Being Merle: The Molecular Genetic Background of the Canine Merle Mutation

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. What Was Known Prior to the Molecular Genetic Identification of the Merle Mutation
3. The Molecular Genetics of Merle Mutation
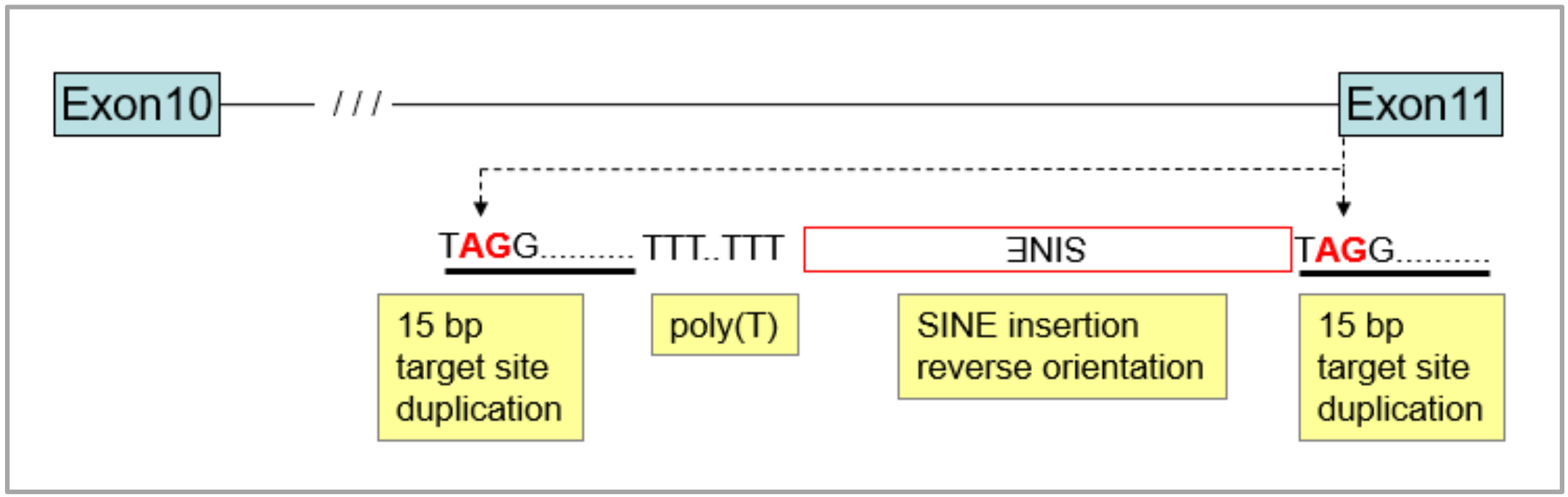
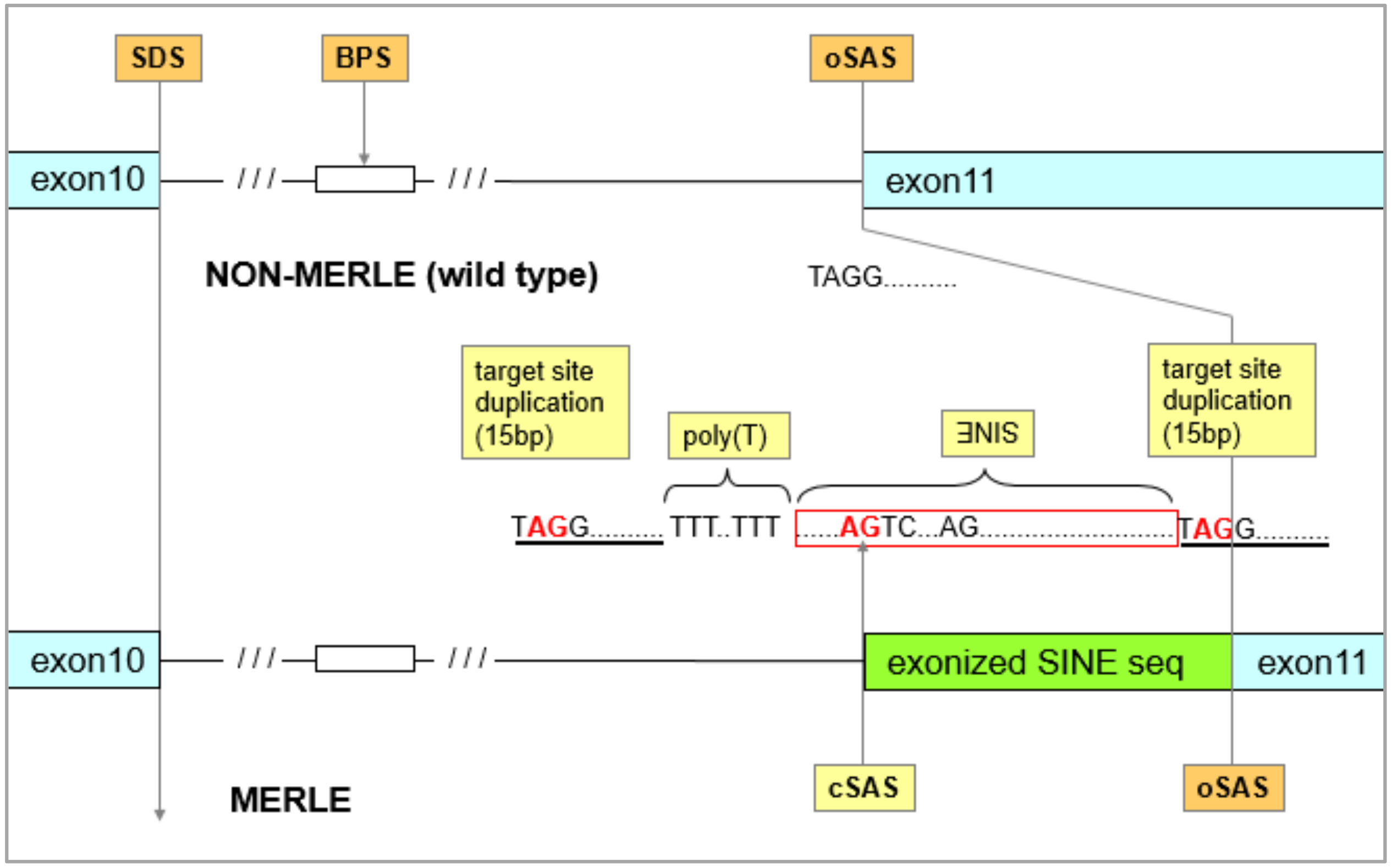
3.1. Identification of the Merle-SINE (Short Interspersed Nuclear Element) Mutation and Its Mode of Action
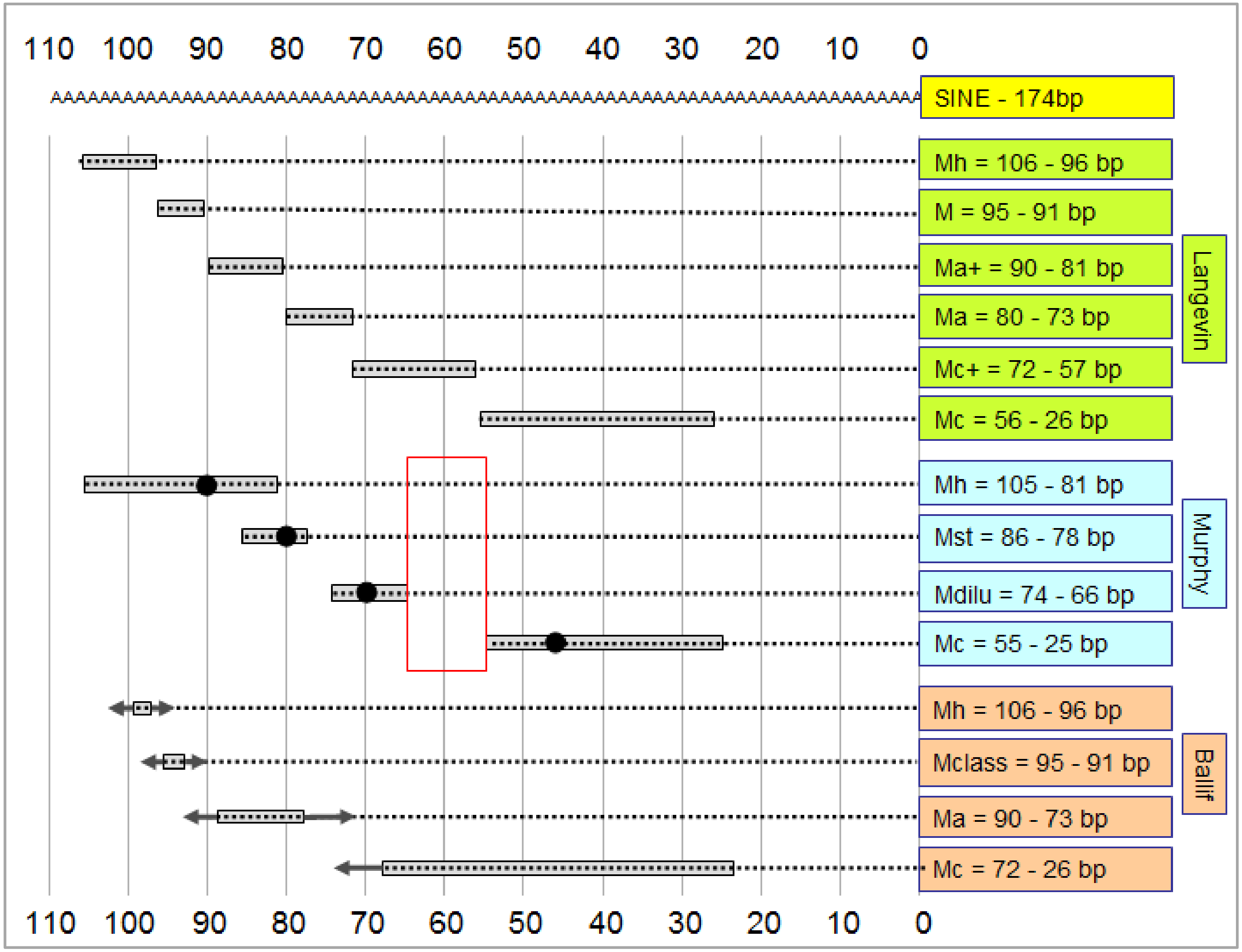
3.2. How is the Length of the Poly(T) Sequence Able to Generate Diverse Merle Phenotypes?
3.2.1. Other SINE Mutations in the Dog Genome
3.2.2. PMEL Mutations in Other Species
4. Correlating the Merle Genotype with the Phenotype
- Phenotype categories were set up by breeders prior to the molecular genetic identification of the merle mutation. As such, these categories might differ substantially from each other by country, breeds or kennel