C.; Brown, G.W. A genome-wide screen for methyl methanesulfonate-sensitive mutants reveals genes required for S phase progression in the presence of DNA damage. Proc. Natl. Acad. Sci. USA 2002, 99, 16934–16939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Rodríguez, N.; Wong, R.P.; Ulrich, H.D. Functions of ubiquitin and SUMO in DNA replication and replication stress. Front. Genet. 2016, 7, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaillard, H.; Santos-Pereira, J.M.; Aguilera, A. The Nup84 complex coordinates the DNA damage response to warrant genome integrity. Nucleic Acids Res. 2019, 47, 4054–4067. [Google Scholar] [CrossRef] [PubMed]
- Olivieri, M.; Cho, T.; Alvarez-Quilon, A.; Li, K.; Schellenberg, M.J.; Zimmermann, M.; Hustedt, N.; Rossi, S.E.; Adam, A.; Melo, H.; et al. A genetic map of the response to DNA damage in human cells. BioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- Loeillet, S.; Palancade, B.; Cartron, M.; Thierry, A.; Richard, G.F.; Dujon, B.; Doye, V.; Nicolas, A. Genetic network interactions among replication, repair and nuclear pore deficiencies in yeast. DNA Repair 2005, 4, 459–468. [Google Scholar] [CrossRef]
- Paulsen, R.D.; Soni, D.V.; Wollman, R.; Hahn, A.T.; Yee, M.C.; Guan, A.; Hesley, J.A.; Miller, S.C.; Cromwell, E.F.; Solow-Cordero, D.E.; et al. A Genome-wide siRNA Screen Reveals Diverse Cellular Processes and Pathways that Mediate Genome Stability. Mol. Cell 2009, 35, 228–239. [Google Scholar] [CrossRef] [Green Version]
- Hochstrasser, M. Minireview SP-RING for SUMO: New Functions Bloom for a Ubiquitin-like Protein. Cell 2001, 107, 5–8. [Google Scholar] [CrossRef] [Green Version]
- Watts, F.Z.; Skilton, A.; Ho, Y.; Boyd, L.K.; Trickey, M.A.M.; Gardner, L.; Ogi, F.X.; Outwin, E.A. The role of Schizosaccharomyces pombe SUMO ligases in genome stability. Biochem. Soc. Trans. 2007, 35, 1379–1384. [Google Scholar] [CrossRef] [Green Version]
- De Piccoli, G.; Torres-Rosell, J.; Aragón, L. The unnamed complex: What do we know about Smc5-Smc6? Chromosome Res. 2009, 17, 251–263. [Google Scholar] [CrossRef]
- Zhao, X.; Wu, C.Y. Mlp-dependent anchorage and stabilization of a desumoylating enzyme is required to prevent clonal lethality. J. Cell Biol. 2004, 167, 605–611. [Google Scholar] [CrossRef]
- Prudden, J.; Pebernard, S.; Raffa, G.; Slavin, D.A.; Perry, J.J.P.; Tainer, J.A.; McGowan, C.H.; Boddy, M.N. SUMO-targeted ubiquitin ligases in genome stability. EMBO J. 2007, 26, 4089–4101. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Leverson, J.D.; Hunter, T. Conserved function of RNF4 family proteins in eukaryotes: Targeting a ubiquitin ligase to SUMOylated proteins. EMBO J. 2007, 26, 4102–4112. [Google Scholar] [CrossRef] [Green Version]
- Nagai, S.; Dubrana, K.; Tsai-Pflugfelder, M.; Davidson, M.B.; Roberts, T.M.; Brown, G.W.; Varela, E.; Hediger, F.; Gasser, S.M.; Krogan, N.J. Functional targeting of DNA damage to a nuclear pore-associated SUMO-dependent ubiquitin ligase. Science 2008, 322, 597–602. [Google Scholar] [CrossRef] [Green Version]
- Galanty, Y.; Belotserkovskaya, R.; Coates, J.; Jackson, S.P. RNF4, a SUMO-targeted ubiquitin E3 ligase, promotes DNA double-strand break repair. Genes Dev. 2012, 26, 1179–1195. [Google Scholar] [CrossRef] [Green Version]
- Sriramachandran, A.M.; Dohmen, R.J. SUMO-targeted ubiquitin ligases. Biochem. Biophys. Acta 2014, 1843, 75–85. [Google Scholar] [CrossRef] [Green Version]
- Cremona, C.A.; Sarangi, P.; Yang, Y.; Hang, L.E.; Rahman, S.; Zhao, X. Extensive DNA Damage-Induced Sumoylation Contributes to Replication and Repair and Acts in Addition to the Mec1 Checkpoint. Mol. Cell 2012, 45, 422–432. [Google Scholar] [CrossRef] [Green Version]
