Precision and Personalized Medicine: How Genomic Approach Improves the Management of Cardiovascular and Neurodegenerative Disease


 ,
,  ,
,  ,
, 
Abstract
: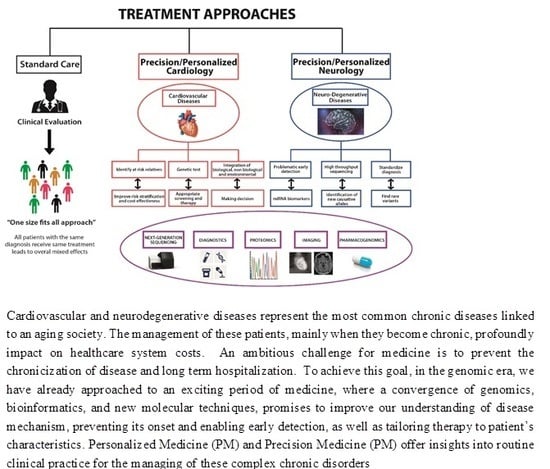
1. Introduction
2. What Is the Difference between Personalized Medicine and Precision Medicine
Precision vs. Personalized Medicine: What Is the Difference?
3. Brief View of Sequencing Eras
4. NGS Applications in Precision Medicine
4.1. Neurodegenerative Diseases
4.2. Cardiovascular Diseases
4.3. Calcific Disease of Aortic Valve
4.4. Dilated Cardiomyopathy
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Roser, M.; Ortiz-Ospina, E.; Ritchie, H. Life Expectancy; Our World in Data: Oxford, UK, 2013. [Google Scholar]
- United Nations. World Population Ageing 2015; Department of Economic and Social Affairs, Population Division: New York, NY, USA, 2013. [Google Scholar]
- Crimmins, E.M. Trends in the health of the elderly. Annu. Rev. Public Health 2004, 25, 79–98. [Google Scholar] [CrossRef] [PubMed]
- Beard, J.; Biggs, S.; Bloom, D.E.; Fried, L.P.; Hogan, P.R.; Kalache, A.; Olshansky, S.J. Global Population Ageing: Peril or Promise? Program on the Global Demography of Aging: Harvard, MA, USA, 2012. [Google Scholar]
- Yenilmez, M.I. Economic and social consequences of population aging the dilemmas and opportunities in the twenty-first century. Appl. Res. Qual. Life 2015, 10, 735–752. [Google Scholar] [CrossRef]
- Atella, V.; Mortari, A.P.; Kopinska, J.; Belotti, F.; Lapi, F.; Cricelli, C.; Fontana, L. Trends in age-related disease burden and healthcare utilization. Aging Cell 2019, 18, e12861. [Google Scholar] [CrossRef]
- Busse, R.; Blümel, M. Tackling Chronic Disease in Europe: Strategies, Interventions and Challenges; WHO Regional Office Europe: København, Denmark, 2010. [Google Scholar]
- Fried, L.P.; Paccaud, F. The Public Health Needs for an Ageing Society. Public Health Rev. 2011, 32, 351–355. [Google Scholar] [CrossRef] [Green Version]
- Miller, A.R.; Tucker, C. Privacy protection, personalized medicine, and genetic testing. Manag. Sci. 2018, 64, 4648–4668. [Google Scholar] [CrossRef] [Green Version]
- National Research Council. Toward Precision Medicine: Building a Knowledge Network for Biomedical Research and a New Taxonomy of Disease; National Academies Press: Washington, DC, USA, 2011. [Google Scholar]
- Tuena, C.; Semonella, M.; Fernández-Álvarez, J.; Colombo, D.; Cipresso, P. Predictive Precision Medicine: Towards the Computational Challenge, in P5 eHealth: An. Agenda for the Health Technologies of the Future; Springer: Berlin/Heidelberg, Germany, 2020; pp. 71–86. [Google Scholar]
- United Nations, Department of Economic and Social Affairs, Population Division. World Population Ageing 2015 Report; ST/ESA/SER. A/390; Department of Economic and Social Affairs, Population Division: New York, NY, USA, 2015. [Google Scholar]
- Kennedy, B.K.; Berger, S.L.; Brunet, A.; Campisi, J.; Cuervo, A.M.; Epel, E.S.; Franceschi, C.; Lithgow, G.J.; Morimoto, R.I.; Pessin, J.E.; et al. Aging: A common driver of chronic diseases and a target for novel interventions. Cell 2014, 159, 709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Currie, G.; Delles, C. Precision Medicine and Personalized Medicine in Cardiovascular Disease, in Sex.-Specific Analysis of Cardiovascular Function; Springer: Berlin/Heidelberg, Germany, 2018; pp. 589–605. [Google Scholar]
- Cappelletti, P. Medicina di precisione e medicina di laboratorio. Riv. Ital. Della Med. Lab.-Ital. J. Lab. Med. 2016, 12, 129–133. [Google Scholar] [CrossRef] [Green Version]
- Gavan, S.P.; Thompson, A.J.; Payne, K. The economic case for precision medicine. Expert Rev. Precis. Med. Drug Dev. 2018, 3, 1–9. [Google Scholar] [CrossRef]
- Jain, K.K. From Molecular Diagnostics to Personalized Medicine; Taylor & Francis: Abingdon, UK, 2002. [Google Scholar]
- Sykiotis, G.P.; Kalliolias, G.D.; Papavassiliou, A.G. Pharmacogenetic principles in the Hippocratic writings. J. Clin. Pharm. 2005, 45, 1218–1220. [Google Scholar] [CrossRef]
- Pray, L. Personalized Medicine: Hope or Hype. Nat. Educ. 2008, 1, 72. [Google Scholar]
- Savoia, C.; Volpe, M.; Grassi, G.; Borghi, C.; Rosei, E.A.; Touyz, R.M. Personalized medicine—A modern approach for the diagnosis and management of hypertension. Clin. Sci. (Lond.) 2017, 131, 2671–2685. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Bamakan, S.M.H.; Qu, Q.; Li, S. Learning for Personalized Medicine: A Comprehensive Review from a Deep Learning Perspective. IEEE Rev. Biomed. Eng. 2018, 12, 194–208. [Google Scholar] [CrossRef] [PubMed]
- Akhondzadeh, S. Personalized medicine: A tailor made medicine. Avicenna J. Med. Biotechnol. 2014, 6, 191. [Google Scholar] [PubMed]
- Di Sanzo, M.; Cipolloni, L.; Borro, M.; La Russa, R.; Santurro, A.; Scopetti, M.; Simmaco, M.; Frati, P. Clinical applications of personalized medicine: A new paradigm and challenge. Curr. Pharm. Biotechnol. 2017, 18, 194–203. [Google Scholar] [CrossRef]
- Vogenberg, F.R.; Barash, C.I.; Pursel, M. Personalized medicine: Part 1: Evolution and development into theranostics. Pharm. Ther. 2010, 35, 560–576. [Google Scholar]
- Leopold, J.A.; Loscalzo, J. Emerging Role of Precision Medicine in Cardiovascular Disease. Circ. Res. 2018, 122, 1302–1315. [Google Scholar] [CrossRef]
- Maier, M. Personalized medicine-a tradition in general practice! Eur. J. Gen. Pract. 2019, 25, 63–64. [Google Scholar] [CrossRef]
- Healthcare, T.I. Executive Office of the President President’s Council of Advisors on Science and Technology. 2010. Available online: http://www.whitehouse.gov/ostp/pcast (accessed on 29 June 2020).
