DNA Methylation Dysfunction in Chronic Kidney Disease

Abstract
:1. Introduction
2. Chronic Kidney Disease: A Worldwide Pandemic
3. Anemia and Erythropoietin in CKD
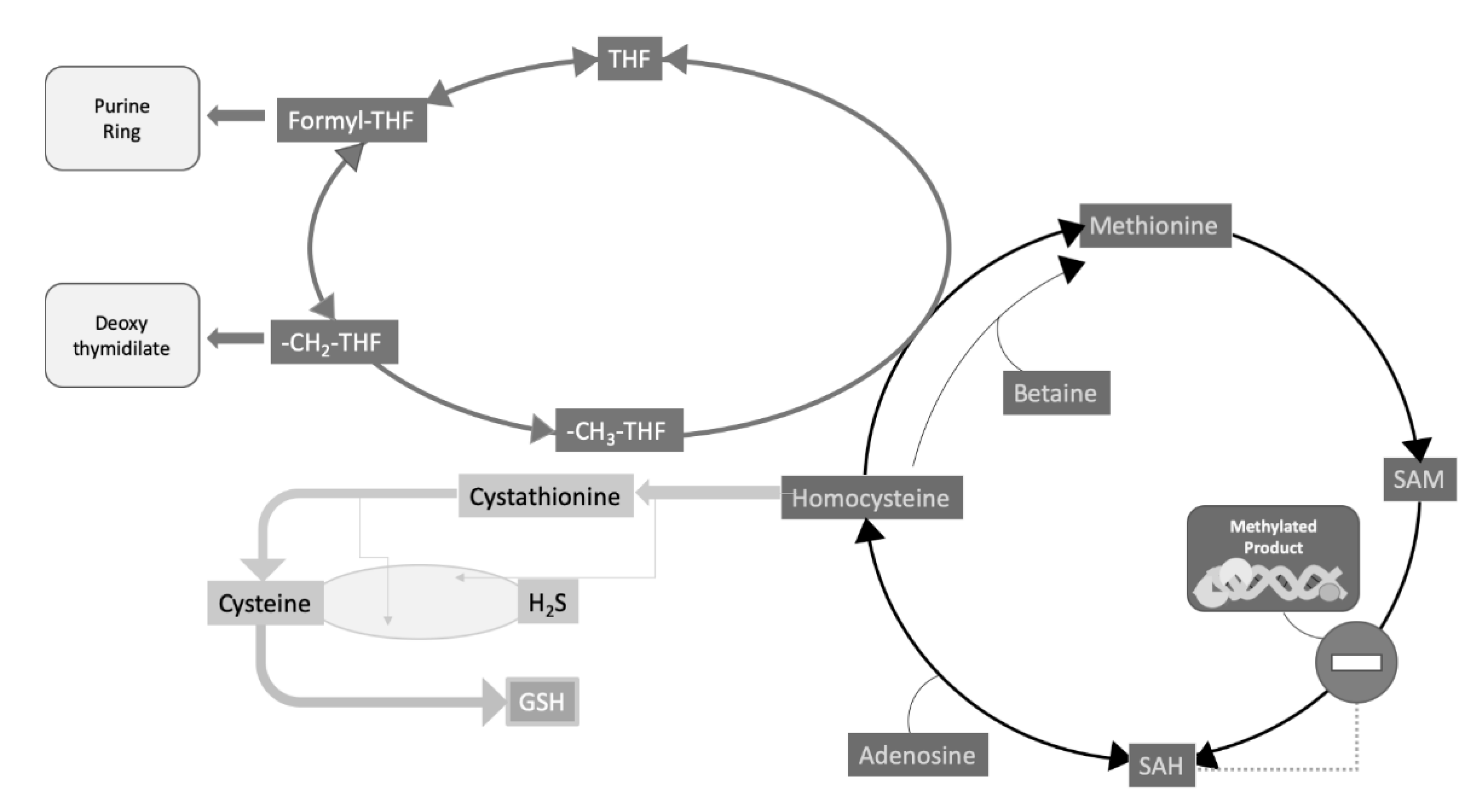
4. CKD and Cardiovascular Risk Increase: The Case for Hyperhomocysteinemia and Folate Intake, Relevant to DNA Methylation
5. DNA Methylation as a Potential Marker of Kidney Function, Inflammation and Fibrosis and CKD Progression
6. DNA Methylation as a Potential Therapeutic Target and to Prevent CKD Progression
7. Onconephrology: A New Frontier in Medicine
8. Synopsis and Conclusive Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Inker, L.A.; Astor, B.C.; Fox, C.H.; Isakova, T.; Lash, J.P.; Peralta, C.A.; Tamura, M.K.; Feldman, H.I. KDOQI US commentary on the 2012 KDIGO clinical practice guideline for the evaluation and management of CKD. Am. J. Kidney Dis. 2014, 63, 713–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levey, A.S.; Coresh, J. Chronic kidney disease. Lancet 2012, 379, 165–180. [Google Scholar] [CrossRef]
- Coresh, J.; Selvin, E.; Stevens, L.A.; Manzi, J.; Kusek, J.W.; Eggers, P.; Van Lente, F.; Levey, A.S. Prevalence of chronic kidney disease in the United States. JAMA 2007, 298, 2038–2047. [Google Scholar] [CrossRef] [Green Version]
- Chronic Kidney Disease—World Kidney Day. Available online: https://www.worldkidneyday.org/facts/chronic-kidney-disease/ (accessed on 13 May 2020).