- Soustelle, C.; Vernis, L.; Freon, K.; Reynaud-Angelin, A.; Chanet, R.; Fabre, F.; Heude, M. A New Saccharomyces cerevisiae Strain with a Mutant Smt3-Deconjugating Ulp1 Protein Is Affected in DNA Replication and Requires Srs2 and Homologous Recombination for Its Viability. Mol. Cell. Biol. 2004, 24, 5130–5143. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Contreras, A.J.; Ruppen, I.; Nieto-Soler, M.; Murga, M.; Rodriguez-Acebes, S.; Remeseiro, S.; Rodrigo-Perez, S.; Rojas, A.M.; Mendez, J.; Muñoz, J.; et al. A Proteomic Characterization of Factors Enriched at Nascent DNA Molecules. Cell Rep. 2013, 34, 1105–1116. [Google Scholar] [CrossRef] [Green Version]
- Hoege, C.; Pfander, B.; Moldovan, G.L.; Pyrowolakis, G.; Jentsch, S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 2002, 419, 135–141. [Google Scholar] [CrossRef]
- Golebiowski, F.; Matic, I.; Tatham, M.H.; Cole, C.; Yin, Y.; Nakamura, A.; Cox, J.; Barton, G.J.; Mann, M.; Hay, R.T. System-Wide Changes to SUMO Modifications in Response to Heat Shock. Sci. Signal. 2009, 2, 72. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Blobel, G. A SUMO ligase is part of a nuclear multiprotein complex that affects DNA repair and chromosomal organization. Proc. Natl. Acad. Sci. USA 2005, 102, 4777–4782. [Google Scholar] [CrossRef] [Green Version]
- Branzei, D.; Sollier, J.; Liberi, G.; Zhao, X.; Maeda, D.; Seki, M.; Enomoto, T.; Ohta, K.; Foiani, M. Ubc9- and Mms21-Mediated Sumoylation Counteracts Recombinogenic Events at Damaged Replication Forks. Cell 2006, 127, 509–522. [Google Scholar] [CrossRef] [Green Version]
- Sacher, M.; Pfander, B.; Hoege, C.; Jentsch, S. Control of Rad52 recombination activity by double-strand break-induced SUMO modification. Nat. Cell Biol. 2006, 8, 1284–1290. [Google Scholar] [CrossRef]
- Saponaro, M.; Callahan, D.; Zheng, X.; Krejci, L.; Haber, J.E.; Klein, H.L.; Liberi, G. Cdk1 targets Srs2 to complete synthesis-dependent strand annealing and to promote recombinational repair. PLoS Genet. 2010, 6. [Google Scholar] [CrossRef] [Green Version]
- Albuquerque, C.P.; Wang, G.; Lee, N.S.; Kolodner, R.D.; Putnam, C.D.; Zhou, H. Distinct SUMO ligases cooperate with Esc2 and Slx5 to suppress duplication-mediated genome rearrangements. PLoS Genet. 2013, 9. [Google Scholar] [CrossRef]
- Bologna, S.; Altmannova, V.; Valtorta, E.; Koenig, C.; Liberali, P.; Gentili, C.; Anrather, D.; Ammerer, G.; Pelkmans, L.; Krejci, L.; et al. Sumoylation regulates EXO1 stability and processing of DNA damage. Cell Cycle 2015, 14, 2439–2450. [Google Scholar] [CrossRef] [Green Version]
- Thu, Y.M.; Van Riper, S.K.; Higgins, A.; Zhang, T.; Becker, J.R.; Markowski, T.W.; Nguyen, H.D.; Griffin, T.J.; Bielinsky, A. Slx5/Slx8 promotes replication stress tolerance by facilitating mitotic progression. Cell Rep. 2016, 15, 1254–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, X.A.; Dion, V.; Gasser, S.M.; Freudenreich, C.H. Regulation of recombination at yeast nuclear pores controls repair and triplet repeat stability. Genes Dev. 2015, 29, 1006–1017. [Google Scholar] [CrossRef] [Green Version]
- Aguilera, P.; Whalen, J.; Minguet, C.; Churikov, D.; Freudenreich, C.; Simon, M.N.; Géli, V. The nuclear pore complex prevents sister chromatid recombination during replicative senescence. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Lamm, N.; Masamsetti, V.P.; Read, M.N.; Biro, M.; Cesare, A.J. ATR and mTOR regulate F-actin to alter nuclear architecture and repair replication stress. BioRxiv 2018. [Google Scholar] [CrossRef] [Green Version]