- Abrahams, E.; Silver, M. The Case for Personalized Medicine; SAGE Publications: Newbury Park, CA, USA, 2009. [Google Scholar]
- Terry, S.F. Obama’s Precision Medicine Initiative. Genet. Test. Mol. Biomark. 2015, 19, 113–114. [Google Scholar] [CrossRef] [Green Version]
- Mills, J.R. Precision Medicine-Right Treatment, Right Patient, Right Time, Wrong Approach? Clin. Chem. 2017, 63, 928–929. [Google Scholar] [CrossRef]
- Olivier, M.; Asmis, R.; Hawkins, G.A.; Howard, T.D.; Cox, L.A. The Need for Multi-Omics Biomarker Signatures in Precision Medicine. Int. J. Mol. Sci. 2019, 20, 4781. [Google Scholar] [CrossRef] [Green Version]
- Ashley, E.A. Towards precision medicine. Nat. Rev. Genet. 2016, 17, 507–522. [Google Scholar] [CrossRef]
- Au, R.; Ritchie, M.; Hardy, S.; Ang, T.F.A.; Lin, H. Aging Well: Using Precision to Drive down Costs and Increase Health Quality. Adv. Geriatr. Med. Res. 2019, 1, 1. [Google Scholar]
- Tan, L.; Jiang, T.; Tan, L.; Yu, J. Toward precision medicine in neurological diseases. Ann. Transl. Med. 2016, 4, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sequencing, H.G. Finishing the euchromatic sequence of the human genome. Nature 2004, 431, 931–945. [Google Scholar]
- Heather, J.M.; Chain, B. The sequence of sequencers: The history of sequencing DNA. Genomics 2016, 107, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Rothberg, J.M.; Leamon, J.H. The development and impact of 454 sequencing. Nat. Biotechnol. 2008, 26, 1117–1124. [Google Scholar] [CrossRef] [PubMed]
- Shendure, J.; Ji, H. Next-generation DNA sequencing. Nat. Biotechnol. 2008, 26, 1135–1145. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef] [PubMed]
- Kalayinia, S.; Goodarzynejad, H.; Maleki, M.; Mahdieh, N. Next generation sequencing applications for cardiovascular disease. Ann. Med. 2018, 50, 91–109. [Google Scholar] [CrossRef] [PubMed]
- Anasykhova, Y.; Barbitoff, Y.A.; A Serebryakova, E.; Katserov, D.; Glotov, A.S. Recent advances and perspectives in next generation sequencing application to the genetic research of type 2 diabetes. World J. Diabetes 2019, 10, 376–395. [Google Scholar] [CrossRef]
- Guerreiro, R.; Bras, J.; Hardy, J.; Singleton, A. Next generation sequencing techniques in neurological diseases: Redefining clinical and molecular associations. Hum. Mol. Genet. 2014, 23, R47–R53. [Google Scholar] [CrossRef] [Green Version]
- Suwinski, P.; Ong, C.; Ling, M.H.T.; Poh, Y.M.; Khan, A.M.; Ong, H.S. Advancing Personalized Medicine Through the Application of Whole Exome Sequencing and Big Data Analytics. Front. Genet. 2019, 10, 49. [Google Scholar] [CrossRef] [Green Version]
- Lohmann, K.; Klein, C. Next generation sequencing and the future of genetic diagnosis. Neurotherapeutics 2014, 11, 699–707. [Google Scholar] [CrossRef] [Green Version]
- Hegde, M.; Santani, A.; Mao, R.; Ferreira-Gonzalez, A.; Weck, K.E.; Voelkerding, K.V. Development and Validation of Clinical Whole-Exome and Whole-Genome Sequencing for Detection of Germline Variants in Inherited Disease. Arch. Pathol. Lab. Med. 2017, 141, 798–805. [Google Scholar] [CrossRef] [Green Version]
- Park, S.T.; Kim, J. Trends in Next-Generation Sequencing and a New Era for Whole Genome Sequencing. Int. Neurourol. J. 2016, 20 (Suppl. 2), S76–S83. [Google Scholar] [CrossRef] [Green Version]
- Pasipoularides, A. The new era of whole-exome sequencing in congenital heart disease: Brand-new insights into rare pathogenic variants. J. Thorac. Dis. 2018, 10 (Suppl. 17), S1923–S1929. [Google Scholar] [CrossRef]
- Marques-Matos, C.; Alonso, I.; Leão, M. Diagnostic yield of next-generation sequencing applied to neurological disorders. J. Clin. Neurosci. 2019, 67, 14–18. [Google Scholar] [CrossRef]
- Leproust, E. Target enrichment strategies for next generation sequencing. MLO Med. Lab. Obs. 2012, 44, 26–27. [Google Scholar]
- Gerdes, L.; Iwobi, A.; Busch, U.; Pecoraro, S. Optimization of digital droplet polymerase chain reaction for quantification of genetically modified organisms. Biomol. Detect. Quantif. 2016, 7, 9–20. [Google Scholar] [CrossRef] [Green Version]
- Niedzicka, M.; Fijarczyk, A.; Dudek, K.; Stuglik, M.; Babik, W. Molecular Inversion Probes for targeted resequencing in non-model organisms. Sci. Rep. 2016, 6, 24051. [Google Scholar] [CrossRef] [PubMed]