- Kidney Disease Statistics for the United States—NIDDK—NIH. Available online: https://www.niddk.nih.gov/health-information/health-statistics/kidney-disease (accessed on 13 May 2020).
- Agarwal, R. Defining end-stage renal disease in clinical trials: A framework for adjudication. Nephrol. Dial. Transplant. 2016, 31, 864–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, K.; Kumagai, N.; Suzuki, N. Alteration of the DNA Methylation Signature of Renal Erythropoietin-Producing Cells Governs the Sensitivity to Drugs Targeting the Hypoxia-Response Pathway in Kidney Disease Progression. Front. Genet. 2019, 10, 1134. [Google Scholar] [CrossRef] [PubMed]
- Gansevoort, R.T.; Correa-Rotter, R.; Hemmelgarn, B.R.; Jafar, T.H.; Heerspink, H.J.; Mann, J.F.; Matsushita, K.; Wen, C.P. Chronic kidney disease and cardiovascular risk: Epidemiology, mechanisms, and prevention. Lancet 2013, 382, 339–352. [Google Scholar] [CrossRef]
- Sterns, R.H.; Rojas, M.; Bernstein, M.P.; ChennupatIon, S. Exchange Resins for the Treatment of Hyperkalemia: Are They Safe and Effective? J. Am. Soc. Nephrol. 2010, 21, 733–735. [Google Scholar] [CrossRef] [Green Version]
- Kawabata, H.; Iwatani, H.; Yamamichi, Y.; Shirahase, K.; Nagai, N.; Isaka, Y. Tolvaptan Efficiently Reduces Intracellular Fluid: Working Toward a Potential Treatment Option for Cellular Edema. Intern. Med. 2019, 58, 639–642. [Google Scholar] [CrossRef] [Green Version]
- Vargas-Santos, A.B.; Neogi, T. Management of Gout and Hyperuricemia in CKD. Am. J. Kidney Dis. 2017, 70, 422–439. [Google Scholar] [CrossRef]
- Kalani, L.R. Metabolic Acidosis and Subclinical Metabolic Acidosis in CKD. J. Am. Soc. Nephrol. 2018, 29, 376–382. [Google Scholar]
- Filopanti, M.; Corbetta, S.; Barbieri, A.M.; Spada, A. Pharmacology of the calcium sensing receptor. Clin. Cases Miner. Bone Metab. 2013, 10, 162–165. [Google Scholar] [PubMed]
- Wenger, R.H.; Kvietikova, I.; Rolfs, A.; Camenisch, G.; Gassmann, M. Oxygen-regulated erythropoietin gene expression is dependent on a CpG methylation-free hypoxia-inducible factor-1 DNA-binding site. Eur. J. Biochem. 1998, 253, 771–777. [Google Scholar] [CrossRef] [Green Version]
- Yin, H.; Blanchard, K.L. DNA methylation represses the expression of the human erythropoietin gene by two different mechanisms. Blood 2000, 95, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.T.; Yang, C.C.; Pan, S.Y.; Chou, Y.H.; Chang, F.C.; Lai, C.F. DNA methyltransferase inhibition restores erythropoietin production in fibrotic murine kidneys. J. Clin. Investig. 2016, 126, 721–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratcliffe, P.J. HIF-1 and HIF-2: Working alone or together in hypoxia? J. Clin. Investig. 2007, 117, 862–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foley, R.N.; Parfrey, P.S.; Sarnak, M.J. Epidemiology of cardiovascular disease in chronic renal disease. J. Am. Soc. Nephrol. 1998, 9, S16–S23. [Google Scholar] [CrossRef]
- Foley, R.N. Anaemia: Cardiovascular adaptations and maladaptive responses in chronic kidney disease. Nephrol. Dial. Transplant. 2002, 17, 32–34. [Google Scholar] [CrossRef] [Green Version]
- Perna, A.F.; Ingrosso, D. Homocysteine and chronic kidney disease: An ongoing narrative. J. Nephrol. 2019, 32, 673–675. [Google Scholar] [CrossRef] [Green Version]
- Zavadáková, P.; Fowler, B.; Suormala, T.; Novotna, Z.; Mueller, P.; Hennermann, J.B.; Zeman, J.; Vilaseca, M.A.; Vilarinho, L.; Gutsche, S.; et al. cblE type of homocystinuria due to methionine synthase reductase deficiency: Functional correction by minigene expression. Hum. Mutat. 2005, 25, 239–247. [Google Scholar] [CrossRef]
- Müller, T.; Werne, B.; Fowler, B.; Kuhn, W. Nigral endothelial dysfunction, homocysteine, and Parkinson’s disease. Lancet 1999, 354, 126–127. [Google Scholar] [CrossRef]