clubs, since judges at competitions might perform the phenotyping based on different considerations. Thus, the basis of phenotyping (i.e., the rate of merle coloration) would be very difficult to standardize globally, even if pictures were available for all animals.
- Determination of the poly(T) sequence has a certain error rate, which tends to increase slightly with the length of the repeat. Overall precision of the genotyping results can be improved using several technical replicates [1].
5. Merle at the Cellular Level
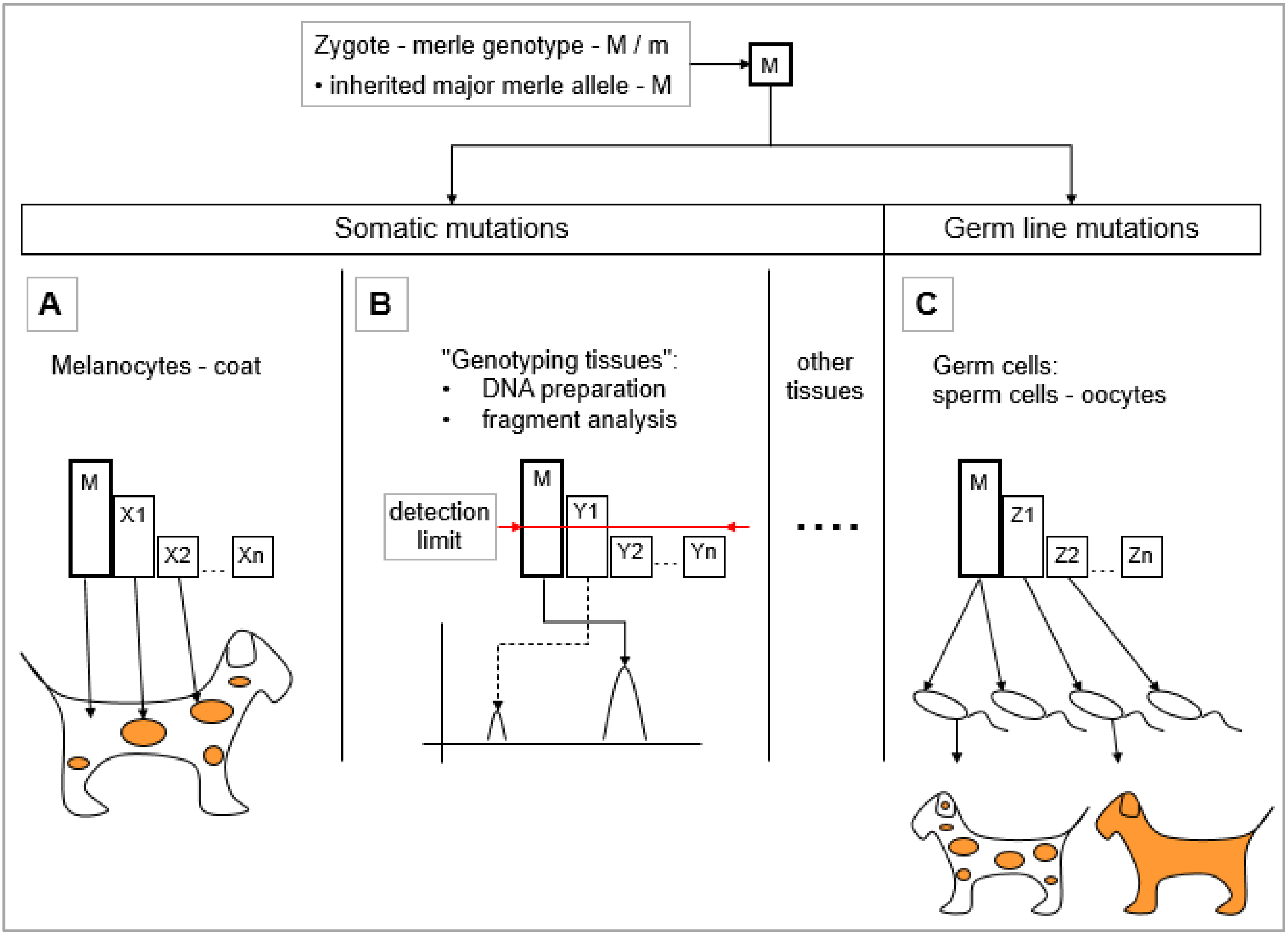
5.1. Somatic Mosaicism of Merle Mutants
5.1.1. Assumptions Regarding the Development of Merle Coat Color Patterns—Somatic Mutations in the Melanocytes
5.1.2. Somatic Mutations Resulting Mosaicism in the ‘Genotyping Tissues’
5.2. Effect of the Germ Line Mosaicism on the Merle Pattern
