The Role of BMI1 in Late-Onset Sporadic Alzheimer’s Disease

{kind=link}
Abstract
:1. Aging as the Number One Risk Factor of Alzheimer’s Disease
2. BMI1
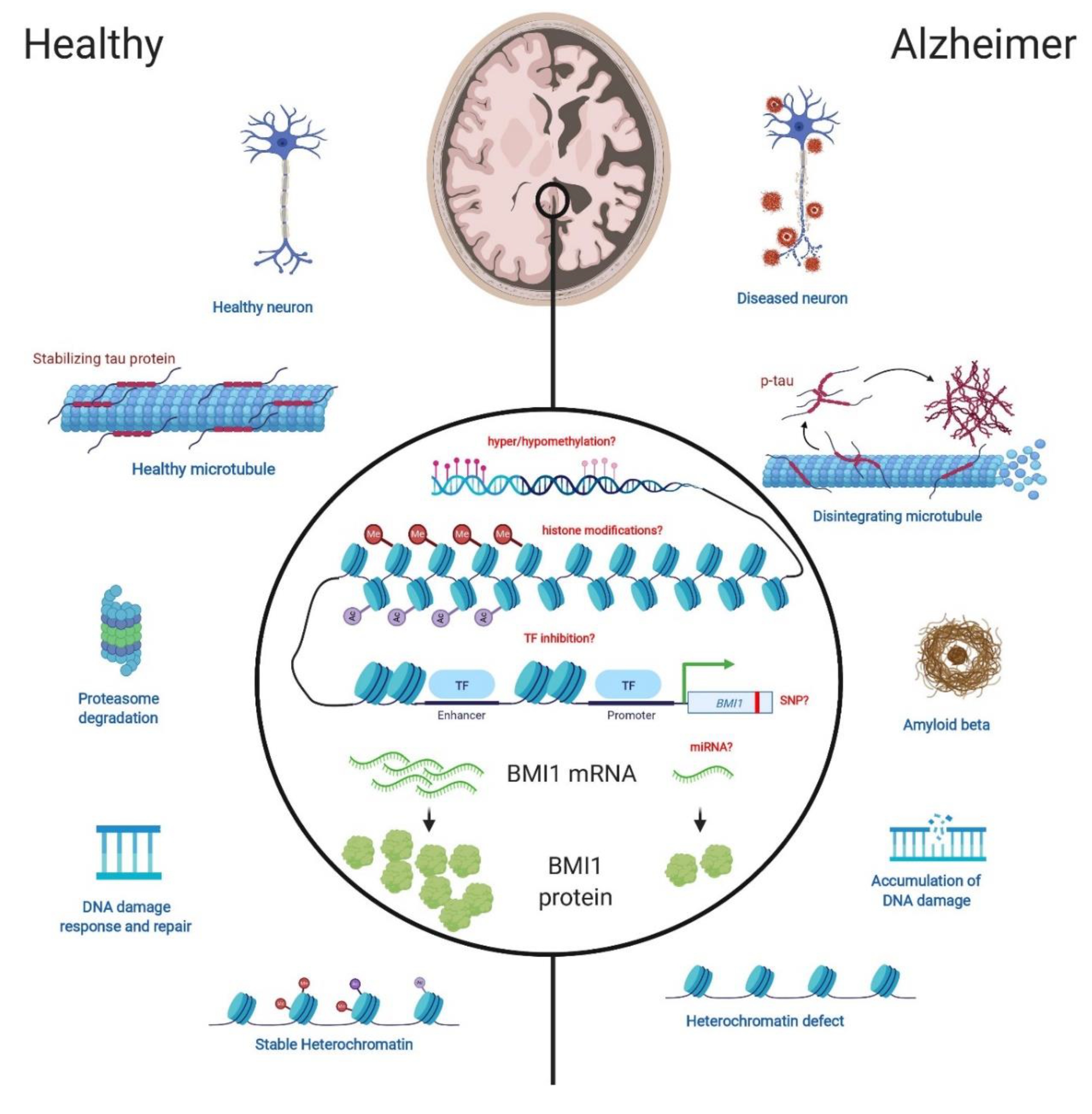
2.1. B-Cell Specific Moloney Murine Leukemia Virus Integration Site 1
2.2. Reduced BMI1 Expression is Associated with Aging
3. Neurodegenerative Hallmarks of Alzheimer’s Disease
3.1. Histopathology
3.2. BMI1 is Reduced in LOAD
4. Models of BMI1-Deficiency Recapitulate LOAD
5. Epigenetics
5.1. PSEN1
5.2. SIRT2
5.3. LRP6
5.4. REST
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Shao, W.; Peng, D.; Wang, X. Genetics of Alzheimer’s disease: From pathogenesis to clinical usage. J. Clin. Neurosci. 2017, 45, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Giau, V.V.; Bagyinszky, E.; Yang, Y.S.; Youn, Y.C.; An, S.S.A.; Kim, S.Y. Genetic analyses of early-onset Alzheimer’s disease using next generation sequencing. Sci. Rep. 2019, 9, 8368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández, M.V.; Kim, J.H.; Budde, J.P.; Black, K.; Medvedeva, A.; Saef, B.; Deming, Y.; Del-Aguila, J.; Ibañez, L.; Dube, U.; et al. Analysis of neurodegenerative Mendelian genes in clinically diagnosed Alzheimer Disease. PLoS Genet. 2017, 13, e1007045. [Google Scholar] [CrossRef] [Green Version]
- Giau, V.; Senanarong, V.; Bagyinszky, E.; An, S.; Kim, S. Analysis of 50 neurodegenerative genes in clinically diagnosed early-onset Alzheimer’s disease. IJMS 2019, 20, 1514. [Google Scholar] [CrossRef] [Green Version]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef] [PubMed]
- Maynard, S.; Fang, E.F.; Scheibye-Knudsen, M.; Croteau, D.L.; Bohr, V.A. DNA damage, DNA repair, aging, and neurodegeneration. Cold Spring Harb. Perspect Med. 2015, 5, a025130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharya, R.; Banerjee Mustafi, S.; Street, M.; Dey, A.; Dwivedi, S.K.D. Bmi-1: At the crossroads of physiological and pathological biology. Genes Dis. 2015, 2, 225–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.-C.; Li, C.-L.; Cui, J.; Jiao, M.; Wu, T.; Jing, L.; Nan, K.-J. BMI-1, a promising therapeutic target for human cancer. Oncol. Lett. 2015, 10, 583–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chittock, E.C.; Latwiel, S.; Miller, T.C.R.; Müller, C.W. Molecular architecture of polycomb repressive complexes. Biochem. Soc. Trans. 2017, 45, 193–205. [Google Scholar] [CrossRef] [Green Version]
- Blackledge, N.P.; Fursova, N.A.; Kelley, J.R.; Huseyin, M.K.; Feldmann, A.; Klose, R.J. PRC1 catalytic activity is central to polycomb system function. Mol. Cell 2020, 77, 857–874.e9. [Google Scholar] [CrossRef] [Green Version]
- Tamburri, S.; Lavarone, E.; Fernández-Pérez, D.; Conway, E.; Zanotti, M.; Manganaro, D.; Pasini, D. Histone H2AK119 mono-ubiquitination is essential for polycomb-mediated transcriptional repression. Mol. Cell 2020, 77, 840–856.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobs, J.J.L.; Kieboom, K.; Marino, S.; DePinho, R.A.; van Lohuizen, M. The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature 1999, 397, 164–168. [Google Scholar] [CrossRef] [PubMed]