- Perna, A.F.; Di Nunzio, A.; Amoresano, A.; Pane, F.; Fontanarosa, C.; Pucci, P.; Vigorito, C.; Cirillo, G.; Zacchia, M.; Trepiccione, F.; et al. Divergent behavior of hydrogen sulfide pools and of the sulfur metabolite lanthionine, a novel uremic toxin, in dialysis patients. Biochimie 2016, 126, 97–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stampfer, M.J.; Malinow, M.R.; Willett, W.C.; Newcomer, L.M.; Upson, B.; Ullmann, D.; Tishler, P.V.; Hennekens, C.H. A prospective study of plasma homocyst(e)ine and risk of myocardial infarction in US physicians. JAMA 1992, 268, 877–881. [Google Scholar] [CrossRef] [PubMed]
- Robinson, K.; Gupta, A.; Dennis, V.; Arheart, K.; Chaudhary, D.; Green, R.; Vigo, P.; Mayer, E.L.; Selhub, J.; Kutner, M.; et al. Hyperhomocysteinemia confers an independent increased risk of atherosclerosis in end-stage renal disease and is closely linked to plasma folate and pyridoxine concentrations. Circulation 1996, 94, 2743–2748. [Google Scholar] [CrossRef] [PubMed]
- Perna, A.F.; Lanza, D.; Sepe, I.; Conzo, G.; Altucci, L.; Ingrosso, D. Altered folate receptor 2 expression in uraemic patients on haemodialysis: Implications for folate resistance. Nephrol. Dial. Transplant. 2013, 28, 1214–1224. [Google Scholar] [CrossRef] [Green Version]
- Huo, Y.; Li, J.; Qin, X.; Huang, Y.; Wang, X.; Gottesman, R.F.; Tang, G.; Wang, B.; Chen, D.; He, M.; et al. CSPPT Investigators. Efficacy of folic acid therapy in primary prevention of stroke among adults with hypertension in China: The CSPPT randomized clinical trial. JAMA 2015, 313, 1325–1335. [Google Scholar] [CrossRef] [PubMed]
- Ingrosso, D.; Cimmino, A.; Perna, A.F.; Masella, L.; De Santo, N.G.; De Bonis, M.L.; Vacca, M.; D’Esposito, M.; D’Urso, M.; Galletti, P.; et al. Folate treatment and unbalanced methylation and changes of allelic expression induced by hyperhomocysteinaemia in patients with uraemia. Lancet 2003, 361, 1693–1699. [Google Scholar] [CrossRef]
- Yang, J.; Fang, P.; Yu, D.; Zhang, L.; Zhang, D.; Jiang, X.; Yang, W.Y.; Bottiglieri, T.; Kunapuli, S.P.; Yu, J.; et al. Chronic Kidney Disease Induces Inflammatory CD40+ Monocyte Differentiation via Homocysteine Elevation and DNA Hypomethylation. Circ. Res. 2016, 119, 1226–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perna, A.F.; Ingrosso, D.; Zappia, V.; Galletti, P.; Capasso, G.; De Santo, N.G. Enzymatic Methyl Esterification of Erythrocyte Membrane Proteins Is Impaired in Chronic Renal Failure. Evidence for High Levels of the Natural Inhibitor S-adenosylhomocysteine. J. Clin. Investig. 1993, 91, 2497–2503. [Google Scholar]
- Wang, H.; Yoshizumi, M.; Lai, K.; Tsai, J.C.; Perrella, M.A.; Haber, E.; Lee, M.E. Inhibition of growth and p21ras methylation in vascular endothelial cells by homocysteine but not cysteine. J. Biol. Chem. 1997, 272, 25380–25385. [Google Scholar] [CrossRef] [Green Version]
- van Guldener, C.; Kulik, W.; Berger, R.; Dijkstra, D.A.; Jakobs, C.; Reijngoud, D.J.; Donker, A.J.; Stehouwer, C.D.; De Meer, K. Homocysteine and methionine metabolism in ESRD: A stable isotope study. Kidney Int. 1999, 56, 1064–1071. [Google Scholar] [CrossRef] [Green Version]
- Yi, P.; Melnyk, S.; Pogribna, M.; Pogribny, I.P.; Hine, R.J.; James, S.J. Increase in plasma homocysteine associated with parallel increases in plasma S-adenosylhomocysteine and lymphocyte DNA hypomethylation. J. Biol. Chem. 2000, 275, 29318–29323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perna, A.F.; Ingrosso, D.; De Santo, N.G.; Galletti, P.; Brunone, M.; Zappia, V. Metabolic consequences of folate-induced reduction of hyperhomocysteinemia in uremia. J. Am. Soc. Nephrol. 1997, 8, 1899–1905. [Google Scholar] [PubMed]
