High Rates of Three Common GJB2 Mutations c.516G>C, c.-23+1G>A, c.235delC in Deaf Patients from Southern Siberia Are Due to the Founder Effect

 , , , and
, , , and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. STRs and SNPs Genotyping
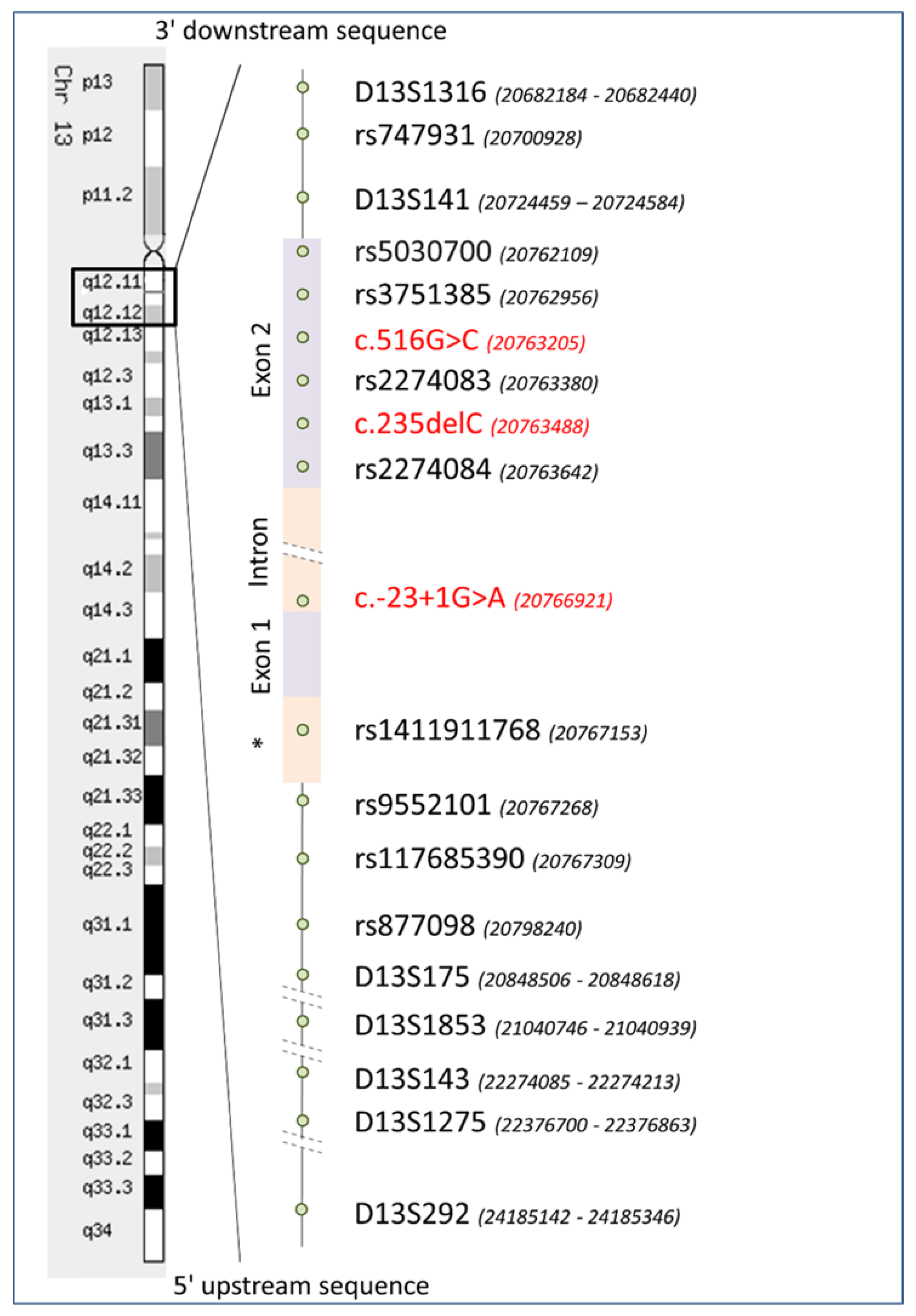
2.3. Reconstruction of STR and SNP Haplotypes
2.4. Estimation of Mutations Age
2.5. Statistical Analysis
3. Results
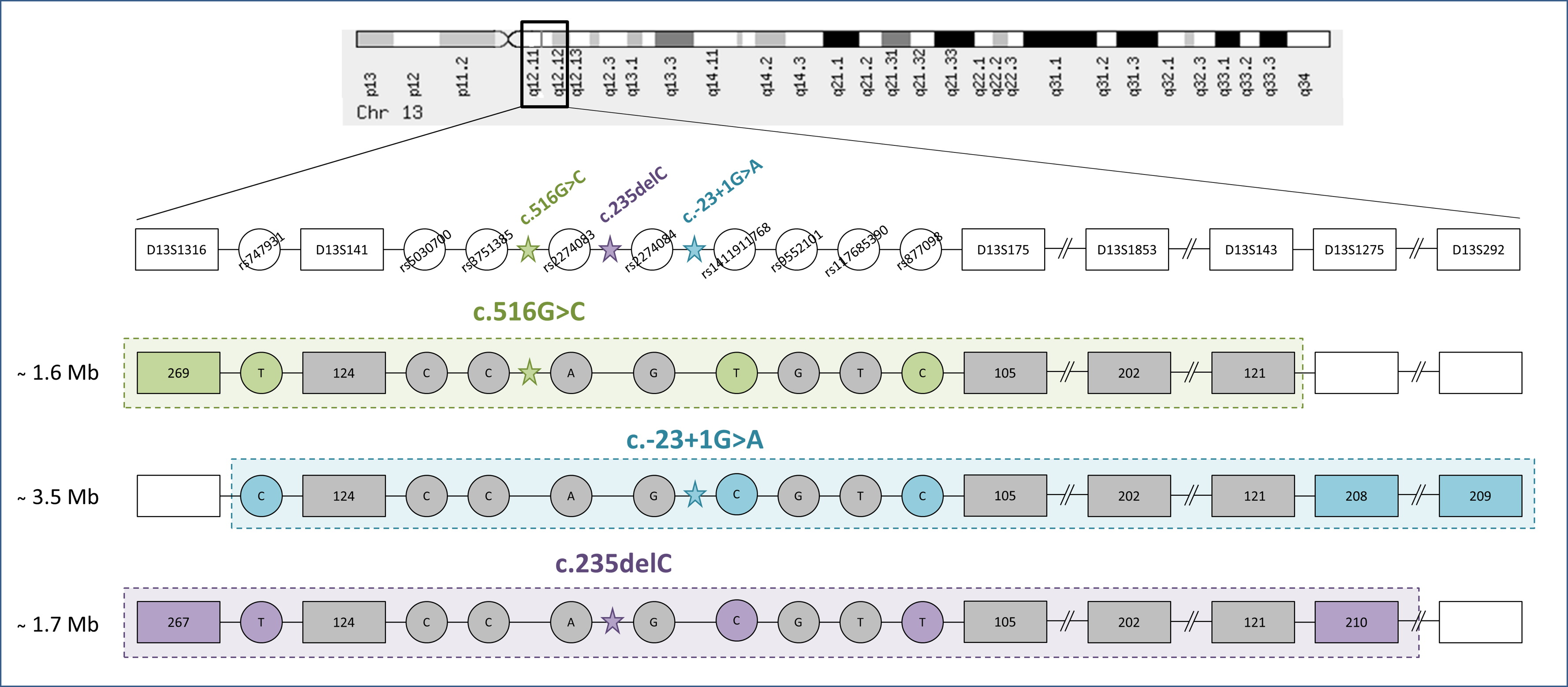
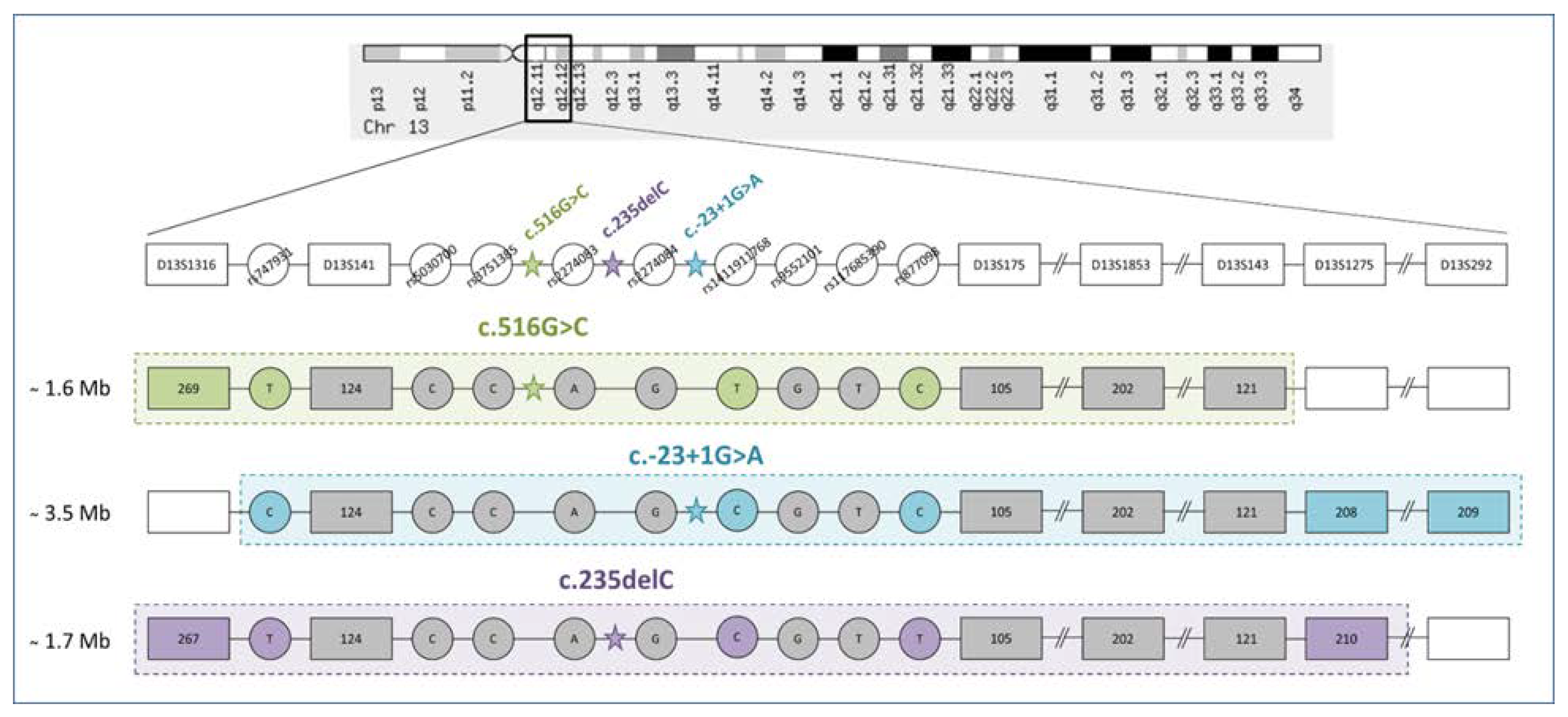
3.1. STR Haplotypes
3.2. SNP Haplotypes
3.3. Age of Mutations c.516G>C, c.-23+1G>A, and c.235delC
4. Discussion
4.1. Ethnic History of Tuvinians and Altaians
4.2. Common Haplotypes for c.516G>C, c.-23+1G>A, and c.235delC
4.3. The c.516G>C Mutation
4.4. The c.-23+1G>A Mutation
4.5. The c.235delC Mutation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Del Castillo, F.J.; del Castillo, I. DFNB1 non-syndromic hearing impairment: Diversity of mutations and associated phenotypes. Front. Mol. Neurosci. 2017, 10, 428. [Google Scholar] [CrossRef] [Green Version]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Evans, K.; Hayden, M.; Heywood, S.; Hussain, M.; Phillips, A.D.; Cooper, D.N. The human gene mutation database: Towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Qual. Life Res. 2017, 136, 665–677. [Google Scholar] [CrossRef] [Green Version]
- Mahdieh, N.; Rabbani, B. Statistical study of 35delG mutation of GJB2 gene: A meta-analysis of carrier frequency. Int. J. Audiol. 2009, 48, 363–370. [Google Scholar] [CrossRef]
- Yao, J.; Lu, Y.; Wei, Q.; Cao, X.; Xing, G. A systematic review and meta-analysis of 235delC mutation of GJB2 gene. J. Transl. Med. 2012, 10, 136. [Google Scholar] [CrossRef] [Green Version]
- Chan, D.K.; Chang, K.W. GJB2-associated hearing loss: Systematic review of worldwide prevalence, genotype, and auditory phenotype: Systematic review of Cx-26-associated hearing loss. Laryngoscope 2014, 124, E34–E53. [Google Scholar] [CrossRef]
- Gasparini, P.; Rabionet, R.; Barbujani, G.; Melchionda, S.; Petersen, M.; Brøndum-Nielsen, K.; Metspalu, A.; Oitmaa, E.; Pisano, M.; Fortina, M.; et al. High carrier frequency of the 35delG deafness mutation in European populations. Eur. J. Hum. Genet. 2000, 8, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Park, H.-J.; Hahn, S.H.; Chun, Y.-M.; Park, K.; Kim, H.-N. Connexin26 mutations associated with nonsyndromic hearing loss. Laryngoscope 2000, 110, 1535–1538. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Xia, X.; Ke, X.; Ouyang, X.; Du, L.; Liu, Y.; Angeli, S.; Telischi, F.F.; Nance, W.E.; Balkany, T.; et al. The prevalence of connexin 26 (GJB2) mutations in the Chinese population. Qual. Life Res. 2002, 111, 394–397. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuka, A.; Yuge, I.; Kimura, S.; Namba, A.; Abe, S.; Van Laer, L.; Van Camp, G.; Usami, S. GJB2 deafness gene shows a specific spectrum of mutations in Japan, including a frequent founder mutation. Qual. Life Res. 2003, 112, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Posukh, O.L.; Pallares-Ruiz, N.; Tadinova, V.; Osipova, L.P.; Claustres, M.; Roux, A.-F. First molecular screening of deafness in the Altai Republic population. BMC Med. Genet. 2005, 6, 12. [Google Scholar] [CrossRef] [Green Version]
- Dai, P.; Yu, F.; Han, M.; Liu, X.Z.; Wang, G.; Li, Q.; Yuan, Y.; Liu, X.; Huang, D.; Kang, D.; et al. GJB2 mutation spectrum in 2063 Chinese patients with nonsyndromic hearing impairment. J. Transl. Med. 2009, 7, 26. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.Y.; Kim, A.R.; Han, K.H.; Kim, M.Y.; Jeon, E.-H.; Koo, J.-W.; Oh, S.H.; Choi, B.Y. Residual hearing in dfnb1 deafness and its clinical implication in a Korean population. PLoS ONE 2015, 10, e0125416. [Google Scholar] [CrossRef] [PubMed]
- Tsukada, K.; Nishio, S.Y.; Hattori, M.; Usami, S. Ethnic-specific spectrum of GJB2 and SLC26A4 mutations: Their origin and a literature review. Ann. Otol. Rhinol. Laryngol. 2015, 124 (Suppl. 1), 61S–76S. [Google Scholar] [CrossRef]
- Erdenechuluun, J.; Lin, Y.-H.; Ganbat, K.; Bataakhuu, D.; Makhbal, Z.; Tsai, C.-Y.; Lin, Y.-H.; Chan, Y.-H.; Hsu, C.-J.; Hsu, W.-C.; et al. Unique spectra of deafness-associated mutations in Mongolians provide insights into the genetic relationships among Eurasian populations. PLoS ONE 2018, 13, e0209797. [Google Scholar] [CrossRef] [Green Version]
- Morell, R.; Kim, H.J.; Hood, L.J.; Goforth, L.; Friderici, K.; Fisher, R.; Van Camp, G.; Berlin, C.I.; Oddoux, C.; Ostrer, H.; et al. Mutations in the connexin 26 Gene (GJB2) among Ashkenazi Jews with nonsyndromic recessive deafness. New. Engl. J. Med. 1998, 339, 1500–1505. [Google Scholar] [CrossRef] [PubMed]
- Sobe, T.; Erlich, P.; Berry, A.; Korostichevsky, M.; Vreugde, S.; Avraham, K.B.; Bonné-Tamir, B.; Shohat, M. High frequency of the deafness-associated 167delT mutation in the connexin 26 (GJB2) gene in Israeli Ashkenazim. Am. J. Med. Genet. 1999, 86, 499–500. [Google Scholar] [CrossRef]
- Hamelmann, C.; Amedofu, G.K.; Albrecht, K.; Muntau, B.; Gelhaus, A.; Brobby, G.W.; Horstmann, R.D. Pattern of connexin 26 (GJB2) mutations causing sensorineural hearing impairment in Ghana. Hum. Mutat. 2001, 18, 84–85. [Google Scholar] [CrossRef] [PubMed]