- Bruggeman, S.W.M. Ink4a and Arf differentially affect cell proliferation and neural stem cell self-renewal in Bmi1-deficient mice. Genes Dev. 2005, 19, 1438–1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molofsky, A.V. Bmi-1 promotes neural stem cell self-renewal and neural development but not mouse growth and survival by repressing the p16Ink4a and p19Arf senescence pathways. Genes Dev. 2005, 19, 1432–1437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Hajjar, J.; Chatoo, W.; Hanna, R.; Nkanza, P.; Tétreault, N.; Tse, Y.C.; Wong, T.P.; Abdouh, M.; Bernier, G. Heterochromatic genome instability and neurodegeneration sharing similarities with Alzheimer’s disease in old Bmi1+/− mice. Sci. Rep. 2019, 9, 594. [Google Scholar] [CrossRef] [PubMed]
- Abdouh, M.; Hanna, R.; El Hajjar, J.; Flamier, A.; Bernier, G. the polycomb repressive complex 1 protein BMI1 is required for constitutive heterochromatin formation and silencing in mammalian somatic cells. J. Biol. Chem. 2016, 291, 182–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Facchino, S.; Abdouh, M.; Chatoo, W.; Bernier, G. BMI1 confers radioresistance to normal and cancerous neural stem cells through recruitment of the DNA damage response machinery. J. Neurosci. 2010, 30, 10096–10111. [Google Scholar] [CrossRef]
- Ismail, I.H.; Andrin, C.; McDonald, D.; Hendzel, M.J. BMI1-mediated histone ubiquitylation promotes DNA double-strand break repair. J. Cell Biol. 2010, 191, 45–60. [Google Scholar] [CrossRef]
- Chatoo, W.; Abdouh, M.; David, J.; Champagne, M.-P.; Ferreira, J.; Rodier, F.; Bernier, G. The polycomb group gene Bmi1 regulates antioxidant defenses in neurons by repressing p53 pro-oxidant activity. J. Neurosci. 2009, 29, 529–542. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Cao, L.; Chen, J.; Song, S.; Lee, I.H.; Quijano, C.; Liu, H.; Keyvanfar, K.; Chen, H.; Cao, L.-Y.; et al. Bmi1 regulates mitochondrial function and the DNA damage response pathway. Nature 2009, 459, 387–392. [Google Scholar] [CrossRef]
- Itahana, K.; Zou, Y.; Itahana, Y.; Martinez, J.-L.; Beausejour, C.; Jacobs, J.J.L.; van Lohuizen, M.; Band, V.; Campisi, J.; Dimri, G.P. Control of the replicative life span of human fibroblasts by p16 and the polycomb protein Bmi-1. MCB 2003, 23, 389–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdouh, M.; Chatoo, W.; El Hajjar, J.; David, J.; Ferreira, J.; Bernier, G. Bmi1 is down-regulated in the aging brain and displays antioxidant and protective activities in neurons. PLoS ONE 2012, 7, e31870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bloom, G.S. Amyloid-β and Tau: The trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 2014, 71, 505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef] [PubMed]
- Kametani, F.; Hasegawa, M. Reconsideration of amyloid hypothesis and Tau hypothesis in Alzheimer’s disease. Front. Neurosci. 2018, 12, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makin, S. The amyloid hypothesis on trial. Nature 2018, 559, S4–S7. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.-C.; Oelze, B.; Schumacher, A. Age-specific epigenetic drift in late-onset Alzheimer’s disease. PLoS ONE 2008, 3, e2698. [Google Scholar] [CrossRef] [Green Version]