- Loehrer, F.M.; Angst, C.P.; Brunner, F.P.; Haefeli, W.E.; Fowler, B. Evidence for disturbed S-adenosylmethionine: S-adenosylhomocysteine ratio in patients with end-stage renal failure: A cause for disturbed methylation reactions? Nephrol. Dial. Transplant. 1998, 13, 656–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laukkanen, M.O.; Mannermaa, S.; Hiltunen, M.O.; Aittomäki, S.; Airenne, K.; Jänne, J.; Ylä-Herttuala, S. Local hypomethylation in atherosclerosis found in rabbit ec-sod gene. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 2171–2178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiltunen, M.O.; Turunen, M.P.; Häkkinen, T.P.; Rutanen, J.; Hedman, M.; Mäkinen, K.; Turunen, A.-M.; Aalto-Setälä, K.; Ylä-Herttuala, S. DNA hypomethylation and methyltransferase expression in atherosclerotic lesions. Vasc. Med. 2002, 7, 5–11. [Google Scholar] [CrossRef] [Green Version]
- Friso, S.; Choi, S.W.; Girelli, D.; Mason, J.B.; Dolnikowski, G.G.; Bagley, P.J.; Olivieri, O.; Jacques, P.F.; Rosenberg, I.H.; Corrocher, R.; et al. A common mutation in the 5,10-methylenetetrahydrofolate reductase gene affects genomic DNA methylation through an interaction with folate status. Proc. Natl. Acad. Sci. USA 2002, 99, 5606–5611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulrey, C.L.; Liu, L.; Andrews, L.G.; Tollefsbol, T.O. The impact of metabolism on DNA methylation. Hum. Mol. Genet. 2005, 14, R139–R147. [Google Scholar] [CrossRef]
- Schwahn, B.C.; Chen, Z.; Laryea, M.D.; Wendel, U.; Lussier-Cacan, S.; Genest, J., Jr.; Mar, M.-H.; Zeisel, S.H.; Castro, C.; Garrow, T.; et al. Homocysteine-betaine interactions in a murine model of 5,10-methylenetetrahydrofolate reductase deficiency. FASEB J. 2003, 17, 512–514. [Google Scholar] [CrossRef]
- Devlin, A.M.; Bottiglieri, T.; Domann, F.E.; Lentz, S.R. Tissue-specific changes in H19 methylation and expression in mice with hyperhomocysteinemia. J. Biol. Chem. 2005, 280, 25506–25511. [Google Scholar] [CrossRef] [Green Version]
- Grenz, A.; Hermes, M.; Hammel, P.; Roll, J.B.; Osswald, H.; Kloor, D. Hyperhomocysteinemia is associated with decreased erythropoietin expression in rats. Cell Physiol. Biochem. 2010, 26, 449–456. [Google Scholar] [CrossRef]
- Sun, C.Y.; Chang, S.C.; Wu, M.S. Suppression of Klotho expression by protein-bound uremic toxins is associated with increased DNA methyltransferase expression and DNA hypermethylation. Kidney Int. 2012, 81, 640–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, S. Perspectives on the biological function and enzymology of protein carboxyl methylation reactions in eucaryotic and procaryotic cells. Adv. Exp. Med. Biol. 1988, 231, 213–328. [Google Scholar] [PubMed]
- Petrossian, T.; Clarke, S. Bioinformatic Identification of Novel Methyltransferases. Epigenomics 2009, 1, 163–175. [Google Scholar] [CrossRef] [Green Version]
- Ingrosso, D.; Fowler, A.V.; Bleibaum, J.; Clarke, S. Sequence of the D-aspartyl/L-isoaspartyl protein methyltransferase from human erythrocytes. Common sequence motifs for protein, DNA, RNA, and small molecule S-adenosylmethionine-dependent methyltransferases. J. Biol. Chem. 1989, 264, 20131–20139. [Google Scholar] [PubMed]
- Perna, A.F.; Ingrosso, D.; Satta, E.; Lombardi, C.; Galletti, P.; D’Aniello, A.; De Santo, N.G. Plasma protein aspartyl damage is increased in hemodialysis patients: Studies on causes and consequences. J. Am. Soc. Nephrol. 2004, 15, 2747–2754. [Google Scholar] [CrossRef] [Green Version]
- Ingrosso, D.; Cimmino, A.; D’Angelo, S.; Alfinito, F.; Zappia, V.; Galletti, P. Protein methylation as a marker of aspartate damage in glucose-6-phosphate dehydrogenase-deficient erythrocytes: Role of oxidative stress. Eur. J. Biochem. 2002, 269, 2032–2039. [Google Scholar] [CrossRef]
- Richart, L.; Margueron, R. Drugging histone methyltransferases in cancer. Curr. Opin. Chem. Biol. 2020, 56, 51–62. [Google Scholar] [CrossRef]
- Wei, X.; Yi, X.; Zhu, X.-H.; Jiang, D.-S. Histone Methylation and Vascular Biology. Clin. Epigenetics 2020, 12, 30. [Google Scholar] [CrossRef] [Green Version]
- WHO-The Top 10 Causes of Death. Available online: https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death (accessed on 13 May 2020).