- Figueroa-Ildefonso, E.; Bademci, G.; Rajabli, F.; Cornejo-Olivas, M.; Villanueva, R.D.C.; Badillo-Carrillo, R.; Inca-Martinez, M.; Neyra, K.M.; Sineni, C.; Tekin, M.; et al. Identification of main genetic causes responsible for non-syndromic hearing loss in a Peruvian population. Genes 2019, 10, 581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minárik, G.; Ferák, V.; Feráková, E.; Ficek, A.; Poláková, H.; Kadasi, L. High frequency of GJB2 mutation W24X among Slovak Romany (Gypsy) patients with non-syndromic hearing loss (NSHL). Gen. Physiol. Biophys. 2003, 22, 549–556. [Google Scholar]
- Ramshankar, M.; Girirajan, S.; Dagan, O.; Ravi, S.; Jalvi, R.; Rangasayee, R.; Avraham, K.B.; Anand, A. Contribution of connexin26 (GJB2) mutations and founder effect to non-syndromic hearing loss in India. J. Med. Genet. 2003, 40, e68. [Google Scholar] [CrossRef] [Green Version]
- Álvarez, A.; Del Castillo, I.; Villamar, M.; Aguirre, L.A.; Gonzalez-Neira, A.; López-Nevot, A.; Moreno-Pelayo, M.A.; Moreno, F. High prevalence of theW24X mutation in the gene encoding connexin-26 (GJB2) in Spanish Romani (gypsies) with autosomal recessive non-syndromic hearing loss. Am. J. Med. Genet. Part. A 2005, 137, 255–258. [Google Scholar] [CrossRef] [PubMed]
- Barashkov, N.A.; Dzhemilev, U.M.; Fedorova, S.A.; Teryutin, F.M.; Posukh, O.L.; Fedotova, E.E.; Lobov, S.; Khusnutdinova, E. Autosomal recessive deafness 1A (DFNB1A) in Yakut population isolate in Eastern Siberia: Extensive accumulation of the splice site mutation IVS1+1G>A in GJB2 gene as a result of founder effect. J. Hum. Genet. 2011, 56, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Carranza, C.; Menendez, I.; Herrera, M.; Castellanos, P.; Amado, C.; Maldonado, F.; Rosales, L.; Escobar, N.; Guerra, M.; Alvarez, D.; et al. A Mayan founder mutation is a common cause of deafness in Guatemala. Clin. Genet. 2015, 89, 461–465. [Google Scholar] [CrossRef] [PubMed]
- Van Laer, L.; Coucke, P.; Mueller, R.F.; Caethoven, G.; Flothmann, K.; Prasad, S.D.; Chamberlin, G.P.; Houseman, M.; Taylor, G.R.; van De Heyning, C.; et al. A common founder for the 35delG GJB2 gene mutation in connexin 26 hearing impairment. J. Med. Genet. 2001, 38, 515–518. [Google Scholar] [CrossRef] [Green Version]
- Tekin, M.; Akar, N.; Cin, Ş.; Blanton, S.; Xia, X.; Liu, X.; Nance, W.; Pandya, A. Connexin 26 (GJB2) mutations in the Turkish population: Implications for the origin and high frequency of the 35delG mutation in Caucasians. Qual. Life Res. 2001, 108, 385–389. [Google Scholar] [CrossRef]