- Samorodnitsky, E.; Datta, J.; Jewell, B.M.; Hagopian, R.; Miya, J.; Wing, M.R.; Damodaran, S.; Lippus, J.M.; Reeser, J.W.; Bhatt, D.; et al. Comparison of custom capture for targeted next-generation DNA sequencing. J. Mol. Diagn. 2015, 17, 64–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaudin, M.; Desnues, C. Hybrid Capture-Based Next Generation Sequencing and Its Application to Human Infectious Diseases. Front. Microbiol. 2018, 9, 2924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersen, B.-S.; Fredrich, B.; Hoeppner, M.P.; Ellinghaus, D.; Franke, A. Opportunities and challenges of whole-genome and -exome sequencing. BMC Genet. 2017, 18, 14. [Google Scholar] [CrossRef] [Green Version]
- Bevan, C.L.; Brooke, G.N.; Rj, P.; Sarah, E.; Eg, S. Exome sequencing explained: A practical guide to its clinical application. Brief. Funct. Genom. 2016, 15, 374–384. [Google Scholar]
- Rabbani, B.; Tekin, M.; Mahdieh, N. The promise of whole-exome sequencing in medical genetics. J. Hum. Genet. 2014, 59, 5–15. [Google Scholar] [CrossRef]
- Dong, L.; Wang, W.; Li, A.; Kansal, R.; Chen, Y.; Chen, H.; Li, X. Clinical Next Generation Sequencing for Precision Medicine in Cancer. Curr. Genom. 2015, 16, 253–263. [Google Scholar] [CrossRef] [Green Version]
- Harper, A.; Parikh, V.N.; Goldfeder, R.L.; Caleshu, C.; Ashley, E.A. Delivering clinical grade sequencing and genetic test interpretation for cardiovascular medicine. Circ. Cardiovasc. Genet. 2017, 10, e001221. [Google Scholar] [CrossRef] [Green Version]
- Van Dijk, E.; Jaszczyszyn, Y.; Naquin, D.; Thermes, C. The Third Revolution in Sequencing Technology. Trends Genet. 2018, 34, 666–681. [Google Scholar] [CrossRef]
- Rhoads, A.; López-Baucells, A. PacBio Sequencing and Its Applications. Genom. Proteom. Bioinform. 2015, 13, 278–289. [Google Scholar] [CrossRef] [Green Version]
- De Lannoy, C.; De Ridder, D.; Risse, J. The long reads ahead: De novo genome assembly using the MinION. F1000Research 2017, 6, 1083. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.H.; E Pearson, C. Disease-associated repeat instability and mismatch repair. DNA Repair (Amst) 2016, 38, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Philip, N.; Rodrigues, K.F.; William, T.; John, D.V. Whole genome sequencing of Mycobacterium tuberculosis SB24 isolated from Sabah, Malaysia. Genom. Data 2016, 9, 137–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gitler, A.D.; Dhillon, P.; Shorter, J. Neurodegenerative disease: Models, mechanisms, and a new hope. Dis. Model. Mech. 2017, 10, 499–502. [Google Scholar] [CrossRef] [Green Version]
- Tysnes, O.-B.; Storstein, A. Epidemiology of Parkinson’s disease. J. Neural Transm. (Vienna) 2017, 124, 901–905. [Google Scholar] [CrossRef]
- Reitz, C.; Brayne, C.; Mayeux, R. Epidemiology of Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006239. [Google Scholar] [CrossRef]
- Simon, D.K.; Tanner, C.M.; Brundin, P. Parkinson Disease Epidemiology, Pathology, Genetics, and Pathophysiology. Clin. Geriatr. Med. 2020, 36, 1–12. [Google Scholar] [CrossRef]
- Bekris, L.M.; Yu, C.-E.; Bird, T.D.; Tsuang, D.W. Genetics of Alzheimer disease. J. Geriatr. Psychiatry Neurol. 2010, 23, 213–227. [Google Scholar] [CrossRef] [Green Version]
- Klein, C.; Westenberger, A. Genetics of Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a008888. [Google Scholar] [CrossRef] [Green Version]
- Jiang, T.; Yu, J.; Tian, Y.; Tan, L. Epidemiology and etiology of Alzheimer’s disease: From genetic to non-genetic factors. Curr. Alzheimer Res. 2013, 10, 852–867. [Google Scholar] [CrossRef]
- Gatz, M.; Reynolds, C.A.; Fratiglioni, L.; Johansson, B.; Mortimer, J.A.; Berg, S.; Fiske, A.; Pedersen, N.L. Role of genes and environments for explaining Alzheimer disease. Arch. Gen. Psychiatry 2006, 63, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Montine, T.J.; Montine, K.S. Precision medicine: Clarity for the clinical and biological complexity of Alzheimer’s and Parkinson’s diseases. J. Exp. Med. 2015, 212, 601–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borrageiro, G.; Haylett, W.; Seedat, S.; Kuivaniemi, H.; Bardien, S. A review of genome-wide transcriptomics studies in Parkinson’s disease. Eur. J. Neurosci. 2018, 47, 1–16. [Google Scholar] [CrossRef]
- Chew, G.; Petretto, E. Transcriptional Networks of Microglia in Alzheimer’s Disease and Insights into Pathogenesis. Genes 2019, 10, 798. [Google Scholar] [CrossRef] [Green Version]
- Dube, U.; Dian, T.; Del-Aguila, J.L.; Li, Z.; Budde, J.P.; Jiang, S.; Hsu, S.; Ibanez, L.; Fernandez, M.V.; Farias, F.; et al. An atlas of cortical circular RNA expression in Alzheimer disease brains demonstrates clinical and pathological associations. Nat. Neurosci. 2019, 22, 1903–1912. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Initiative, F.A.D.N.; Miller, J.E.; Byun, S.; Kim, D.; Risacher, S.L.; Saykin, A.J.; Lee, Y.; Nho, K. Identification of exon skipping events associated with Alzheimer’s disease in the human hippocampus. BMC Med. Genom. 2019, 12 (Suppl. 1), 13. [Google Scholar] [CrossRef] [Green Version]