- Mastroeni, D.; Grover, A.; Delvaux, E.; Whiteside, C.; Coleman, P.D.; Rogers, J. Epigenetic changes in Alzheimer’s disease: Decrements in DNA methylation. Neurobiol. Aging 2010, 31, 2025–2037. [Google Scholar] [CrossRef] [Green Version]
- Coppieters, N.; Dieriks, B.V.; Lill, C.; Faull, R.L.M.; Curtis, M.A.; Dragunow, M. Global changes in DNA methylation and hydroxymethylation in Alzheimer’s disease human brain. Neurobiol. Aging 2014, 35, 1334–1344. [Google Scholar] [CrossRef] [PubMed]
- Lashley, T.; Gami, P.; Valizadeh, N.; Li, A.; Revesz, T.; Balazs, R. Alterations in global DNA methylation and hydroxymethylation are not detected in Alzheimer’s disease: Global DNA methylation in AD. Neuropathol. Appl. Neurobiol. 2015, 41, 497–506. [Google Scholar] [CrossRef]
- Bradley-Whitman, M.A.; Lovell, M.A. Epigenetic changes in the progression of Alzheimer’s disease. Mech. Ageing Dev. 2013, 134, 486–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Francesco, A.; Arosio, B.; Falconi, A.; Micioni Di Bonaventura, M.V.; Karimi, M.; Mari, D.; Casati, M.; Maccarrone, M.; D’Addario, C. Global changes in DNA methylation in Alzheimer’s disease peripheral blood mononuclear cells. Brain Behav. Immun. 2015, 45, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Frost, B.; Hemberg, M.; Lewis, J.; Feany, M.B. Tau promotes neurodegeneration through global chromatin relaxation. Nat. Neurosci. 2014, 17, 357–366. [Google Scholar] [CrossRef] [Green Version]
- Weissman, L.; Jo, D.-G.; Sorensen, M.M.; de Souza-Pinto, N.C.; Markesbery, W.R.; Mattson, M.P.; Bohr, V.A. Defective DNA base excision repair in brain from individuals with Alzheimer’s disease and amnestic mild cognitive impairment. Nucleic Acids Res. 2007, 35, 5545–5555. [Google Scholar] [CrossRef]
- Rodier, F.; Coppé, J.-P.; Patil, C.K.; Hoeijmakers, W.A.M.; Muñoz, D.P.; Raza, S.R.; Freund, A.; Campeau, E.; Davalos, A.R.; Campisi, J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 2009, 11, 973–979. [Google Scholar] [CrossRef] [PubMed]
- Züchner, S.; Gilbert, J.R.; Martin, E.R.; Leon-Guerrero, C.R.; Xu, P.-T.; Browning, C.; Bronson, P.G.; Whitehead, P.; Schmechel, D.E.; Haines, J.L.; et al. Linkage and association study of late-onset Alzheimer disease families linked to 9p21.3. Ann. Hum. Genet. 2008, 72, 725–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lars Rödel, T.A.; Gärtner, U.; Holzer, M. Expression of the cyclin-dependent kinase inhibitor p16 in Alzheimer’s disease. NeuroReport 1996, 7, 3047–3050. [Google Scholar] [CrossRef]
- Arendt, T.; Holzer, M.; Gärtner, U. Neuronal expression of cycline dependent kinase inhibitors of the INK4 family in Alzheimer’s disease. J. Neural Transm. 1998, 105, 949–960. [Google Scholar] [CrossRef]