- Shahin, H.; Walsh, T.; Sobe, T.; Lynch, E.; King, M.-C.; Avraham, K.B.; Kanaan, M. Genetics of congenital deafness in the Palestinian population: Multiple connexin 26 alleles with shared origins in the Middle East. Qual. Life Res. 2002, 110, 284–289. [Google Scholar] [CrossRef]
- Rothrock, C.R.; Murgia, A.; Sartorato, E.L.; Leonardi, E.; Wei, S.; Lebeis, S.L.; Yu, L.E.; Elfenbein, J.L.; Fisher, R.A.; Friderici, K.H. Connexin 26 35delG does not represent a mutational hotspot. Qual. Life Res. 2003, 113, 18–23. [Google Scholar] [CrossRef]
- Balci, B.; Gerçeker, F.O.; Aksoy, S.; Sennaroğlu, G.; Kalay, E.; Sennaroğlu, L.; Dinçer, P. Identification of an ancestral haplotype of the 35delG mutation in the GJB2 (connexin 26) gene responsible for autosomal recessive non-syndromic hearing loss in families from the Eastern Black Sea Region in Turkey. Turk. J. Pediatr. 2005, 47, 213–221. [Google Scholar]
- Belguith, H.; Hajji, S.; Salem, N.; Charfeddine, I.; Lahmar, I.; Amor, M.B.; Ouldim, K.; Chouery, E.; Driss, N.; Drira, M.; et al. Analysis of GJB2 mutation: Evidence for a Mediterranean ancestor for the 35delG mutation. Clin. Genet. 2005, 68, 188–189. [Google Scholar] [CrossRef]
- Tekin, M.; Boğoçlu, G.; Arican, S.; Orman, M.; Tastan, H.; Elsayed, S.; Akar, N. Evidence for single origins of 35delG and delE120 mutations in the GJB2 gene in Anatolia. Clin. Genet. 2004, 67, 31–37. [Google Scholar] [CrossRef]
- Abidi, O.; Boulouiz, R.; Nahili, H.; Imken, L.; Rouba, H.; Chafik, A.; Barakat, A. The analysis of three markers flanking GJB2 gene suggests a single origin of the most common 35delG mutation in the Moroccan population. Biochem. Biophys. Res. Commun. 2008, 377, 971–974. [Google Scholar] [CrossRef]
- Kokotas, H.; Van Laer, L.; Grigoriadou, M.; Iliadou, V.; Economides, J.; Pomoni, S.; Pampanos, A.; Eleftheriades, N.; Ferekidou, E.; Korres, S.; et al. Strong linkage disequilibrium for the frequent GJB2 35delG mutation in the Greek population. Am. J. Med. Genet. Part A 2008, 146, 2879–2884. [Google Scholar] [CrossRef] [PubMed]
- Kokotas, H.; Grigoriadou, M.; Villamar, M.; Giannoulia-Karantana, A.; Del Castillo, I.; Petersen, M.B. Hypothesizing an ancient greek origin of the GJB2 35delG Mutation: Can science meet history? Genet. Test. Mol. Biomark. 2010, 14, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Dzhemileva, L.U.; Posukh, O.L.; Barashkov, N.A.; Fedorova, S.A.; Teryutin, F.M.; Akhmetova, V.L.; Khidiyatova, I.M.; Khusainova, R.I.; Lobov, S.L.; Khusnutdinova, E.K. Haplotype diversity and reconstruction of ancestral haplotype associated with the c.35delG Mutation in the GJB2 (Cx26) gene among the Volgo-Ural populations of Russia. Acta Naturae 2011, 3, 52–63. [Google Scholar] [CrossRef]