- Alaaeddine, R.; Fayad, M.; Nehme, E.; Bahmad, H.F.; Kobeissy, F. The Emerging Role of Proteomics in Precision Medicine: Applications in Neurodegenerative Diseases and Neurotrauma. Adv. Exp. Med. Biol. 2017, 1007, 59–70. [Google Scholar]
- Strafella, C.; Caputo, V.; Galota, M.R.; Zampatti, S.; Marella, G.; Mauriello, S.; Cascella, R.; Giardina, E. Application of Precision Medicine in Neurodegenerative Diseases. Front. Neurol. 2018, 9, 701. [Google Scholar] [CrossRef] [Green Version]
- Hampel, H.; O’Bryant, S.; Castrillo, J.; Ritchie, C.; Rojkova, K.; Broich, K.; Benda, N.; Nisticò, R.; Frank, R.; Dubois, B.; et al. PRECISION MEDICINE—The Golden Gate for Detection, Treatment and Prevention of Alzheimer’s Disease. J. Prev. Alzheimers Dis. 2016, 3, 243–259. [Google Scholar]
- Giardina, E.; Caltagirone, C.F. The IRCCS Network of Neuroscience and Neurorehabilitation: The Italian Platform for Care and Research about Neurodegenerative Disorders; Wiley: Hoboken, NJ, USA, 2018. [Google Scholar]
- Nuzziello, N.; Ciaccia, L.; Liguori, M. Precision Medicine in Neurodegenerative Diseases: Some Promising Tips Coming from the microRNAs’ World. Cells 2019, 9, 75. [Google Scholar] [CrossRef] [Green Version]
- Alieva, A.K.; Filatova, E.V.; Karabanov, A.V.; Illarioshkin, S.N.; Limborska, S.A.; Shadrina, M.I.; Slominsky, P.A. miRNA expression is highly sensitive to a drug therapy in Parkinson’s disease. Parkinsonism Relat. Disord. 2015, 21, 72–74. [Google Scholar] [CrossRef]
- Margis, R.; Rieder, C.R. Identification of blood microRNAs associated to Parkinsonĭs disease. J. Biotechnol. 2011, 152, 96–101. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.-N.; Wang, Y.-J.; Wang, H.; Song, L.; Chen, Y.; Wang, J.-L.; Ye, Y.; Jiang, B. The Anti-dementia Effects of Donepezil Involve miR-206-3p in the Hippocampus and Cortex. Biol. Pharm. Bull. 2017, 40, 465–472. [Google Scholar] [CrossRef] [Green Version]
- Tian, N.; Cao, Z.; Zhang, Y. MiR-206 decreases brain-derived neurotrophic factor levels in a transgenic mouse model of Alzheimer’s disease. Neurosci. Bull. 2014, 30, 191–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruts, M.; Theuns, J.; Van Broeckhoven, C. Locus-specific mutation databases for neurodegenerative brain diseases. Hum. Mutat. 2012, 33, 1340–1344. [Google Scholar] [CrossRef] [Green Version]
- Cruchaga, C.; Karch, C.; Jin, S.C.; Benitez, B.A.; Cai, Y.; Guerreiro, R.; Harari, O.; Norton, J.; Budde, J.P. UK Brain Expression Consortium (UKBEC); et al. Rare coding variants in the phospholipase D3 gene confer risk for Alzheimer’s disease. Nature 2014, 505, 550–554. [Google Scholar] [CrossRef] [Green Version]
- Jonsson, T.; Stefansson, K. TREM2 and neurodegenerative disease. N. Engl. J. Med. 2013, 369, 1568–1569. [Google Scholar]
- Bravo, P.; Darvish, H.; Tafakhori, A.; Azcona, L.J.; Johari, A.H.; Jamali, F.; Paisán-Ruiz, C. Molecular characterization of PRKN structural variations identified through whole-genome sequencing. Mol. Genet. Genom. Med. 2018, 6, 1243–1248. [Google Scholar] [CrossRef] [Green Version]
- Nicolas, A.; Kenna, K.P.; Renton, A.E.; Ticozzi, N.; Faghri, F.; Chia, R.; Dominov, J.A.; Kenna, B.J.; Nalls, M.A.; Keagle, P.; et al. Genome-wide Analyses Identify KIF5A as a Novel ALS Gene. Neuron 2018, 97, 1268–1283.e6. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.E.; Shivakumar, M.K.; Lee, Y.; Han, S.; Horgousluoglu, E.; Risacher, S.L.; Saykin, A.J.; Nho, K.; Kim, D.; Initiative, A.D.N.; et al. Rare variants in the splicing regulatory elements of EXOC3L4 are associated with brain glucose metabolism in Alzheimer’s disease. BMC Med. Genom. 2018, 11 (Suppl. 3), 76. [Google Scholar] [CrossRef]
- Zhao, L.; He, Z.; Zhang, D.; Wang, G.T.; Renton, A.E.; Vardarajan, B.N.; Nothnagel, M.; Goate, A.M.; Mayeux, R.; Leal, S.M. A Rare Variant Nonparametric Linkage Method for Nuclear and Extended Pedigrees with Application to Late-Onset Alzheimer Disease via WGS Data. Am. J. Hum. Genet. 2019, 105, 822–835. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, E.; Darvish, H.; Mesias, R.; Taghavi, S.; Firouzabadi, S.G.; Walker, R.H.; Tafakhori, A.; Paisán-Ruiz, C. Identification of a Large DNAJB2 Deletion in a Family with Spinal Muscular Atrophy and Parkinsonism. Hum. Mutat. 2016, 37, 1180–1189. [Google Scholar] [CrossRef] [Green Version]
- Butcher, N.J.; Merico, D.; Zarrei, M.; Ogura, L.; Marshall, C.R.; Chow, E.W.C.; Lang, A.E.; Scherer, S.W.; Bassett, A.S. Whole-genome sequencing suggests mechanisms for 22q11. 2 deletion-associated Parkinson’s disease. PLoS ONE 2017, 12, e0173944. [Google Scholar] [CrossRef]