- Shen, W.; Gao, C.; Cueto, R.; Liu, L.; Fu, H.; Shao, Y.; Yang, W.Y.; Fang, P.; Choi, E.T.; Wu, Q.; et al. Homocysteine-methionine cycle is a metabolic sensor system controlling methylation-regulated pathological signaling. Redox Biol. 2020, 28, 101322–101334. [Google Scholar] [CrossRef]
- Outteryck, O.; de Sèze, J.; Stojkovic, T.; Cuisset, J.M.; Dobbelaere, D.; Delalande, S.; Lacour, A.; Cabaret, M.; Lepoutre, A.C.; Deramecourt, V.; et al. Methionine synthase deficiency: A rare cause of adult-onset leukoencephalopathy. Neurology 2012, 79, 386–388. [Google Scholar] [CrossRef]
- Perna, A.F.; Ingrosso, D.; Satta, E.; Romano, M.; Cimmino, A.; Galletti, P.; Zappia, V.; De Santo, N.G. Metabolic consequences of hyperhomocysteinemia in uremia. Am. J. Kidney Dis. 2001, 38, S85–S90. [Google Scholar] [CrossRef]
- Li, Y.; Spence, J.D.; Wang, X.; Huo, Y.; Xu, X.; Qin, X. Effect of Vitamin B 12 Levels on the Association Between Folic Acid Treatment and CKD Progression: A Post Hoc Analysis of a Folic Acid Interventional Trial. Am. J. Kidney Dis. 2020, 75, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.Y. Does Vitamin B12 Delay CKD Progression? Am. J. Kidney Dis. 2020, 75, 317–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rooney, M.; Bottiglieri, T.; Wasek-Patterson, B.; McMahon, A.; Hughes, C.F.; McCann, A.; Horigan, G.; Strain, J.J.; McNulty, H.; Ward, M. Impact of the MTHFR C677T polymorphism on one-carbon metabolites: Evidence from a randomised trial of riboflavin supplementation. Biochimie 2020, 173, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Amenyah, S.D.; McMahon, A.; Ward, M.; Deane, J.; McNulty, H.; Hughes, C.F.; Strain, J.J.; Horigan, G.; Purvis, J.; Walsh, C.P.; et al. Riboflavin supplementation alters global and gene-specific DNA methylation in adults with the MTHFR 677 TT genotype. Biochimie 2020, 173, 17–26. [Google Scholar] [CrossRef]
- Efimova, O.A.; Koltsova, A.S.; Krapivin, M.I.; Tikhonov, A.V.; Pendina, A.A. Environmental Epigenetics and Genome Flexibility: Focus on 5-Hydroxymethylcytosine. Int. J. Mol. Sci. 2020, 21, 3223. [Google Scholar] [CrossRef]
- Solary, E.; Bernard, O.A.; Tefferi, A.; Fuks, F.; Vainchenker, W. The Ten-Eleven Translocation-2 (TET2) gene in hematopoiesis and hematopoietic diseases. Leukemia 2014, 28, 485–496. [Google Scholar] [CrossRef]
- Nakajima, H.; Kunimoto, H. TET2 as an epigenetic master regulator for normal and malignant hematopoiesis. Cancer Sci. 2014, 105, 1093–1099. [Google Scholar] [CrossRef] [Green Version]
- Williams, K.; Christensen, J.; Helin, K. DNA methylation: TET proteins—Guardians of CpG islands? EMBO Rep. 2012, 13, 28–35. [Google Scholar] [CrossRef] [Green Version]
- Brocato, J.; Costa, M. Basic mechanics of DNA methylation and the unique landscape of the DNA methylome in metal-induced carcinogenesis. Crit. Rev. Toxicol. 2013, 43, 493–514. [Google Scholar] [CrossRef] [Green Version]
- Niedzwiecki, M.M.; Liu, X.; Hall, M.N.; Thomas, T.; Slavkovich, V.; Ilievski, V.; Levy, D.; Alam, S.; Siddique, A.B.; Parvez, F.; et al. Sex-specific associations of arsenic exposure with global DNA methylation and hydroxymethylation in leukocytes: Results from two studies in Bangladesh. Cancer Epidemiol. Biomark. Prev. 2015, 24, 1748–1757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inker, L.A.; Schmid, C.H.; Tighiouart, H.; Eckfeldt, J.H.; Feldman, H.I.; Greene, T.; Kusek, J.W.; Manzi, J.; Van Lente, F.; Zhang, Y.L.; et al. CKD-EPI Investigators. Estimating glomerular filtration rate from serum creatinine and cystatin C. N. Engl. J. Med. 2012, 367, 20–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-β: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Morgado-Pascual, J.L.; Marchant, V.; Rodrigues-Diez, R.; Dolade, N.; Suarez-Alvarez, B.; Kerr, B.; Valdivielso, J.M.; Ruiz-Ortega, M.; Rayego-Mateos, S. Epigenetic Modification Mechanisms Involved in Inflammation and Fibrosis in Renal Pathology. Mediat. Inflamm. 2018, 2018, 2931049. [Google Scholar] [CrossRef] [Green Version]