- Germ cell volume: Germ cells may be examined in mass, as one sample in a volume as a ‘genotyping tissue’. Practically speaking, this denotes a DNA preparation from a sperm volume, as this approach does not work with oocytes. Fragment analysis can be performed to determine the major allele in addition to those mutant alleles which arose from cell divisions during the early stages. Since alleles that represent smaller proportions of the whole mutant cell population may remain under the detection limit, this analysis provides only a partial picture of the germ line mosaicism.
- Individual sperm typing: By genotyping a high number of individual germ cells for the length of the merle-SINE insertion [63], a more precise estimate can be obtained regarding the level of germ line mosaicism, individual mutations, and their actual proportion. However, this approach is suitable only for research purposes, and is not practical for the testing of individual families, because it is both expensive and labor-intensive.
- Mutant spectra comparison from ‘genotyping tissues’ within the family: The genotyping of that parent carrying the M mutation along with the offspring from ‘genotyping tissue’ is an indirect way for deducing the inherited major allele in every single progeny, and thus, assessing the derived mutant alleles. If the fragment analysis of ‘genotyping tissue’ identifies a merle-SINE insertion having the same poly(T) size to be the major allele typically with the highest peak intensity in the parent-offspring pair, then the progeny has inherited the paternal, non-mutant allele in the germ line. However, if the original parental M allele is not present in the offspring and it possesses a longer or shorter allele instead, then it must be a germ line mutation, insomuch that the parentage is indeed certain. The recent, aforementioned articles applied this approach for the study of germ line mosaicism [1,2,3].