- Vardarajan, B.N.; Ghani, M.; Kahn, A.; Sheikh, S.; Sato, C.; Barral, S.; Lee, J.H.; Cheng, R.; Reitz, C.; Lantigua, R.; et al. Rare coding mutations identified by sequencing of Alzheimer disease genome-wide association studies loci. Ann. Neurol. 2015, 78, 487–498. [Google Scholar] [CrossRef]
- Germer, E.L.; Imhoff, S.; Vilariño-Güell, C.; Kasten, M.; Seibler, P.; Brüggemann, N.; Klein, C.; Trinh, J. International Parkinson’s Disease Genomics Consortium. The Role of Rare Coding Variants in Parkinson’s Disease GWAS Loci. Front. Neurol. 2019, 10, 1284. [Google Scholar] [CrossRef] [Green Version]
- Blanckenberg, J.; Bardien, S.; Glanzmann, B.; Okubadejo, N.U.; Carr, J. The prevalence and genetics of Parkinson’s disease in sub-Saharan Africans. J. Neurol. Sci. 2013, 335, 22–25. [Google Scholar] [CrossRef]
- Oluwole, O.G.; Kuivaniemi, H.; Abrahams, S.; Haylett, W.L.; Vorster, A.A.; Van Heerden, C.J.; Kenyon, C.P.; Tabb, D.L.; Fawale, M.B.; Sunmonu, T.A.; et al. Targeted next-generation sequencing identifies novel variants in candidate genes for Parkinson’s disease in Black South African and Nigerian patients. BMC Med. Genet. 2020, 21, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gialluisi, A.; Reccia, M.G.; Tirozzi, A.; Nutile, T.; Lombardi, A.; De Sanctis, C.; Varanese, S.; Pietracupa, S.; Modugno, N.; Simeone, A.; et al. Whole Exome Sequencing study of Parkinson Disease and related endophenotypes in the Italian Population. Front. Neurol. 2020, 10, 1362. [Google Scholar] [CrossRef] [Green Version]
- Jansen, I.E.; Ye, H.; Heetveld, S.; Lechler, M.C.; Michels, H.; Seinstra, R.I.; Lubbe, S.J.; Drouet, V.; Lesage, S. International Parkinson’s Disease Genetics Consortium (IPGDC); et al. Discovery and functional prioritization of Parkinson’s disease candidate genes from large-scale whole exome sequencing. Genome Biol. 2017, 18, 22. [Google Scholar] [CrossRef] [Green Version]
- Kessler, T.; Vilne, B.; Schunkert, H. The impact of genome-wide association studies on the pathophysiology and therapy of cardiovascular disease. EMBO Mol. Med. 2016, 8, 688–701. [Google Scholar] [CrossRef]
- Arking, D.; Chakravarti, A. Understanding cardiovascular disease through the lens of genome-wide association studies. Trends Genet. 2009, 25, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Barrett, J.C.; Dunham, I.; Birney, E. Using human genetics to make new medicines. Nat. Rev. Genet. 2015, 16, 561–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myocardial Infarction Genetics Consortium. Genome-wide association of early-onset myocardial infarction with single nucleotide polymorphisms and copy number variants. Nat. Genet. 2009, 41, 334–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abifadel, M.; Varret, M.; Rabes, J.-P.; Allard, D.; Ouguerram, K.; Devillers, M.; Cruaud, C.; Benjannet, S.; Wickham, L.; Erlich, D.; et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat. Genet. 2003, 34, 154–156. [Google Scholar] [CrossRef]
- Cohen, J.C.; Boerwinkle, E.; Mosley, T.H.; Hobbs, H.H. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N. Engl. J. Med. 2006, 354, 1264–1272. [Google Scholar] [CrossRef]
- Teslovich, T.M.; Musunuru, K.; Smith, A.V.; Edmondson, A.C.; Stylianou, I.M.; Koseki, M.; Pirruccello, J.P.; Ripatti, S.; Chasman, D.I.; Willer, C.J.; et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature 2010, 466, 707–713. [Google Scholar] [CrossRef]
- Peden, J.F.; Hopewell, J.C.; Saleheen, D.; Chambers, J.C.; Hager, J.; Soranzo, N.; Collins, R.; Danesh, J.; Elliott, P. The Coronary Artery Disease (C4D) Genetics Consortium; et al. A genome-wide association study in Europeans and South Asians identifies five new loci for coronary artery disease. Nat. Genet. 2011, 43, 339–344. [Google Scholar]
- Smith, N.L.; Felix, J.F.; Morrison, A.; Demissie, S.; Glazer, N.; Loehr, L.R.; Cupples, L.A.; Dehghan, A.; Lumley, T.; Rosamond, W.D.; et al. Association of genome-wide variation with the risk of incident heart failure in adults of European and African ancestry: A prospective meta-analysis from the cohorts for heart and aging research in genomic epidemiology (CHARGE) consortium. Circ. Cardiovasc. Genet. 2010, 3, 256–266. [Google Scholar] [CrossRef]
- Villard, E.; Perret, C.; Gary, F.; Proust, C.; Dilanian, G.; Hengstenberg, C.; Ruppert, V.; Arbustini, E.; Wichter, T.; Germain, M.; et al. A genome-wide association study identifies two loci associated with heart failure due to dilated cardiomyopathy. Eur. Heart J. 2011, 32, 1065–1076. [Google Scholar] [CrossRef] [Green Version]