- Whalen, J.M.; Dhingra, N.; Wei, L.; Zhao, X.; Freudenreich, C.H. Relocation of collapsed forks to the nuclear pore complex depends on sumoylation of DNA repair proteins and permits Rad51 association. Cell Rep. 2020, 31, 107635. [Google Scholar] [CrossRef] [PubMed]
- Lambert, S.; Watson, A.; Sheedy, D.M.; Martin, B.; Carr, A.M. Gross Chromosomal Rearrangements and Elevated Recombination at an Inducible Site-Specific Replication Fork Barrier. Cell 2005, 121, 689–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ait Saada, A.; Teixeira-Silva, A.; Iraqui, I.; Costes, A.; Hardy, J.; Paoletti, G.; Fréon, K.; Lambert, S.A.E. Unprotected Replication Forks Are Converted into Mitotic Sister Chromatid Bridges. Mol. Cell 2017, 66, 398–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khadaroo, B.; Teixeira, M.T.; Luciano, P.; Eckert-Boulet, N.; Germann, S.M.; Simon, M.N.; Gallina, I.; Abdallah, P.; Gilson, E.; Géli, V.; et al. The DNA damage response at eroded telomeres and tethering to the nuclear pore complex. Nat. Cell Biol. 2009, 11, 980–987. [Google Scholar] [CrossRef] [PubMed]
- Ohki, R.; Ishikawa, F. Telomere-bound TRF1 and TRF2 stall the replication fork at telomeric repeats. Nucleic Acids Res. 2004, 32, 1627–1637. [Google Scholar] [CrossRef] [PubMed]
- Edwards, D.N.; Machwe, A.; Wang, Z.; Orren, D.K. Intramolecular telomeric G-quadruplexes dramatically inhibit DNA synthesis by replicative and translesion polymerases, revealing their potential to lead to genetic change. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [Green Version]
- Rhodes, D.; Lipps, H.J. Survey and summary G-quadruplexes and their regulatory roles in biology. Nucleic Acids Res. 2015, 43, 8627–8637. [Google Scholar] [CrossRef] [Green Version]
- Jay, K.A.; Smith, D.L.; Blackburn, E.H. Early Loss of Telomerase Action in Yeast Creates a Dependence on the DNA Damage Response Adaptor Proteins. Mol. Cell. Biol. 2016, 36, 1908–1919. [Google Scholar] [CrossRef] [Green Version]
- Lisby, M.; Géli, V. DNA damage response to eroded telomeres. Cell Cycle 2009, 8, 3614–3618. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.C.; Stone, S.; Hoatlin, M.E.; Gautier, J. Fanconi anemia proteins stabilize replication forks. DNA Repair 2008, 7, 1973–1981. [Google Scholar] [CrossRef] [Green Version]
- Sirbu, B.M.; McDonald, W.H.; Dungrawala, H.; Badu-Nkansah, A.; Kavanaugh, G.M.; Chen, Y.; Tabb, D.L.; Cortez, D. Identification of proteins at active, stalled, and collapsed replication forks using isolation of proteins on nascent DNA (iPOND) coupled with mass spectrometry. J. Biol. Chem. 2013, 288, 31458–31467. [Google Scholar] [CrossRef] [Green Version]
- Kalocsay, M.; Hiller, N.J.; Jentsch, S. Chromosome-wide Rad51 Spreading and SUMO-H2A.Z-Dependent Chromosome Fixation in Response to a Persistent DNA Double-Strand Break. Mol. Cell 2009, 33, 335–343. [Google Scholar]
- Oza, P.; Jaspersen, S.L.; Miele, A.; Dekker, J.; Peterson, C.L. Mechanisms that regulate localization of a DNA double-strand break to the nuclear periphery. Genes Dev. 2009, 23, 912–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horigome, C.; Oma, Y.; Konishi, T.; Schmid, R.; Marcomini, I.; Hauer, M.H.; Dion, V.; Harata, M.; Gasser, S.M. SWR1 and INO80 Chromatin Remodelers Contribute to DNA Double-Strand Break Perinuclear Anchorage Site Choice. Mol. Cell 2014, 55, 626–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, T.; Spatola, B.; Delabaere, L.; Bowlin, K.; Hopp, H.; Kunitake, R.; Karpen, G.H.; Chiolo, I. Heterochromatic breaks move to the nuclear periphery to continue recombinational repair. Nat. Cell Biol. 2015, 17, 1404–1411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amaral, S.; Ryu, T.; Li, X.; Chiolo, I. Nuclear Dynamics of Heterochromatin Repair. Trends Genet. 2017, 33, 86–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horigome, C.; Unozawa, E.; Ooki, T.; Kobayashi, T. Ribosomal RNA gene repeats associate with the nuclear pore complex for maintenance after DNA damage. PLoS Genet. 2019, 15, e1008103. [Google Scholar] [CrossRef] [PubMed]