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Murphy, S.C.; Evans, J.M.; Tsai, K.L.; Clark, L.A. Length variations within the Merle retrotransposon of canine PMEL: Correlating genotype with phenotype. Mob. DNA 2018, 9, 26. [Google Scholar] [CrossRef] [PubMed]
- Ballif, B.C.; Ramirez, C.J.; Carl, C.R.; Sundin, K.; Krug, M.; Zahand, A.; Shaffer, L.G.; Flores-Smith, H. The PMEL Gene and Merle in the Domestic Dog: A Continuum of Insertion Lengths Leads to a Spectrum of Coat Color Variations in Australian Shepherds and Related Breeds. Cytogenet. Genome Res. 2018, 156, 22–34. [Google Scholar] [CrossRef] [PubMed]
- Langevin, M.; Synkova, H.; Jancuskova, T.; Pekova, S. Merle phenotypes in dogs-SILV SINE insertions from Mc to Mh. PLoS ONE 2018, 13, e0198536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelles, Z.; Maróti-Agóts, Á.; Gáspárdy, A.; Zöldág, L.; Zenke, P. A rejtett cifra szín molekuláris genetikai módszerekkel való kimutatása mudi fajtában. Magy. Állatorvosok Lapja 2018, 140, 121–127. [Google Scholar]
- Pelles, Z.; Gaspardy, A.; Zoldag, L.; Lenart, X.; Ninausz, N.; Varga, L.; Zenke, P. Merle allele variations in the Mudi dog breed and their effects on phenotypes. Acta Vet. Hung. 2019, 67, 159–173. [Google Scholar] [CrossRef]
- Clark, L.A.; Wahl, J.M.; Rees, C.A.; Murphy, K.E. Retrotransposon insertion in SILV is responsible for merle patterning of the domestic dog. Proc. Natl. Acad. Sci. USA 2006, 103, 1376–1381. [Google Scholar] [CrossRef] [Green Version]
- Clark, L.A.; Wahl, J.M.; Rees, C.A.; Strain, G.M.; Cargill, E.J.; Vanderlip, S.L.; Murphy, K.E. Canine SINEs and Their Effects on Phenotypes of the Domestic Dog. In Genomics of Disease; Gustafson, J.P., Tayler, J., Stacey, G., Eds.; Springer Science+Business Media LLC: New York, NY, USA, 2008; pp. 79–87. [Google Scholar]
- Mitchell, A.L. Dominant dilution and other color factors in Collie dogs. J. Hered. 1935, 26, 425–430. [Google Scholar] [CrossRef]
- Kaelin, C.B.; Barsh, G.S. Genetics of pigmentation in dogs and cats. Annu. Rev. Anim. Biosci. 2013, 1, 125–156. [Google Scholar] [CrossRef]
- Hedan, B.; Corre, S.; Hitte, C.; Dreano, S.; Vilboux, T.; Derrien, T.; Denis, B.; Galibert, F.; Galibert, M.D.; Andre, C. Coat colour in dogs: Identification of the merle locus in the Australian shepherd breed. BMC Vet. Res. 2006, 2, 9. [Google Scholar] [CrossRef]
- Everts, R.E.; Rothuizen, J.; van Oost, B.A. Identification of a premature stop codon in the melanocyte-stimulating hormone receptor gene (MC1R) in Labrador and Golden retrievers with yellow coat colour. Anim. Genet. 2000, 31, 194–199. [Google Scholar] [CrossRef]
- Newton, J.M.; Wilkie, A.L.; He, L.; Jordan, S.A.; Metallinos, D.L.; Holmes, N.G.; Jackson, I.J.; Barsh, G.S. Melanocortin 1 receptor variation in the domestic dog. Mamm. Genome 2000, 11, 24–30. [Google Scholar] [CrossRef]
- Schmutz, S.M.; Berryere, T.G. Genes affecting coat colour and pattern in domestic dogs: A review. Anim. Genet. 2007, 38, 539–549. [Google Scholar] [CrossRef]
- Schmutz, S.M.; Dreger, D.L. Genetic Interactions Among Three Pigmentation Loci in Domestic Dogs. In Proceedings of the 10th World Congress of Genetics Applied to Livestock Production, Vancouver, BC, Canada, 17–22 August 2014. [Google Scholar]
- Candille, S.I.; Kaelin, C.B.; Cattanach, B.M.; Yu, B.; Thompson, D.A.; Nix, M.A.; Kerns, J.A.; Schmutz, S.M.; Millhauser, G.L.; Barsh, G.S. A -defensin mutation causes black coat color in domestic dogs. Science 2007, 318, 1418–1423. [Google Scholar] [CrossRef] [Green Version]
- Durig, N.; Letko, A.; Lepori, V.; Rasouliha, S.H.; Loechel, R.; Kehl, A.; Hytonen, M.K.; Lohi, H.; Mauri, N.; Dietrich, J.; et al. Two MC1R loss-of-function alleles in cream-coloured Australian Cattle Dogs and white Huskies. Anim. Genet. 2018, 49, 284–290. [Google Scholar] [CrossRef] [Green Version]