- Walsh, R.; Exome Aggregation Consortium; Thomson, K.L.; Ware, J.S.; Funke, B.; Woodley, J.; McGuire, K.J.; Mazzarotto, F.; Blair, E.; Seller, A.; et al. Reassessment of Mendelian gene pathogenicity using 7855 cardiomyopathy cases and 60,706 reference samples. Genet. Med. 2017, 19, 192–203. [Google Scholar] [CrossRef] [Green Version]
- Hershberger, R.E.; Hedges, D.J.; Morales, A. Dilated cardiomyopathy: The complexity of a diverse genetic architecture. Nat. Rev. Cardiol. 2013, 10, 531–547. [Google Scholar] [CrossRef]
- Jabbari, J.; Olesen, M.S.; Yuan, L.; Nielsen, J.B.; Liang, B.; Macri, V.; Christophersen, I.E.; Nielsen, N.; Sajadieh, A.; Ellinor, P.T.; et al. Common and rare variants in SCN10A modulate the risk of atrial fibrillation. Circ. Cardiovasc. Genet. 2015, 8, 64–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roselli, C. Atrial fibrillation. Nat. Genet. 2018, 50, 1225–1233. [Google Scholar] [CrossRef]
- Chiu, C.; Bagnall, R.D.; Ingles, J.; Yeates, L.; Kennerson, M.; Donald, J.A.; Jormakka, M.; Lind, J.M.; Semsarian, C. Mutations in alpha-actinin-2 cause hypertrophic cardiomyopathy: A genome-wide analysis. J. Am. Coll. Cardiol. 2010, 55, 1127–1135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumgartner, H.; Falk, V.; Bax, J.J.; De Bonis, M.; Hamm, C.; Holm, P.J.; Lansac, E.; Muñoz, D.R.; Rosenhek, R.; Sjögren, J.; et al. 2017 ESC/EACTS Guidelines for the management of valvular heart disease. Eur. Heart J. 2017, 38, 2739–2791. [Google Scholar] [CrossRef] [PubMed]
- Small, A.; Kiss, D.; Giri, J.; Anwaruddin, S.; Siddiqi, H.; Guerraty, M.; Chirinos, J.A.; Ferrari, G.; Rader, D. Biomarkers of Calcific Aortic Valve Disease. Arter. Thromb. Vasc. Biol. 2017, 37, 623–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cowell, S.J.; E Newby, D.; Burton, J.; White, A.; Northridge, D.B.; A Boon, N.; Reid, J. Aortic valve calcification on computed tomography predicts the severity of aortic stenosis. Clin. Radiol. 2003, 58, 712–716. [Google Scholar] [CrossRef]
- Cueff, C.; Cimadevilla, C.; Himbert, D.; Tubach, F.; Duval, X.; Iung, B.; Enriquez-Sarano, M.; Vahanian, A.; Messika-Zeitoun, D.; Serfaty, J.-M.; et al. Measurement of aortic valve calcification using multislice computed tomography: Correlation with haemodynamic severity of aortic stenosis and clinical implication for patients with low ejection fraction. Heart 2011, 97, 721–726. [Google Scholar] [CrossRef]
- Capoulade, R.; Chan, K.L.; Yeang, C.; Mathieu, P.; Bossé, Y.; Dumesnil, J.G.; Tam, J.W.; Teo, K.K.; Mahmut, A.; Yang, X.; et al. Oxidized Phospholipids, Lipoprotein(a), and Progression of Calcific Aortic Valve Stenosis. J. Am. Coll. Cardiol. 2015, 66, 1236–1246. [Google Scholar] [CrossRef] [Green Version]
- Stewart, B.F.; Siscovick, D.; Lind, B.K.; Gardin, J.M.; Gottdiener, J.S.; Smith, V.E.; Kitzman, D.W.; Otto, C.M. Clinical factors associated with calcific aortic valve disease. Cardiovasc. Health Study. J. Am. Coll. Cardiol. 1997, 29, 630–634. [Google Scholar] [CrossRef] [Green Version]
- Thanassoulis, G.; Campbell, C.Y.; Owens, D.; Smith, J.G.; Smith, A.V.; Peloso, G.M.; Kerr, K.F.; Pechlivanis, S.; Budoff, M.J.; Harris, T.B.; et al. Genetic associations with valvular calcification and aortic stenosis. N. Engl. J. Med. 2013, 368, 503–512. [Google Scholar] [CrossRef] [Green Version]
- Emdin, C.A.; Khera, A.V.; Natarajan, P.; Klarin, D.; Won, H.-H.; Peloso, G.M.; Stitziel, N.O.; Nomura, A.; Zekavat, S.M.; Bick, A.; et al. Phenotypic Characterization of Genetically Lowered Human Lipoprotein(a) Levels. J. Am. Coll. Cardiol. 2016, 68, 2761–2772. [Google Scholar] [CrossRef]
- Garg, V.; Muth, A.N.; Ransom, J.F.; Schluterman, M.K.; Barnes, R.; King, I.N.; Grossfeld, P.D.; Srivastava, D. Mutations in NOTCH1 cause aortic valve disease. Nature 2005, 437, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Theodoris, C.; Li, M.; White, M.P.; Liu, L.; He, D.; Pollard, K.S.; Bruneau, B.G.; Srivastava, D. Human disease modeling reveals integrated transcriptional and epigenetic mechanisms of NOTCH1 haploinsufficiency. Cell 2015, 160, 1072–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, Q.; Song, R.; Ao, L.; Weyant, M.J.; Lee, J.; Xu, D.; Fullerton, D.A.; Meng, X. Notch1 promotes the pro-osteogenic response of human aortic valve interstitial cells via modulation of ERK1/2 and nuclear factor-κB activation. Arter. Thromb. Vasc. Biol. 2013, 33, 1580–1590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lahaye, S.; Lincoln, J.; Garg, V. Genetics of valvular heart disease. Curr. Cardiol. Rep. 2014, 16, 487. [Google Scholar] [CrossRef] [Green Version]