- Tsouroula, K.; Furst, A.; Rogier, M.; Heyer, V.; Maglott-Roth, A.; Ferrand, A.; Reina-San-Martin, B.; Soutoglou, E. Temporal and Spatial Uncoupling of DNA Double Strand Break Repair Pathways within Mammalian Heterochromatin. Mol. Cell 2016, 63, 293–305. [Google Scholar] [CrossRef] [Green Version]
- Horigome, C.; Bustard, D.E.; Marcomini, I.; Delgoshaie, N.; Tsai-Pflugfelder, M.; Cobb, J.A.; Gasser, S.M. PolySUMOylation by Siz2 and Mms21 triggers relocation of DNA breaks to nuclear pores through the Slx5/Slx8 STUbL. Genes Dev. 2016, 8, 931–935. [Google Scholar] [CrossRef] [Green Version]
- Ryu, T.; Bonner, M.; Chiolo, I. Cervantes and Quijote protect heterochromatin from aberrant recombination and lead the way to the periphery. Nucleus 2016, 7, 485–497. [Google Scholar] [CrossRef] [Green Version]
- Churikov, D.; Charifi, F.; Eckert-Boulet, N.; Silva, S.; Simon, M.N.; Lisby, M.; Géli, V. SUMO-Dependent Relocalization of Eroded Telomeres to Nuclear Pore Complexes Controls Telomere Recombination. Cell Rep. 2016, 15, 1242–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatham, M.H.; Jaffray, E.; Vaughan, O.A.; Desterro, J.M.P.; Botting, C.H.; Naismith, J.H.; Hay, R.T. Polymeric Chains of SUMO-2 and SUMO-3 are Conjugated to Protein Substrates by SAE1/SAE2 and Ubc9. J. Biol. Chem. 2001, 276, 35368–35374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bylebyl, G.R.; Belichenko, I.; Johnson, E.S. The SUMO Isopeptidase Ulp2 Prevents Accumulation of SUMO Chains in Yeast. J. Biol. Chem. 2003, 278, 44113–44120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Y.; Kerscher, O.; Kroetz, M.B.; McConchie, H.F.; Sung, P.; Hochstrasser, M. The yeast Hex3·Slx8 heterodimer is a ubiquitin ligase stimulated by substrate sumoylation. J. Biol. Chem. 2007, 282, 34176–34184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Y.; Rubenstein, E.M.; Matt, T.; Hochstrasser, M. SUMO-independent in vivo activity of a SUMO-targeted ubiquitin ligase toward a short-lived transcription factor. Genes Dev. 2010, 24, 893–903. [Google Scholar] [CrossRef] [Green Version]
- Silva, S.; Altmannova, V.; Eckert-Boulet, N.; Kolesar, P.; Gallina, I.; Hang, L.; Chung, I.; Arneric, M.; Zhao, X.; Buron, L.D.; et al. SUMOylation of Rad52-Rad59 synergistically change the outcome of mitotic recombination. DNA Repair 2016, 42, 11–25. [Google Scholar] [CrossRef] [Green Version]
- Dhingra, N.; Wei, L.; Zhao, X. Replication protein A (RPA) SUMOylation positively influences the DNA damage checkpoint response in yeast. J. Biol. Chem. 2018, 294, 2690–2699. [Google Scholar] [CrossRef] [Green Version]
- Lindroos, H.B.; Ström, L.; Itoh, T.; Katou, Y.; Shirahige, K.; Sjögren, C. Chromosomal Association of the Smc5/6 Complex Reveals that It Functions in Differently Regulated Pathways. Mol. Cell 2006, 22, 755–767. [Google Scholar] [CrossRef]
- Chiolo, I.; Minoda, A.; Colmenares, S.U.; Polyzos, A.; Costes, S.V.; Karpen, G.H. Double-strand breaks in heterochromatin move outside of a dynamic HP1a domain to complete recombinational repair. Cell 2011, 144, 732–744. [Google Scholar] [CrossRef] [Green Version]