- Hurtado Del Pozo, C.; Garreta, E.; Izpisuɐa Belmonte, J.C.; Montserrat, N. Modeling epigenetic modifications in renal development and disease with organoids and genome editing. Dis. Model. Mech. 2018, 11, dmm035048. [Google Scholar] [CrossRef] [Green Version]
- Zawada, A.M.; Rogacev, K.S.; Hummel, B.; Grün, O.S.; Friedrich, A.; Rotter, B.; Winter, P.; Geisel, J.; Fliser, D.; Heine, G.H. SuperTAG methylation-specific digital karyotyping reveals uremia-induced epigenetic dysregulation of atherosclerosis-related genes. Circ. Cardiovasc. Genet. 2012, 5, 611–620. [Google Scholar] [CrossRef] [Green Version]
- Chu, A.Y.; Tin, A.; Schlosser, P.; Ko, Y.A.; Qiu, C.; Yao, C.; Joehanes, R.; Grams, M.E.; Liang, L.; Gluck, C.A.; et al. Epigenome-wide association Studies identify DNA methylation associated with kidney function. Nat. Commun. 2017, 8, 1286. [Google Scholar] [CrossRef]
- Perna, A.F.; Pizza, A.; Di Nunzio, A.; Bellantone, R.; Raffaelli, M.; Cicchella, T.; Conzo, G.; Santini, L.; Zacchia, M.; Trepiccione, F.; et al. ADAM17, a New Player in the Pathogenesis of Chronic Kidney Disease-Mineral and Bone Disorder. J. Ren. Nutr. 2017, 27, 453–457. [Google Scholar] [CrossRef]
- Yin, S.; Zhang, Q.; Yang, J.; Lin, W.; Li, Y.; Chen, F.; Cao, W. TGFβ-incurred epigenetic aberrations of miRNA and DNA methyltransferase suppress Klotho and potentiate renal fibrosis. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 1207–1216. [Google Scholar] [CrossRef]
- Gluck, C.; Qiu, C.; Han, S.Y.; Palmer, M.; Park, J.; Ko, Y.-A.; Guan, Y.; Sheng, X.; Hanson, R.L.; Huang, J.; et al. Kidney cytosine methylation changes improve renal function decline estimation in patients with diabetic kidney disease. Nat. Commun. 2019, 10, 2461. [Google Scholar] [CrossRef]
- Sun, J.; Wang, Y.; Cui, W.; Lou, Y.; Sun, G.; Zhang, D.; Miao, L. Role of Epigenetic Histone Modifications in Diabetic Kidney Disease Involving Renal Fibrosis. J. Diabetes Res. 2017, 2017, 7242384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smyth, L.J.; Duffy, S.; Maxwell, A.P.; McKnight, A.J. Genetic and epigenetic factors influencing chronic kidney disease. Am. J. Physiol. Ren. Physiol. 2014, 307, F757–F776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- CanɁadas-Garre, M.; Anderson, K.; McGoldrick, J.; Maxwell, A.P.; McKnight, A.J. Genomic approaches in the search for molecular biomarkers in chronic kidney disease. J. Transl. Med. 2018, 16, 292. [Google Scholar] [CrossRef] [PubMed]
- OMIM.org—UROMODULIN; UMOD *191845. Available online: https://omim.org/entry/191845 (accessed on 14 May 2020).
- OMIM.org—# 167030; Nephrolithiasis, Calcium Oxalate. Available online: https://omim.org/entry/167030 (accessed on 14 May 2020).
- Shroom Family Member 3—* 604570. Available online: https://omim.org/entry/604570?search=SHROOM3&highlight=shroom3 (accessed on 14 May 2020).
- GeneCards—The Human Gene Database—ELMO1 Gene. Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=ELMO1 (accessed on 14 May 2020).