- Jaiswal, S.; Natarajan, P.; Silver, A.J.; Gibson, C.J.; Bick, A.; Shvartz, E.; McConkey, M.; Gupta, N.; Gabriel, S.; Ardissino, D.; et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Berglund, G.; Elmstähl, S.; Janzon, L.; A Larsson, S. The Malmo Diet and Cancer Study. Design and feasibility. J. Intern. Med. 1993, 233, 45–51. [Google Scholar] [PubMed]
- Muntendam, P.; McCall, C.; Sanz, J.; Falk, E.; Fuster, V. The BioImage Study: Novel approaches to risk assessment in the primary prevention of atherosclerotic cardiovascular disease—study design and objectives. Am. Heart J. 2010, 160, 49–57.e1. [Google Scholar] [CrossRef]
- Fuster, J.J.; MacLauchlan, S.; Zuriaga, M.A.; Polackal, M.N.; Ostriker, A.C.; Chakraborty, R.; Wu, C.-L.; Sano, S.; Muralidharan, S.; Rius, C.; et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 2017, 355, 842–847. [Google Scholar] [CrossRef] [Green Version]
- Li, X.F.; Wang, Y.; Zheng, D.-D.; Xu, H.-X.; Wang, T.; Pan, M.; Shi, J.-H.; Zhu, J.-H. M1 macrophages promote aortic valve calcification mediated by microRNA-214/TWIST1 pathway in valvular interstitial cells. Am. J. Transl. Res. 2016, 8, 5773–5783. [Google Scholar]
- Aquila, G.; Fortini, C.; Pannuti, A.; Delbue, S.; Pannella, M.; Morelli, M.B.; Caliceti, C.; Castriota, F.; De Mattei, M.; Ongaro, A.; et al. Distinct gene expression profiles associated with Notch ligands Delta-like 4 and Jagged1 in plaque material from peripheral artery disease patients: A pilot study. J. Transl. Med. 2017, 15, 98. [Google Scholar] [CrossRef] [PubMed]
- Akula, M.K.; Shi, M.; Jiang, Z.; Foster, C.E.; Miao, D.; Li, A.S.; Zhang, X.; Gavin, R.M.; Forde, S.D.; Germain, G.; et al. Control of the innate immune response by the mevalonate pathway. Nat. Immunol. 2016, 17, 922–929. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Lan, Y.; Schwartz-Orbach, L.; Korol, E.; Tahiliani, M.; Evans, T.; Goll, M.G. Overlapping Requirements for Tet2 and Tet3 in Normal Development and Hematopoietic Stem Cell Emergence. Cell Rep. 2015, 12, 1133–1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bondue, A.; Arbustini, E.; Bianco, A.M.; Ciccarelli, M.; Dawson, D.K.; De Rosa, M.; Hamdani, N.; Hilfiker-Kleiner, D.; Meder, B.; Leite-Moreira, A.; et al. Complex roads from genotype to phenotype in dilated cardiomyopathy: Scientific update from the Working Group of Myocardial Function of the European Society of Cardiology. Cardiovasc. Res. 2018, 114, 1287–1303. [Google Scholar] [CrossRef] [Green Version]
- McNally, E.; Mestroni, L. Dilated Cardiomyopathy: Genetic Determinants and Mechanisms. Circ. Res. 2017, 121, 731–748. [Google Scholar] [CrossRef]
- Hershberger, R.E.; Pinto, J.R.; Parks, S.B.; Kushner, J.D.; Li, D.; Ludwigsen, S.; Cowan, J.; Morales, A.; Parvatiyar, M.S.; Potter, J.D. Clinical and functional characterization of TNNT2 mutations identified in patients with dilated cardiomyopathy. Circ. Cardiovasc. Genet. 2009, 2, 306–313. [Google Scholar] [CrossRef] [Green Version]
- Kamisago, M.; Sharma, S.D.; DePalma, S.R.; Solomon, S.; Sharma, P.; McDonough, B.; Smoot, L.; Mullen, M.P.; Woolf, P.K.; Wigle, E.D.; et al. Mutations in sarcomere protein genes as a cause of dilated cardiomyopathy. N. Engl. J. Med. 2000, 343, 1688–1696. [Google Scholar] [CrossRef]
- Li, D.; Czernuszewicz, G.Z.; Gonzalez, O.; Tapscott, T.; Karibe, A.; Durand, J.-B.; Brugada, R.; Hill, R.; Gregoritch, J.M.; Anderson, J.L.; et al. Novel cardiac troponin T mutation as a cause of familial dilated cardiomyopathy. Circulation 2001, 104, 2188–2193. [Google Scholar] [CrossRef] [Green Version]
- Hanson, E.L.; Jakobs, P.M.; Keegan, H.; Coates, K.; Bousman, S.; Dienel, N.H.; Litt, M.; Hershberger, R.E. Cardiac troponin T lysine 210 deletion in a family with dilated cardiomyopathy. J. Card Fail. 2002, 8, 28–32. [Google Scholar] [CrossRef]
- Mogensen, J.; Murphy, R.T.; Kubo, T.; Bahl, A.; Moon, J.C.; Klausen, I.C.; Elliott, P.M.; McKenna, W.J. Frequency and clinical expression of cardiac troponin I mutations in 748 consecutive families with hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2004, 44, 2315–2325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charron, P.; Arad, M.; Arbustini, E.; Basso, C.; Bilinska, Z.T.; Elliott, P.; Helio, T.; Keren, A.; McKenna, W.J.; Monserrat, L.; et al. Genetic counselling and testing in cardiomyopathies: A position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2010, 31, 2715–2726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merlo, M.; Cannatà, A.; Gobbo, M.; Stolfo, D.; Elliott, P.M.; Sinagra, G. Evolving concepts in dilated cardiomyopathy. Eur. J. Heart Fail. 2018, 20, 228–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, T.M.; Ware, S.M. Toward Personalized Medicine: Does Genetic Diagnosis of Pediatric Cardiomyopathy Influence Patient Management? Prog. Pediatr. Cardiol. 2015, 39, 43–47. [Google Scholar] [CrossRef] [Green Version]