- Norouzi, V.; Azizi, H.; Fattahi, Z.; Esteghamat, F.; Bazazzadegan, N.; Nishimura, C.; Nikzat, N.; Jalalvand, K.; Kahrizi, K.; Smith, R.J.H.; et al. Did the GJB2 35delG mutation originate in Iran? Am. J. Med. Genet. Part A 2011, 155, 2453–2458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zytsar, M.V.; Barashkov, N.A.; Bady-Khoo, M.S.; Shubina-Olejnik, O.A.; Danilenko, N.G.; Bondar, A.A.; Morozov, I.V.; Solovyev, A.; Danilchenko, V.Y.; Maximov, V.N.; et al. Updated carrier rates for c.35delG (GJB2) associated with hearing loss in Russia and common c.35delG haplotypes in Siberia. BMC Med. Genet. 2018, 19, 138. [Google Scholar] [CrossRef]
- Kudo, T.; Ikeda, K.; Kure, S.; Matsubara, Y.; Oshima, T.; Watanabe, K.-I.; Kawase, T.; Narisawa, K.; Takasaka, T. Novel mutations in the connexin 26 gene (GJB2) responsible for childhood deafness in the Japanese population. Am. J. Med. Genet. 2000, 90, 141–145. [Google Scholar] [CrossRef]
- Yan, D.; Park, H.-J.; Ouyang, X.M.; Pandya, A.; Doi, K.; Erdenetungalag, R.; Du, L.L.; Matsushiro, N.; Nance, W.E.; Griffith, A.J.; et al. Evidence of a founder effect for the 235delC mutation of GJB2 (connexin 26) in east Asians. Qual. Life Res. 2003, 114, 44–50. [Google Scholar] [CrossRef]
- Shinagawa, J.; Moteki, H.; Nishio, S.-Y.; Noguchi, Y.; Usami, S.-I. Haplotype analysis of GJB2 mutations: Founder effect or mutational hot spot? Genes 2020, 11, 250. [Google Scholar] [CrossRef] [Green Version]
- Gallant, E.; Francey, L.J.; Tsai, E.A.; Berman, M.; Zhao, Y.; Fetting, H.; Kaur, M.; Deardorff, M.A.; Wilkens, A.; Clark, D.; et al. Homozygosity for the V37I GJB2 mutation in fifteen probands with mild to moderate sensorineural hearing impairment: Further confirmation of pathogenicity and haplotype analysis in asian populations. Am. J. Med. Genet. Part A 2013, 161, 2148–2157. [Google Scholar] [CrossRef] [Green Version]
- Posukh, O.L.; Zytsar, M.V.; Bady-Khoo, M.S.; Danilchenko, V.Y.; Maslova, E.A.; Barashkov, N.A.; Bondar, A.A.; Morozov, I.V.; Maximov, V.N.; Voevoda, M.I. Unique mutational spectrum of the GJB2 Gene and its pathogenic contribution to deafness in Tuvinians (Southern Siberia, Russia): A high prevalence of rare variant c.516G>C (p.Trp172Cys). Genes 2019, 10, 429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Posukh, O.L.; Institute of Cytology and Genetics, Novosibirsk, Russia. Personal communication, 2019.