- Onishi, A.; Sugiyama, H.; Kitagawa, M.; Yamanari, T.; Tanaka, K.; Ogawa-Akiyama, A.; Kano, Y.; Mise, K.; Tanabe, K.; Morinaga, H.; et al. Urine 5MedC, a Marker of DNA Methylation, in the Progression of Chronic Kidney Disease. Dis. Markers 2019, 2019, 5432453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larkin, B.P.; Glastras, S.J.; Chen, H.; Pollock, C.A.; Saad, S. DNA methylation and the potential role of demethylating agents in prevention of progressive chronic kidney disease. FASEB J. 2018, 32, 5215–5226. [Google Scholar] [CrossRef] [Green Version]
- Fontecha-Barriuso, M.; Martin-Sanchez, D.; Ruiz-Andres, O.; Poveda, J.; Sanchez-Niño, M.D.; Valiño-Rivas, L.; Ruiz-Ortega, M.; Ortiz, A.; Sanz, A.B. Targeting epigenetic DNA and histone modifications to treat kidney disease. Nephrol. Dial. Transplant. 2018, 33, 1875–1886. [Google Scholar] [CrossRef]
- Seibert, F.S.; Sitz, M.; Passfall, J.; Haesner, M.; Laschinski, P.; Buhl, M.; Bauer, F.; Babel, N.; Pagonas, N.; Westhoff, T.H. Prognostic Value of Urinary Calprotectin, NGAL and KIM-1 in Chronic Kidney Disease. Kidney Blood Press Res. 2018, 43, 1255–1262. [Google Scholar] [CrossRef]
- Padmanabhan, B.; Mathur, S.; Manjula, R.; Tripathi, S. Bromodomain and extra-terminal (BET) family proteins: New therapeutic targets in major diseases. J. Biosci. 2016, 41, 295–311. [Google Scholar] [CrossRef]
- Zinellu, A.; Sotgia, S.; Sotgiu, E.; Assaretti, S.; Baralla, A.; Mangoni, A.A.; Satta, A.E.; Carru, C. Cholesterol lowering treatment restores blood global DNA methylation in chronic kidney disease (CKD) patients. Nutr. Metab. Cardiovasc. Dis. 2017, 27, 822–829. [Google Scholar] [CrossRef]
- Chapter 2: Why Do We Need an Onco-Nephrology Curriculum? (by Perazella MA & Rosner MH), Copyright © 2016 by the American Society of Nephrology. Available online: https://www.asn-online.org/education/distancelearning/curricula/onco/Chapter2.pdf (accessed on 14 March 2020).
- Decker, J.; Neuhaus, C.; Macdonald, F.; Brauch, H.; Maher, E.R. Clinical utility gene card for: Von Hippel–Lindau (VHL). Eur. J. Hum. Genet. 2014, 22. [Google Scholar] [CrossRef]
- de Cubas, A.A.; Rathmell, W.K. Epigenetic modifiers: Activities in renal cell carcinoma WK. Nat. Rev. Urol. 2018, 15, 599–614. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.R.; Latif, F. The epigenetic landscape of renal cancer. Nat. Rev. Nephrol. 2017, 13, 47–60. [Google Scholar] [CrossRef]
- Inamura, K. Renal Cell Tumors: Understanding Their Molecular Pathological Epidemiology and the 2016 WHO Classification. Int. J. Mol. Sci. 2017, 18, 2195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joosten, S.C.; Smits, K.M.; Aarts, M.J.; Melotte, V.; Koch, A.; Tjan-Heijnen, V.C.; van Engeland, M. Epigenetics in renal cell cancer: Mechanisms and clinical applications. Nat. Rev. Urol. 2018, 15, 430–451. [Google Scholar] [CrossRef]
- Petrozza, V.; Carbone, A.; Bellissimo, T.; Porta, N.; Palleschi, G.; Pastore, A.L.; Di Carlo, A.; Della Rocca, C.; Fazi, F. Oncogenic MicroRNAs Characterization in Clear Cell Renal Cell Carcinoma. Int. J. Mol. Sci. 2015, 16, 29219–29225. [Google Scholar] [CrossRef] [Green Version]
- NIH-National Cancer Institute. The Cancer Genome Atlas Program. Available online: https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga (accessed on 14 March 2020).