- Flamier, A.; El Hajjar, J.; Adjaye, J.; Fernandes, K.J.; Abdouh, M.; Bernier, G. modeling late-onset sporadic Alzheimer’s disease through BMI1 deficiency. Cell Rep. 2018, 23, 2653–2666. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.D.; Ganat, Y.M.; Kishinevsky, S.; Bowman, R.L.; Liu, B.; Tu, E.Y.; Mandal, P.K.; Vera, E.; Shim, J.; Kriks, S.; et al. Human iPSC-based modeling of late-onset disease via progerin-induced aging. Cell Stem Cell 2013, 13, 691–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berry, B.J.; Smith, A.S.T.; Young, J.E.; Mack, D.L. Advances and current challenges associated with the use of human induced pluripotent stem cells in modeling neurodegenerative disease. Cells Tissues Organs 2018, 205, 331–349. [Google Scholar] [CrossRef] [PubMed]
- Gu, M.; Shen, L.; Bai, L.; Gao, J.; Marshall, C.; Wu, T.; Ding, J.; Miao, D.; Xiao, M. Heterozygous knockout of the Bmi-1 gene causes an early onset of phenotypes associated with brain aging. AGE 2014, 36, 129–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, H.L.; Li, W.; Cheetham, M.E. Molecular chaperones and neuronal proteostasis. Semin. Cell Dev. Biol. 2015, 40, 142–152. [Google Scholar] [CrossRef] [Green Version]
- Stokin, G.B. Axonopathy and transport deficits early in the pathogenesis of Alzheimer’s disease. Science 2005, 307, 1282–1288. [Google Scholar] [CrossRef] [PubMed]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular senescence: Defining a path forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef]
- Qazi, T.J.; Quan, Z.; Mir, A.; Qing, H. Epigenetics in Alzheimer’s disease: Perspective of DNA methylation. Mol. Neurobiol. 2018, 55, 1026–1044. [Google Scholar] [CrossRef]
- Fuso, A.; Seminara, L.; Cavallaro, R.A.; D’Anselmi, F.; Scarpa, S. S-adenosylmethionine/homocysteine cycle alterations modify DNA methylation status with consequent deregulation of PS1 and BACE and beta-amyloid production. Mol. Cell. Neurosci. 2005, 28, 195–204. [Google Scholar] [CrossRef]
- Lin, H.-C.; Hsieh, H.-M.; Chen, Y.-H.; Hu, M.-L. S-Adenosylhomocysteine increases beta-amyloid formation in BV-2 microglial cells by increased expressions of beta-amyloid precursor protein and presenilin 1 and by hypomethylation of these gene promoters. Neurotoxicology 2009, 30, 622–627. [Google Scholar] [CrossRef]
- Fuso, A.; Cavallaro, R.A.; Cavallaroa, R.A.; Nicolia, V.; Scarpa, S. PSEN1 promoter demethylation in hyperhomocysteinemic TgCRND8 mice is the culprit, not the consequence. Curr. Alzheimer Res. 2012, 9, 527–535. [Google Scholar] [CrossRef]
- Monti, N.; Cavallaro, R.A.; Stoccoro, A.; Nicolia, V.; Scarpa, S.; Kovacs, G.G.; Fiorenza, M.T.; Lucarelli, M.; Aronica, E.; Ferrer, I.; et al. CpG and non-CpG Presenilin1 methylation pattern in course of neurodevelopment and neurodegeneration is associated with gene expression in human and murine brain. Epigenetics Off. J. DNA Methylation Soc. 2020. [CrossRef] [Green Version]
- Cacabelos, R.; Carril, J.; Cacabelos, N.; Kazantsev, A.; Vostrov, A.; Corzo, L.; Cacabelos, P.; Goldgaber, D. Sirtuins in Alzheimer’s disease: SIRT2-related GenoPhenotypes and implications for PharmacoEpiGenetics. IJMS 2019, 20, 1249. [Google Scholar] [CrossRef] [Green Version]