- Sponenberg, D.P. Inheritance of the harlequin color in Great Dane dogs. J. Hered. 1985, 76, 224–225. [Google Scholar] [CrossRef]
- Clark, L.A.; Starr, A.N.; Tsai, K.L.; Murphy, K.E. Genome-wide linkage scan localizes the harlequin locus in the Great Dane to chromosome 9. Gene 2008, 418, 49–52. [Google Scholar] [CrossRef] [PubMed]
- Clark, L.A.; Tsai, K.L.; Starr, A.N.; Nowend, K.L.; Murphy, K.E. A missense mutation in the 20S proteasome beta2 subunit of Great Danes having harlequin coat patterning. Genomics 2011, 97, 244–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theos, A.C.; Truschel, S.T.; Raposo, G.; Marks, M.S. The Silver locus product Pmel17/gp100/Silv/ME20: Controversial in name and in function. Pigment. Cell Res. 2005, 18, 322–336. [Google Scholar] [CrossRef] [Green Version]
- Berson, J.F.; Harper, D.C.; Tenza, D.; Raposo, G.; Marks, M.S. Pmel17 initiates premelanosome morphogenesis within multivesicular bodies. Mol. Biol. Cell 2001, 12, 3451–3464. [Google Scholar] [CrossRef] [Green Version]
- Kwon, B.S.; Chintamaneni, C.; Kozak, C.A.; Copeland, N.G.; Gilbert, D.J.; Jenkins, N.; Barton, D.; Francke, U.; Kobayashi, Y.; Kim, K.K. A melanocyte-specific gene, Pmel 17, maps near the silver coat color locus on mouse chromosome 10 and is in a syntenic region on human chromosome 12. Proc. Natl. Acad. Sci. USA 1991, 88, 9228–9232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Esparza, M.; Jimenez-Cervantes, C.; Bennett, D.C.; Lozano, J.A.; Solano, F.; Garcia-Borron, J.C. The mouse silver locus encodes a single transcript truncated by the silver mutation. Mamm. Genome 1999, 10, 1168–1171. [Google Scholar] [CrossRef] [PubMed]
- Brunberg, E.; Andersson, L.; Cothran, G.; Sandberg, K.; Mikko, S.; Lindgren, G. A missense mutation in PMEL17 is associated with the Silver coat color in the horse. BMC Genet. 2006, 7, 46. [Google Scholar] [CrossRef] [PubMed]
- Kerje, S.; Sharma, P.; Gunnarsson, U.; Kim, H.; Bagchi, S.; Fredriksson, R.; Schutz, K.; Jensen, P.; von Heijne, G.; Okimoto, R.; et al. The Dominant white, Dun and Smoky color variants in chicken are associated with insertion/deletion polymorphisms in the PMEL17 gene. Genetics 2004, 168, 1507–1518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kramerov, D.A.; Vassetzky, N.S. SINEs. Wiley Interdiscip. Rev. RNA 2011, 2, 772–786. [Google Scholar] [CrossRef]
- Watt, B.; van Niel, G.; Raposo, G.; Marks, M.S. PMEL: A pigment cell-specific model for functional amyloid formation. Pigment Cell Melanoma Res. 2013, 26, 300–315. [Google Scholar] [CrossRef] [Green Version]
- Raposo, G.; Marks, M.S. Melanosomes-dark organelles enlighten endosomal membrane transport. Nat. Rev. Mol. Cell Biol. 2007, 8, 786–797. [Google Scholar] [CrossRef] [Green Version]
- Hellstrom, A.R.; Watt, B.; Fard, S.S.; Tenza, D.; Mannstrom, P.; Narfstrom, K.; Ekesten, B.; Ito, S.; Wakamatsu, K.; Larsson, J.; et al. Inactivation of Pmel alters melanosome shape but has only a subtle effect on visible pigmentation. PLoS Genet. 2011, 7, e1002285. [Google Scholar] [CrossRef] [Green Version]
- Thiruvenkadan, A.K.; Kandasamy, N.; Panneerselvam, S. Coat colour inheritance in horses. Livest. Sci. 2008, 117, 109–129. [Google Scholar] [CrossRef]
- Minnick, M.F.; Stillwell, L.C.; Heineman, J.M.; Stiegler, G.L. A highly repetitive DNA sequence possibly unique to canids. Gene 1992, 110, 235–238. [Google Scholar] [CrossRef]
- Wang, W.; Kirkness, E.F. Short interspersed elements (SINEs) are a major source of canine genomic diversity. Genome Res. 2005, 15, 1798–1808. [Google Scholar] [CrossRef] [Green Version]