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Bengtsson, B.O.; Thomson, G. Measuring the strength of associations between HLA antigens and diseases. Tissue Antigens 1981, 18, 356–363. [Google Scholar] [CrossRef]
- Reeve, J.P.; Rannala, B. DMLE+: Bayesian linkage disequilibrium gene mapping. Bioinform 2002, 18, 894–895. [Google Scholar] [CrossRef] [Green Version]
- Risch, N.; De Leon, D.; Ozelius, L.; Kramer, P.; Almasy, L.; Singer, B.; Fahn, S.; Breakefield, X.; Bressman, S. Genetic analysis of idiopathic torsion dystonia in Ashkenazi Jews and their recent descent from a small founder population. Nat. Genet. 1995, 9, 152–159. [Google Scholar] [CrossRef]
- Labuda, M.; Labuda, D.; Korab-Laskowska, M.; Cole, D.E.; Zietkiewicz, E.; Weissenbach, J.; Popowska, E.; Pronicka, E.; Root, A.W.; Glorieux, F.H. Linkage disequilibrium analysis in young populations: Pseudo-vitamin D-deficiency rickets and the founder effect in French Canadians. Am. J. Hum. Genet. 1996, 59, 633–643. [Google Scholar] [PubMed]
- Labuda, D.; Zietkiewicz, E.; Labuda, M. The genetic clock and the age of the founder effect in growing populations: A lesson from French Canadians and Ashkenazim. Am. J. Hum. Genet. 1997, 61, 768–771. [Google Scholar] [CrossRef] [Green Version]
- Colombo, R. Age estimate of the N370S mutation causing gaucher disease in Ashkenazi Jews and European populations: A reappraisal of haplotype data. Am. J. Hum. Genet. 2000, 66, 692–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiner, O. Faculty opinions recommendation of mutations of bacteria from virus sensitivity to virus resistance. Fac. Opin. 2010, 28, 491–511. [Google Scholar] [CrossRef]
- Slatkin, M.; Rannala, B. Estimating allele age. Annu. Rev. Genom. Hum. Genet. 2000, 1, 225–249. [Google Scholar] [CrossRef] [Green Version]
- Mongush, M.V. Tuvans of Mongolia and China. Int. J. Cent. Asian Stud. 1996, 1, 225–243. [Google Scholar]
- Chen, Z.; Zhang, Y.; Fan, A.; Zhang, Y.; Wu, Y.; Zhao, Q.; Zhou, Y.; Zhou, C.; Bawudong, M.; Mao, X.; et al. Brief communication: Y-chromosome haplogroup analysis indicates that Chinese Tuvans share distinctive affinity with Siberian Tuvans. Am. J. Phys. Anthr. 2011, 144, 492–497. [Google Scholar] [CrossRef]
- Vainshtein, S.I.; Mannay-Ool, M.H. History of Tyva, 2nd ed.; Science: Novosibirsk, Russia, 2001. (In Russian) [Google Scholar]
- Mannai-ool, M.K.; Tuvan People. The Origin and Formation of the Ethnos; Nauka Publ: Novosibirsk, Russia, 2004; pp. 99–166. (In Russian) [Google Scholar]
- Potapov, L.P. Ethnical structure and origin of Altaians; Nauka: Leningrad, Soviet Union, 1969. [Google Scholar]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the american college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]
- Distefano, M.T.; Hemphill, S.E.; Oza, A.M.; Siegert, R.K.; Grant, A.R.; Hughes, M.Y.; Cushman, B.J.; Azaiez, H.; Booth, K.T.; Chapin, A.; et al. ClinGen expert clinical validity curation of 164 hearing loss gene–disease pairs. Genet. Med. 2019, 21, 2239–2247. [Google Scholar] [CrossRef] [PubMed]
- Tekin, M.; Xia, X.-J.; Erdenetungalag, R.; Cengiz, F.B.; White, T.W.; Radnaabazar, J.; Dangaasuren, B.; Tastan, H.; Nance, W.E.; Pandya, A. GJB2 mutations in Mongolia: Complex alleles, low frequency, and reduced fitness of the deaf. Ann. Hum. Genet. 2010, 74, 155–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Ao, L.; Ding, H.; Zhang, D. Genetic frequencies related to severe or profound sensorineural hearing loss in inner Mongolia autonomous region. Genet. Mol. Boil. 2016, 39, 567–572. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.-L.; Bai-Cheng, X.; Chen, X.-J.; Pan-Pan, B.; Jian-Li, M.; Xiao-Wen, L.; Zhang, Z.-W.; Wan, D.; Zhu, Y.-M.; Guo, Y.-F. Common molecular etiology of patients with nonsyndromic hearing loss in Tibetan, Tu nationality, and Mongolian patients in the northwest of China. Acta Oto Laryngol. 2013, 133, 930–934. [Google Scholar] [CrossRef]