- Malouf, G.G.; Zhang, J.; Yuan, Y.; Compérat, E.; Rouprêt, M.; Cussenot, O.; Chen, Y.; Thompson, E.J.; Tannir, N.M.; Weinstein, J.N.; et al. Characterization of long non-coding RNA transcriptome in clear-cell renal cell carcinoma by next-generation deep sequencing. Mol. Oncol. 2015, 9, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Webster, A.C.; Nagler, E.V.; Morton, R.L.; Masson, P. Chronic Kidney Disease. Lancet 2017, 389, 1238–1252. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complication | Related Lab Markers | Parameters/Symptoms/ Nosological Entities | Prevention/Therapy | Ref. |
|---|---|---|---|---|
| Anemia |
|
| Erythropoietin | [7] |
| Cardiovascular disease (CVD) | Dyslipidemia Hyperhomocysteinemia Other metabolic alterations | CVD events (myocardial infarction, stroke, thrombosis) | Control of cardiovascular risk factors | [8] |
| Electrolyte imbalance |
| Heart dysfunction (ECG abnormalities, arrhythmias) | Specific electrolyte correction (cation exchange resins; diuretics) | [9] |
| Fluid retention |
| Edema |
| [10] |
| Gout | High uric acid (**) |
| Xanthine oxidase inhibitors (allopurinol, febuxostat) | [11] |
| Metabolic acidosis | Arterial Blood Gas (ABG) alterations (low pH, low bicarbonate, low BE, compensatory low paCO2) | Organ injury (headache, chest and bone pain, palpitation, dyspnoea, nausea, vomiting, weakness, and bone pain. anxiety, mental derangements, seizures, coma, heart arrhythmia. | Specific therapy based on correction of pH and bicarbonate alterations. | [12] |
| Mineral and Bone Disease |
|
|
| [13] |
| Group | Disease/Condition/Compound | Pathophysiology | Symptoms/Laboratory Findings |
|---|---|---|---|
| Genetic | |||
| Homocystinuria | CBS mutations/deletions | CVD/ectopia lentis/Severe HHcy (a) | |
| Methionine synthase | CVD/Severe HHcy; low Met | ||
| Other (b) | Severe HHcy (low Met) | ||
| Polymorphysms | MTHFR C677T (c) | (CVD)/ Moderate/Intermediate HHcy (incostant/conditioned) (d; e) | |
| MTHFR A1298C | (CVD)/ Moderate/Intermediate HHcy (incostant/conditioned) (d; e) | ||
| Nutritional deficiency | |||
| Folate | Met-Hcy cycle deficiency | High Hcy (moderate) | |
| B12 | Met-Hcy cycle deficiency | High Hcy (moderate); methylmalonic aciduria; low HoloTC; check also for gastric intrinsic factor (GIF) deficiency | |
| B6 | Transsulfuration deficiency | Drug interaction; alcohol abuse | |
| Drug administration | |||
| Antifolates | Methotrexate (f) | Cancer or inflammatory disease (Psoriasis/arthritis) | |
| Anti-parkinsonian drugs | L-DOPA (g) | Parkinson disease | |
| Acquired | |||
| CKD | Uremic toxins? (h) | see also Table 1 |
| Finding(s) | Reference(s) |
|---|---|
| Ex vivo protein methylation inhibition and decreased intracellular SAM/SAH concentration ratio in CKD | [30] |
| Impaired DNA methylation and vascular endothelial cell growth induced by homocysteine, in vitro | [31] |
| In vivo studies on transmethylation inhibition in CKD | [32,33] |
| Raised blood SAH concentration in CKD patients ex vivo | [34,35] |
| Defective macromolecule methylation and repair associated with growth factors activation, lipid deposition and increased vascular smooth cell proliferation in atherogenesis, ex vivo studies | [36,37] |
| In vivo global DNA hypomethylation in MTHFR C677/ polymorphism (mononuclear blood cells) | [38] |
| In vivo global DNA hypomethylation and response to folate (mononuclear blood cells) | [28] |
| Metabolic effects on DNA methylation. A very exhaustive review article | [39] |
| Mice lacking MTHFR develop severe steatosis and have elevated plasma homocysteine, increased hepatic content of SAH and reduced SAM | [40] |
| HHcy is associated with higher SAH in liver and brain and induces tissue-specific changes in H19 methylation and expression in CBS knockout mice | [41] |
| Severe HHcy affects methylation potential in the renal tissue and lowers erythropoietin expression following CO induced intoxication in rats | [42] |
| Suppression of Klotho expression by protein-bound uremic toxins is associated with increased DNA methyltransferase expression and DNA hypermethylation (in vitro) | [43] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ingrosso, D.; Perna, A.F. DNA Methylation Dysfunction in Chronic Kidney Disease. Genes 2020, 11, 811. https://doi.org/10.3390/genes11070811
Ingrosso D, Perna AF. DNA Methylation Dysfunction in Chronic Kidney Disease. Genes. 2020; 11(7):811. https://doi.org/10.3390/genes11070811
Chicago/Turabian StyleIngrosso, Diego, and Alessandra F. Perna. 2020. "DNA Methylation Dysfunction in Chronic Kidney Disease" Genes 11, no. 7: 811. https://doi.org/10.3390/genes11070811
APA StyleIngrosso, D., & Perna, A. F. (2020). DNA Methylation Dysfunction in Chronic Kidney Disease. Genes, 11(7), 811. https://doi.org/10.3390/genes11070811