- Torres-Rosell, J.; Sunjevaric, I.; De Piccoli, G.; Sacher, M.; Eckert-Boulet, N.; Reid, R.; Jentsch, S.; Rothstein, R.; Aragon, L.; Lisby, M. The Smc5-Smc6 complex and SUMO modification of Rad52 regulates recombinational repair at the ribosomal gene locus. Nat Cell Biol. 2007, 9, 923–931. [Google Scholar] [CrossRef]
- Spector, I.; Shochet, N.R.; Kashman, Y.; Groweiss, A. Latrunculins: Novel marine toxins that disrupt microfilament organization in cultured cells. Science 1983, 219, 493–495. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Jones, G.M.; Prelich, G. Genetic analysis connects SLX5 and SLX8 to the SUMO pathway in Saccharomyces cerevisiae. Genetics 2006, 172, 1499–1509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caridi, C.P.; D’agostino, C.; Ryu, T.; Zapotoczny, G.; Delabaere, L.; Li, X.; Khodaverdian, V.Y.; Amaral, N.; Lin, E.; Rau, A.R.; et al. Nuclear F-actin and myosins drive relocalization of heterochromatic breaks. Nature 2018, 559, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Chung, D.K.C.; Chan, J.N.Y.; Strecker, J.; Zhang, W.; Ebrahimi-Ardebili, S.; Lu, T.; Abraham, K.J.; Durocher, D.; Mekhail, K. Perinuclear tethers license telomeric DSBs for a broad kinesin-and NPC-dependent DNA repair process. Nat. Commun. 2015, 6, 7742. [Google Scholar] [CrossRef] [Green Version]
- Oshidari, R.; Strecker, J.; Chung, D.C.K.; Abraham, K.J.; Chan, J.N.Y.; Damaren, C.J.; Mekhail, K. Nuclear microtubule filaments mediate non-linear directional motion of chromatin and promote DNA repair. Nat. Commun. 2018, 9, 1–11. [Google Scholar] [CrossRef]
- Pyhtila, B.; Rexach, M. A gradient of affinity for the karyopherin Kap95p along the yeast nuclear pore complex. J. Biol. Chem. 2003, 278, 42699–42709. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.M.; Stewart, M. Structural bias for the high-affinity binding of nucleoporin Nup1p to the Saccharomyces cerevisiae importin-beta homologue, Kap95p. J. Mol. Biol. 2005, 349, 515–525. [Google Scholar] [CrossRef]
- Moudry, P.; Lukas, C.; Macurek, L.; Neumann, B.; Heriche, J.K.; Pepperkok, R.; Ellenberg, J.; Hodny, Z.; Lukas, J.; Bartek, J. Nucleoporin NUP153 guards genome integrity by promoting nuclear import of 53BP1. Cell Death Differ. 2012, 19, 798–807. [Google Scholar] [CrossRef] [Green Version]
- Duheron, V.; Nilles, N.; Pecenko, S.; Martinelli, V.; Fahrenkrog, B. Localisation of Nup153 and SENP1 to nuclear pore complexes is required for 53BP1-mediated DNA Double-strand break repair. J. Cell Sci. 2017, 130, 2306–2316. [Google Scholar] [CrossRef] [Green Version]
- Geli, V.; Lisby, M. Recombinational DNA repair is regulated by compartmentalization of DNA lesions at the nuclear pore complex. Bioessays 2015, 37, 1287–1292. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Whalen, J.M.; Freudenreich, C.H. Location, Location, Location: The Role of Nuclear Positioning in the Repair of Collapsed Forks and Protection of Genome Stability. Genes 2020, 11, 635. https://doi.org/10.3390/genes11060635
Whalen JM, Freudenreich CH. Location, Location, Location: The Role of Nuclear Positioning in the Repair of Collapsed Forks and Protection of Genome Stability. Genes. 2020; 11(6):635. https://doi.org/10.3390/genes11060635
Chicago/Turabian StyleWhalen, Jenna M., and Catherine H. Freudenreich. 2020. "Location, Location, Location: The Role of Nuclear Positioning in the Repair of Collapsed Forks and Protection of Genome Stability" Genes 11, no. 6: 635. https://doi.org/10.3390/genes11060635
APA StyleWhalen, J. M., & Freudenreich, C. H. (2020). Location, Location, Location: The Role of Nuclear Positioning in the Repair of Collapsed Forks and Protection of Genome Stability. Genes, 11(6), 635. https://doi.org/10.3390/genes11060635