- Kirkness, E.F.; Bafna, V.; Halpern, A.L.; Levy, S.; Remington, K.; Rusch, D.B.; Delcher, A.L.; Pop, M.; Wang, W.; Fraser, C.M.; et al. The dog genome: Survey sequencing and comparative analysis. Science 2003, 301, 1898–1903. [Google Scholar] [CrossRef] [PubMed]
- Lindblad-Toh, K.; Wade, C.M.; Mikkelsen, T.S.; Karlsson, E.K.; Jaffe, D.B.; Kamal, M.; Clamp, M.; Chang, J.L.; Kulbokas, E.J., 3rd; Zody, M.C.; et al. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature 2005, 438, 803–819. [Google Scholar] [CrossRef]
- Elbarbary, R.A.; Lucas, B.A.; Maquat, L.E. Retrotransposons as regulators of gene expression. Science 2016, 351, aac7247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordaux, R.; Batzer, M.A. The impact of retrotransposons on human genome evolution. Nat. Rev. Genet. 2009, 10, 691–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitz, J. SINEs as driving forces in genome evolution. Genome Dynomes 2012, 7, 92–107. [Google Scholar]
- Schmitz, J.; Brosius, J. Exonization of transposed elements: A challenge and opportunity for evolution. Biochimie 2011, 93, 1928–1934. [Google Scholar] [CrossRef]
- Lin, L.; Faraco, J.; Li, R.; Kadotani, H.; Rogers, W.; Lin, X.; Qiu, X.; de Jong, P.J.; Nishino, S.; Mignot, E. The sleep disorder canine narcolepsy is caused by a mutation in the hypocretin (orexin) receptor 2 gene. Cell 1999, 98, 365–376. [Google Scholar] [CrossRef] [Green Version]
- Keren, H.; Lev-Maor, G.; Ast, G. Alternative splicing and evolution: Diversification, exon definition and function. Nat. Rev. Genet. 2010, 11, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Downs, L.M.; Mellersh, C.S. An Intronic SINE insertion in FAM161A that causes exon-skipping is associated with progressive retinal atrophy in Tibetan Spaniels and Tibetan Terriers. PLoS ONE 2014, 9, e93990. [Google Scholar] [CrossRef]
- Litt, M.; Luty, J.A. A Hypervariable Microsatellite Revealed by In Vitro Amplification of a Dinucleotide Repeat within the Cardiac Muscle Actin Gene. Am. J. Hum. Genet. 1989, 44, 397–401. [Google Scholar]
- Tautz, D. Hypervariability of simple sequences as a general source for polymorphic DNA markers. Nucleic Acids Res. 1989, 17, 6463–6471. [Google Scholar] [CrossRef] [PubMed]
- Buschiazzo, E.; Gemmell, N.J. The rise, fall and renaissance of microsatellites in eukaryotic genomes. Bioessays 2006, 28, 1040–1050. [Google Scholar] [CrossRef] [PubMed]
- Kelkar, Y.D.; Eckert, K.A.; Chiaromonte, F.; Makova, K.D. A matter of life or death: How microsatellites emerge in and vanish from the human genome. Genome Res. 2011, 21, 2038–2048. [Google Scholar] [CrossRef] [Green Version]
- Nadir, E.; Margalit, H.; Gallily, T.; Ben-Sasson, S.A. Microsatellite spreading in the human genome: Evolutionary mechanisms and structural implications. Proc. Natl. Acad. Sci. USA 1996, 93, 6470–6475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, S.; Mishra, R.K.; Singh, L. Genome-wide analysis of microsatellite repeats in humans: Their abundance and density in specific genomic regions. Genome Biol. 2003, 4, R13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grandi, F.C.; Rosser, J.M.; An, W. LINE-1-derived poly(A) microsatellites undergo rapid shortening and create somatic and germline mosaicism in mice. Mol. Biol. Evol. 2013, 30, 503–512. [Google Scholar] [CrossRef]
- Korberg, I.B.; Sundstrom, E.; Meadows, J.R.; Pielberg, G.R.; Gustafson, U.; Hedhammar, A.; Karlsson, E.K.; Seddon, J.; Soderberg, A.; Vila, C.; et al. A simple repeat polymorphism in the MITF-M promoter is a key regulator of white spotting in dogs. PLoS ONE 2014, 9, e104363. [Google Scholar]
- Ellegren, H. Microsatellites: Simple sequences with complex evolution. Nat. Rev. Genet. 2004, 5, 435–445. [Google Scholar] [CrossRef]