- Tsukada, K.; Nishio, S.-Y.; Usami, S.; The Deafness Gene Study Consortium. A large cohort study of GJB2 mutations in Japanese hearing loss patients. Clin. Genet. 2010, 78, 464–470. [Google Scholar] [CrossRef]
- Sirmaci, A.; Akcayoz-Duman, D.; Tekin, M. The c.IVS1+1G>A mutation in the GJB2 gene is prevalent and large deletions involving the GJB6 gene are not present in the Turkish population. J. Genet. 2006, 85, 213–216. [Google Scholar] [CrossRef]
- Seeman, P.; Sakmaryová, I. High prevalence of the IVS 1+1 G to A/GJB2 mutation among Czech hearing impaired patients with monoallelic mutation in the coding region of GJB2: IVS 1 + 1 G to A GJB2 mutation in Czech. Clin. Genet. 2006, 69, 410–413. [Google Scholar] [CrossRef]
- Yuan, Y.; Yu, F.; Wang, G.; Huang, S.; Yu, R.; Zhang, X.; Huang, D.-L.; Han, D.-Y.; Dai, P. Prevalence of the GJB2 IVS1+1G >A mutation in Chinese hearing loss patients with monoallelic pathogenic mutation in the coding region of GJB. J. Transl. Med. 2010, 8, 127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bazazzadegan, N.; Nikzat, N.; Fattahi, Z.; Nishimura, C.; Meyer, N.; Sahraian, S.; Jamali, P.; Babanejad, M.; Kashef, A.; Yazdan, H.; et al. The spectrum of GJB2 mutations in the Iranian population with non-syndromic hearing loss—A twelve year study. Int. J. Pediatr. Otorhinolaryngol. 2012, 76, 1164–1174. [Google Scholar] [CrossRef] [PubMed]
- Barashkov, N.A.; Pshennikova, V.G.; Posukh, O.L.; Teryutin, F.M.; Solovyev, A.V.; Klarov, L.A.; Romanov, G.P.; Gotovtsev, N.; Kozhevnikov, A.A.; Kirillina, E.V.; et al. Spectrum and frequency of the GJB2 gene pathogenic variants in a large cohort of patients with hearing impairment living in a subarctic region of Russia (the Sakha Republic). PLoS ONE 2016, 11, e0156300. [Google Scholar] [CrossRef]
- Solovyev, A.; Barashkov, N.A.; Teryutin, F.M.; Pshennikova, V.G.; Romanov, G.P.; Rafailov, A.M.; Sazonov, N.N.; Dzhemilev, U.M.; Tomsky, M.I.; Posukh, O.L.; et al. Selective Heterozygous advantage of carriers of c.-23+1G>A mutation in GJB2 gene causing autosomal recessive deafness 1A. Bull. Exp. Boil. Med. 2019, 167, 380–383. [Google Scholar] [CrossRef] [PubMed]
- Solovyev, A.V.; Barashkov, N.A.; Bady-Khoo, M.S.; Zytsar, M.V.; Posukh, O.L.; Romanov, G.P.; Rafailov, A.M.; Sazonov, N.N.; Alexeev, A.N.; Dzhemilev, U.M.; et al. Reconstruction of SNP haplotypes with mutation c.-23+1G>A in human gene GJB2 (Chromosome 13) in some populations of Eurasia. Russ. J. Genet. 2017, 53, 936–941. [Google Scholar] [CrossRef]
- Fedorova, S.A.; Reidla, M.; Metspalu, E.; Metspalu, M.; Rootsi, S.; Tambets, K.; Trofimova, N.; Zhadanov, S.I.; Kashani, B.H.; Olivieri, A.; et al. Autosomal and uniparental portraits of the native populations of Sakha (Yakutia): Implications for the peopling of Northeast Eurasia. BMC Evol. Boil. 2013, 13, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Grillo, A.P.; De Oliveira, F.M.; De Carvalho, G.Q.; Medrano, R.F.V.; Da Silva-Costa, S.M.; Sartorato, E.L.; De Oliveira, C.A. Single nucleotide polymorphisms of the GJB2 and GJB6 genes are associated with autosomal recessive nonsyndromic hearing loss. BioMed Res. Int. 2015, 2015, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Parzefall, T.; Lucas, T.; Koenighofer, M.; Ramsebner, R.; Frohne, A.; Czeiger, S.; Baumgartner, W.-D.; Schoefer, C.; Gstoettner, W.; Frei, K. The role of alternative GJB2 transcription in screening for neonatal sensorineural deafness in Austria. Acta Oto Laryngol. 2016, 137, 356–360. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Haplotypes * | Frequency of Haplotypes | x2 | p | |