- Polito, L.; Kehoe, P.G.; Davin, A.; Benussi, L.; Ghidoni, R.; Binetti, G.; Quadri, P.; Lucca, U.; Tettamanti, M.; Clerici, F.; et al. The SIRT2 polymorphism rs10410544 and risk of Alzheimer’s disease in two Caucasian case-control cohorts. Alzheimer’s Dement. 2013, 9, 392–399. [Google Scholar] [CrossRef]
- Wei, W.; Xu, X.; Li, H.; Zhang, Y.; Han, D.; Wang, Y.; Yan, W.; Wang, X.; Zhang, J.; Liu, N.; et al. The SIRT2 polymorphism rs10410544 and risk of Alzheimer’s disease: A Meta-analysis. Neuromol. Med. 2014, 16, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Chen, L.; Zhang, S.; Xie, L. Correlation between SIRT2 3’UTR gene polymorphism and the susceptibility to Alzheimer’s disease. J. Mol. Neurosci. 2020, 70, 878–886. [Google Scholar] [CrossRef] [PubMed]
- Buechler, J.; Salinas, P.C. Deficient Wnt signaling and synaptic vulnerability in Alzheimer’s disease: Emerging roles for the LRP6 receptor. Front. Synaptic Neurosci. 2018, 10, 38. [Google Scholar] [CrossRef] [PubMed]
- De Ferrari, G.V.; Papassotiropoulos, A.; Biechele, T.; Wavrant De-Vrieze, F.; Avila, M.E.; Major, M.B.; Myers, A.; Saez, K.; Henriquez, J.P.; Zhao, A.; et al. Common genetic variation within the low-density lipoprotein receptor-related protein 6 and late-onset Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2007, 104, 9434–9439. [Google Scholar] [CrossRef] [Green Version]
- Alarcón, M.A.; Medina, M.A.; Hu, Q.; Avila, M.E.; Bustos, B.I.; Pérez-Palma, E.; Peralta, A.; Salazar, P.; Ugarte, G.D.; Reyes, A.E.; et al. A novel functional low-density lipoprotein receptor-related protein 6 gene alternative splice variant is associated with Alzheimer’s disease. Neurobiol. Aging 2013, 34, 1709.e9–1709.e18. [Google Scholar] [CrossRef]
- Hwang, J.-Y.; Zukin, R.S. REST, a master transcriptional regulator in neurodegenerative disease. Curr. Opin. Neurobiol. 2018, 48, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Aron, L.; Zullo, J.; Pan, Y.; Kim, H.; Chen, Y.; Yang, T.-H.; Kim, H.-M.; Drake, D.; Liu, X.S.; et al. REST and stress resistance in ageing and Alzheimer’s disease. Nature 2014, 507, 448–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, K.; Feldman, H.M.; Lu, T.; Drake, D.; Lim, E.T.; Ling, K.-H.; Bishop, N.A.; Pan, Y.; Seo, J.; Lin, Y.-T.; et al. REST and neural gene network dysregulation in iPSC models of Alzheimer’s disease. Cell Rep. 2019, 26, 1112–1127.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hogan, R.; Flamier, A.; Nardini, E.; Bernier, G. The Role of BMI1 in Late-Onset Sporadic Alzheimer’s Disease. Genes 2020, 11, 825. https://doi.org/10.3390/genes11070825
Hogan R, Flamier A, Nardini E, Bernier G. The Role of BMI1 in Late-Onset Sporadic Alzheimer’s Disease. Genes. 2020; 11(7):825. https://doi.org/10.3390/genes11070825
Chicago/Turabian StyleHogan, Ryan, Anthony Flamier, Eleonora Nardini, and Gilbert Bernier. 2020. "The Role of BMI1 in Late-Onset Sporadic Alzheimer’s Disease" Genes 11, no. 7: 825. https://doi.org/10.3390/genes11070825
APA StyleHogan, R., Flamier, A., Nardini, E., & Bernier, G. (2020). The Role of BMI1 in Late-Onset Sporadic Alzheimer’s Disease. Genes, 11(7), 825. https://doi.org/10.3390/genes11070825