- Laidlaw, J.; Gelfand, Y.; Ng, K.W.; Garner, H.R.; Ranganathan, R.; Benson, G.; Fondon, J.W., 3rd. Elevated basal slippage mutation rates among the Canidae. J. Hered. 2007, 98, 452–460. [Google Scholar] [CrossRef] [Green Version]
- Dreger, D.L.; Schmutz, S. A SINE Insertion Causes the Blackand-Tan and Saddle Tan Phenotypes in Domestic Dogs. J. Hered. 2011, 102, S11–S18. [Google Scholar] [CrossRef]
- Sutter, N.B.; Bustamante, C.D.; Chase, K.; Gray, M.M.; Zhao, K.; Zhu, L.; Padhukasahasram, B.; Karlins, E.; Davis, S.; Jones, P.G.; et al. A single IGF1 allele is a major determinant of small size in dogs. Science 2007, 316, 112–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray, M.M.; Sutter, N.B.; Ostrander, E.A.; Wayne, R.K. The IGF1 small dog haplotype is derived from Middle Eastern grey wolves. BMC Biol. 2010, 8, 16. [Google Scholar]
- Pele, M.; Tiret, L.; Kessler, J.L.; Blot, S.; Panthier, J.J. SINE exonic insertion in the PTPLA gene leads to multiple splicing defects and segregates with the autosomal recessive centronuclear myopathy in dogs. Hum. Mol. Genet. 2005, 14, 1417–1427. [Google Scholar] [CrossRef] [Green Version]
- Schmutz, S.M.; Dreger, D.L. Interaction of MC1R and PMEL alleles on solid coat colors in Highland cattle. Anim. Genet. 2013, 44, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Philipp, U.; Hamann, H.; Mecklenburg, L.; Nishino, S.; Mignot, E.; Gunzel-Apel, A.R.; Schmutz, S.M.; Leeb, T. Polymorphisms within the canine MLPH gene are associated with dilute coat color in dogs. BMC Genet. 2005, 6, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drogemuller, C.; Philipp, U.; Haase, B.; Gunzel-Apel, A.R.; Leeb, T. A noncoding melanophilin gene (MLPH) SNP at the splice donor of exon 1 represents a candidate causal mutation for coat color dilution in dogs. J. Hered. 2007, 98, 468–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirobe, T. How are proliferation and differentiation of melanocytes regulated? Pigment Cell Melanoma Res. 2011, 24, 462–478. [Google Scholar] [CrossRef]
- Lin, J.Y.; Fisher, D.E. Melanocyte biology and skin pigmentation. Nature 2007, 445, 843–850. [Google Scholar] [CrossRef]
- Cieslak, M.; Reissmann, M.; Hofreiter, M.; Ludwig, A. Colours of domestication. Biol. Rev. Camb. Philos. Soc. 2011, 86, 885–899. [Google Scholar] [CrossRef]
- Karlsson, E.K.; Baranowska, I.; Wade, C.M.; Hillbertz, N.H.S.; Zody, M.C.; Anderson, N.; Biagi, T.M.; Patterson, N.; Pielberg, G.R.; Kulbokas, E.J., 3rd; et al. Efficient mapping of mendelian traits in dogs through genome-wide association. Nat. Genet. 2007, 39, 1321–1328. [Google Scholar] [CrossRef]
- Arnheim, N.; Calabrese, P.; Tiemann-Boege, I. Mammalian meiotic recombination hot spots. Annu. Rev. Genet. 2007, 41, 369–399. [Google Scholar] [CrossRef] [PubMed]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Varga, L.; Lénárt, X.; Zenke, P.; Orbán, L.; Hudák, P.; Ninausz, N.; Pelles, Z.; Szőke, A. Being Merle: The Molecular Genetic Background of the Canine Merle Mutation. Genes 2020, 11, 660. https://doi.org/10.3390/genes11060660
Varga L, Lénárt X, Zenke P, Orbán L, Hudák P, Ninausz N, Pelles Z, Szőke A. Being Merle: The Molecular Genetic Background of the Canine Merle Mutation. Genes. 2020; 11(6):660. https://doi.org/10.3390/genes11060660
Chicago/Turabian StyleVarga, László, Xénia Lénárt, Petra Zenke, László Orbán, Péter Hudák, Nóra Ninausz, Zsófia Pelles, and Antal Szőke. 2020. "Being Merle: The Molecular Genetic Background of the Canine Merle Mutation" Genes 11, no. 6: 660. https://doi.org/10.3390/genes11060660
APA StyleVarga, L., Lénárt, X., Zenke, P., Orbán, L., Hudák, P., Ninausz, N., Pelles, Z., & Szőke, A. (2020). Being Merle: The Molecular Genetic Background of the Canine Merle Mutation. Genes, 11(6), 660. https://doi.org/10.3390/genes11060660