|---|---|---|---|---|
| Mutant Chromosomes | Normal Chromosomes | |||
| Haplotypes for c.516G>C: D13S1316-D13S141-D13S175-D13S1853-D13S143 (~ 1.6 Mb) | ||||
| 269-124-105-204-125 | 0.6786 | 0.0161 | 79 | <10−14 |
| 267-124-105-204-125 | 0.2857 | 0.2979 | 0.0093 | 0.5462 |
| 269-124-105-204-129 | 0.0357 | 0 | 0.67 | 0.1842 |
| other haplotypes | 0 | 0.6860 | - | - |
| Haplotypes for c.-23+1G>A: D13S141-D13S175-D13S1853-D13S143-D13S1275-D13S292 (~ 3.5 Mb) | ||||
| 124-105-204-125-208-209 | 0.8333 | 0.0538 | 53 | <10−8 |
| 124-105-204-125-202-211 | 0.0833 | 0.0108 | 0.66 | 0.1695 |
| 124-105-204-125-210-209 | 0.0833 | 0.0472 | 0.011 | 0.4586 |
| other haplotypes | 0 | 0.8882 | - | - |
| Haplotypes for c.235delC: D13S1316-D13S141-D13S175-D13S1853-D13S143-D13S1275 (~ 1.7 Mb) | ||||
| 267-124-105-204-125-210 | 1.0 | 0 | 103 | <10−11 |
| other haplotypes | 0 | 1.0 | - | - |
| Haplotypes * | Frequency of Haplotypes | x2 | p | |
|---|---|---|---|---|
| Mutant Chromosomes | Normal Chromosomes | |||
| Haplotypes for c.516G>C: rs747931-rs5030700-rs3751385-rs2274083-rs2274084-rs1411911768-rs9552101-rs117685390-rs877098 | ||||
| T-C-C-A-G-T-G-T-C | 1 | 0.0217 | 120 | <10−26 |
| other haplotypes | 0 | 0.9783 | - | - |
| Haplotypes for c.-23+1G>A: rs747931-rs5030700-rs3751385-rs2274083-rs2274084-rs1411911768-rs9552101-rs117685390-rs877098 | ||||
| C-C-C-A-G-C-G-T-C | 0.9167 | 0.0532 | 64 | <10−10 |
| C-C-C-A-G-C-G-T-T | 0.0833 | 0.1540 | 0.047 | 0.4488 |
| other haplotypes | 0 | 0.7928 | - | - |
| Haplotypes for c.235delC: rs747931-rs5030700-rs3751385-rs2274083-rs2274084-rs1411911768-rs9552101-rs117685390-rs877098 | ||||
| T-C-C-A-G-C-G-T-T | 1 | 0.1587 | 26 | 1 |
| other haplotypes | 0 | 0.8413 | - | - |
| Mutation | d | g (95% CI) | Age (95% CI) |
|---|---|---|---|
| c.516G>C | 0.05 0.1 0.2 | 91–180 57–106 31–55 | 2275–4500 years 1425–2650 years 775–1375 years |
| c.-23+1G>A | 0.05 0.1 0.2 | 73–164 42–91 29–54 | 1825–4100 years 1050–2275 years 725–1350 years |
| c.235delC | 0.05 0.1 0.2 | 45–126 34–79 22–46 | 1125–3150 years 850–1975 years 550–1150 years |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zytsar, M.V.; Bady-Khoo, M.S.; Danilchenko, V.Y.; Maslova, E.A.; Barashkov, N.A.; Morozov, I.V.; Bondar, A.A.; Posukh, O.L. High Rates of Three Common GJB2 Mutations c.516G>C, c.-23+1G>A, c.235delC in Deaf Patients from Southern Siberia Are Due to the Founder Effect. Genes 2020, 11, 833. https://doi.org/10.3390/genes11070833
Zytsar MV, Bady-Khoo MS, Danilchenko VY, Maslova EA, Barashkov NA, Morozov IV, Bondar AA, Posukh OL. High Rates of Three Common GJB2 Mutations c.516G>C, c.-23+1G>A, c.235delC in Deaf Patients from Southern Siberia Are Due to the Founder Effect. Genes. 2020; 11(7):833. https://doi.org/10.3390/genes11070833
Chicago/Turabian StyleZytsar, Marina V., Marita S. Bady-Khoo, Valeriia Yu. Danilchenko, Ekaterina A. Maslova, Nikolay A. Barashkov, Igor V. Morozov, Alexander A. Bondar, and Olga L. Posukh. 2020. "High Rates of Three Common GJB2 Mutations c.516G>C, c.-23+1G>A, c.235delC in Deaf Patients from Southern Siberia Are Due to the Founder Effect" Genes 11, no. 7: 833. https://doi.org/10.3390/genes11070833
APA StyleZytsar, M. V., Bady-Khoo, M. S., Danilchenko, V. Y., Maslova, E. A., Barashkov, N. A., Morozov, I. V., Bondar, A. A., & Posukh, O. L. (2020). High Rates of Three Common GJB2 Mutations c.516G>C, c.-23+1G>A, c.235delC in Deaf Patients from Southern Siberia Are Due to the Founder Effect. Genes, 11(7), 833. https://doi.org/10.3390/genes11070833