- Sorani, M.D.; Yue, J.K.; Sharma, S.; Manley, G.T.; Ferguson, A.R.; TRACK TBI Investigators; Cooper, M.S.R.; Dams-O’Connor, K.; Gordon, W.A.; Lingsma, H.F.; et al. Genetic data sharing and privacy. Neuroinformatics 2015, 13, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Thorogood, A.; Dalpé, G.; Knoppers, B.M. Return of individual genomic research results: Are laws and policies keeping step? Eur. J. Hum. Genet. 2019, 27, 535–546. [Google Scholar] [CrossRef] [Green Version]
- Chandrasekaran, A.; Mathaiyan, J.; Davis, S. Ethics of genomic research. Perspect. Clin. Res. 2013, 4, 100–104. [Google Scholar] [CrossRef]
- Issa, A.M. Personalized medicine and the practice of medicine in the 21st century. Mcgill. J. Med. 2007, 10, 53–57. [Google Scholar]
- E Pritchard, D.; Moeckel, F.; Villa, M.S.; Housman, L.T.; Mccarty, C.A.; McLeod, H.L. Strategies for integrating personalized medicine into healthcare practice. Per. Med. 2017, 14, 141–152. [Google Scholar] [CrossRef] [Green Version]
- Iriart, J.A.B. Precision medicine/personalized medicine: A critical analysis of movements in the transformation of biomedicine in the early 21st century. Cad. Saude Publica 2019, 35, e00153118. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
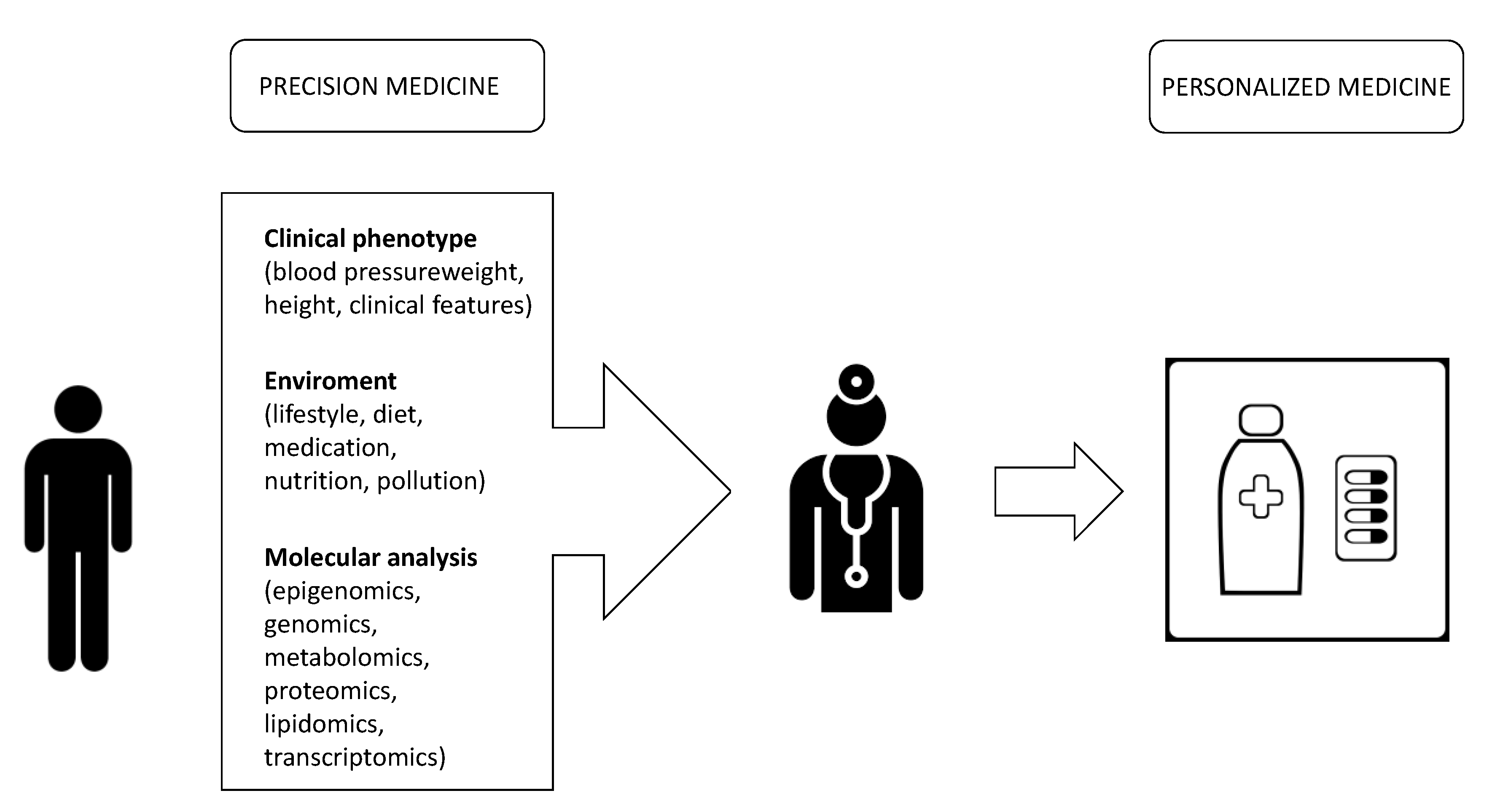{kind=link}
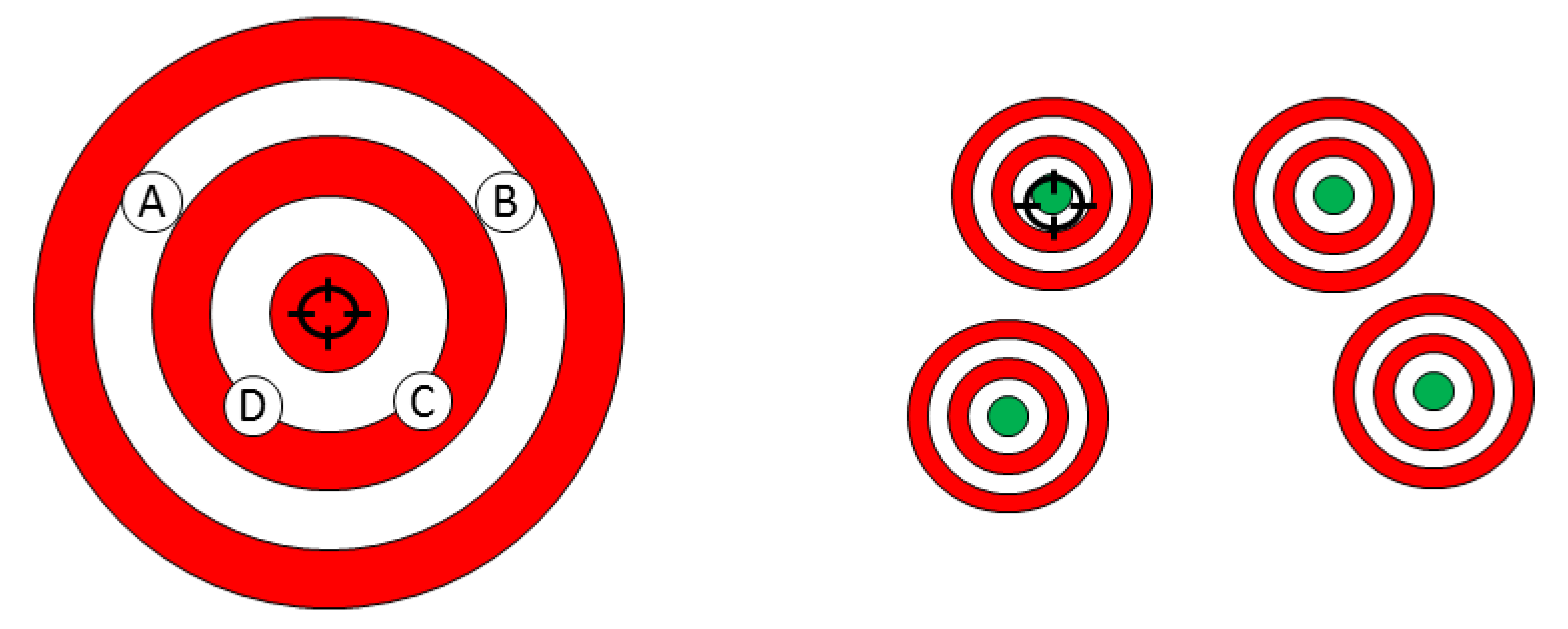{kind=link}
{kind=link}
| Technique | Target Regions | Variants Detected | Advantages | Limitations |
|---|---|---|---|---|
| WGS | Entire genome | ~4,000,000 |
|
|
| WES | 2% of genome | ~20,000 |
|
|
| Target panels | Few genes | Variable: depends on the panel size |
|
|
| Gene/Locus | Disease | Reference |
|---|---|---|
| TREM2 | Alzheimer’s | Jonsson et al., 2013 [88] |
| ABCA7, BIN1, CD2AP, CLU, CR1, EPHA1, MS4A4A/MS4A6A, PICALM | Alzheimer’s | Vardarajan et al., 2015 [90] |
| KIF5A | Alzheimer’s | Nicolas et al., 2018 [91] |
| EXOC3L4 | Alzheimer’s | Miller et al., 2018 [92] |
| PSMF1, PTPN21, ABCA7, ACE, EPHA1, SORL1 | Alzheimer’s | Zhao et al., 2019 [93] |
| DNAJB2, HSJ1 | Parkinson’s | Sanchez et al., 2016 [94] |
| 22q11.2 | Parkinson’s | Butcher et al., 2017 [95] |
| PRKN | Parkinson’s | Bravo et al., 2018 [90] |
| DNAH1, STAB1, ANK2, SH3GL2, NOD2 | Parkinson’s | Germer et al., 2019 [96] |
| Genes | Associated CVD | Reference |
|---|---|---|
| PCSK9 | Myocardial Infarction | Myocardial Infarction Genetics Consortium (2009) [105]; Abifadel M. et al. (2003) [106]; Cohen J.C. et al. (2006) [107]; Teslovich T.M. et al. (2010) [108] |
| PDGFD | Coronary Artery Disease. | Coronary Artery Disease C4D Genetics Consortium (2011) [109] |
| LRIG3 | Congestive Heart Failure | Smith N.L. et al. (2010) [110] |
| ZBTB17 BAG 3 MYBPC3 LMNA PLN | DCM | Villard E. et al. (2011) [111]; Walsh R. et al. (2017) [112]; Hershberger R.E. et al. (2013) [113] |
| PITX2 SCN10A | Atrial Fibrillation | Jabbari J. et al. (2016) [114], Roselli C. et al. (2018) [115] |
| ACTN2 | Hypertrophic Cardiomyopathy | Chiu C. et al. (2010) [116] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Strianese, O.; Rizzo, F.; Ciccarelli, M.; Galasso, G.; D’Agostino, Y.; Salvati, A.; Del Giudice, C.; Tesorio, P.; Rusciano, M.R. Precision and Personalized Medicine: How Genomic Approach Improves the Management of Cardiovascular and Neurodegenerative Disease. Genes 2020, 11, 747. https://doi.org/10.3390/genes11070747
Strianese O, Rizzo F, Ciccarelli M, Galasso G, D’Agostino Y, Salvati A, Del Giudice C, Tesorio P, Rusciano MR. Precision and Personalized Medicine: How Genomic Approach Improves the Management of Cardiovascular and Neurodegenerative Disease. Genes. 2020; 11(7):747. https://doi.org/10.3390/genes11070747
Chicago/Turabian StyleStrianese, Oriana, Francesca Rizzo, Michele Ciccarelli, Gennaro Galasso, Ylenia D’Agostino, Annamaria Salvati, Carmine Del Giudice, Paola Tesorio, and Maria Rosaria Rusciano. 2020. "Precision and Personalized Medicine: How Genomic Approach Improves the Management of Cardiovascular and Neurodegenerative Disease" Genes 11, no. 7: 747. https://doi.org/10.3390/genes11070747
APA StyleStrianese, O., Rizzo, F., Ciccarelli, M., Galasso, G., D’Agostino, Y., Salvati, A., Del Giudice, C., Tesorio, P., & Rusciano, M. R. (2020). Precision and Personalized Medicine: How Genomic Approach Improves the Management of Cardiovascular and Neurodegenerative Disease. Genes, 11(7), 747. https://doi.org/10.3390/genes11070747