The Gut Microbiota Profile in Children with Prader–Willi Syndrome

 , , ,
, , ,  , , ,
, , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. Assessments
2.3. Fecal Sample Collection, DNA Sequencing, and Microbiome Analysis
2.4. Statistical Analyses
3. Results
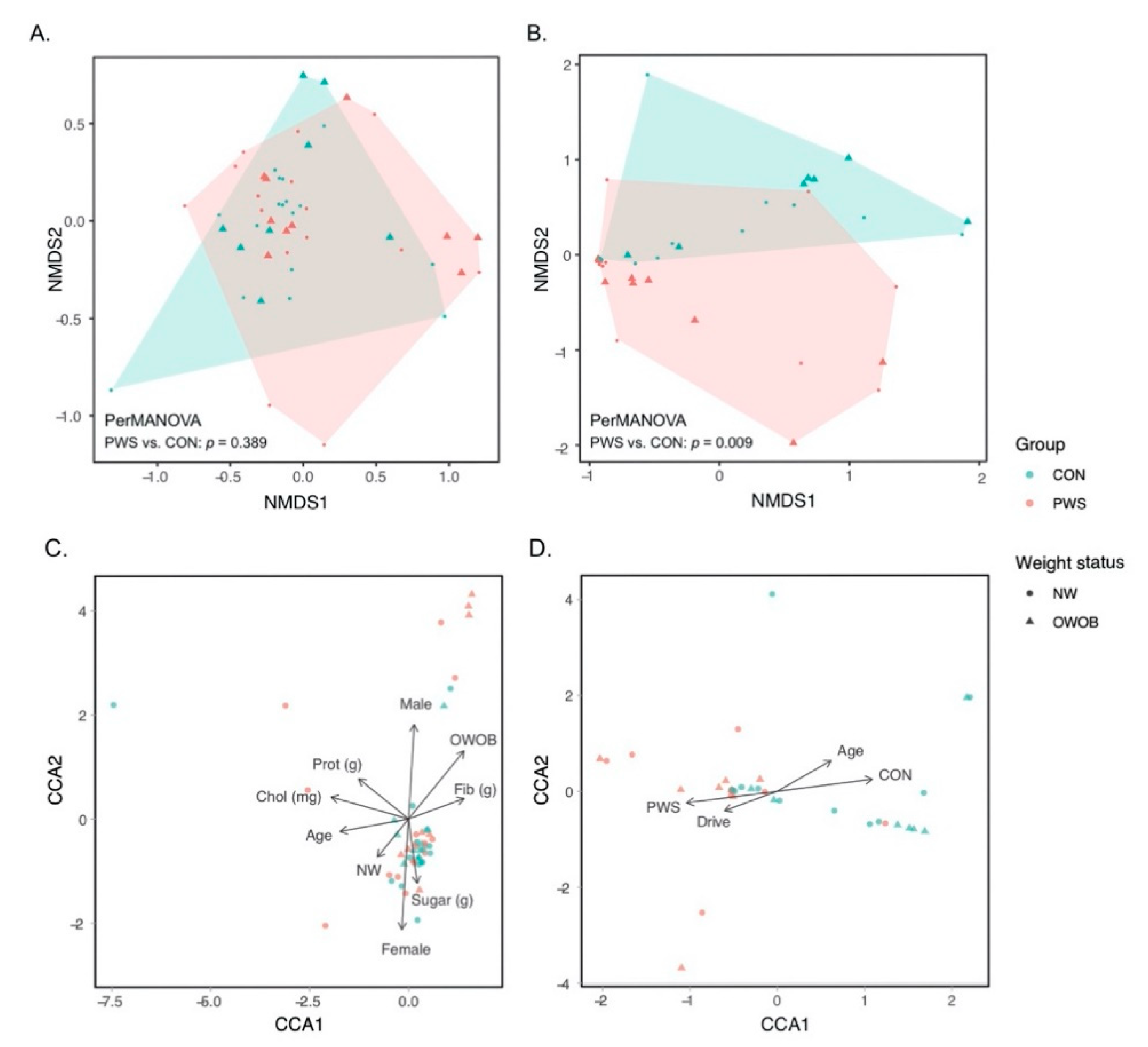
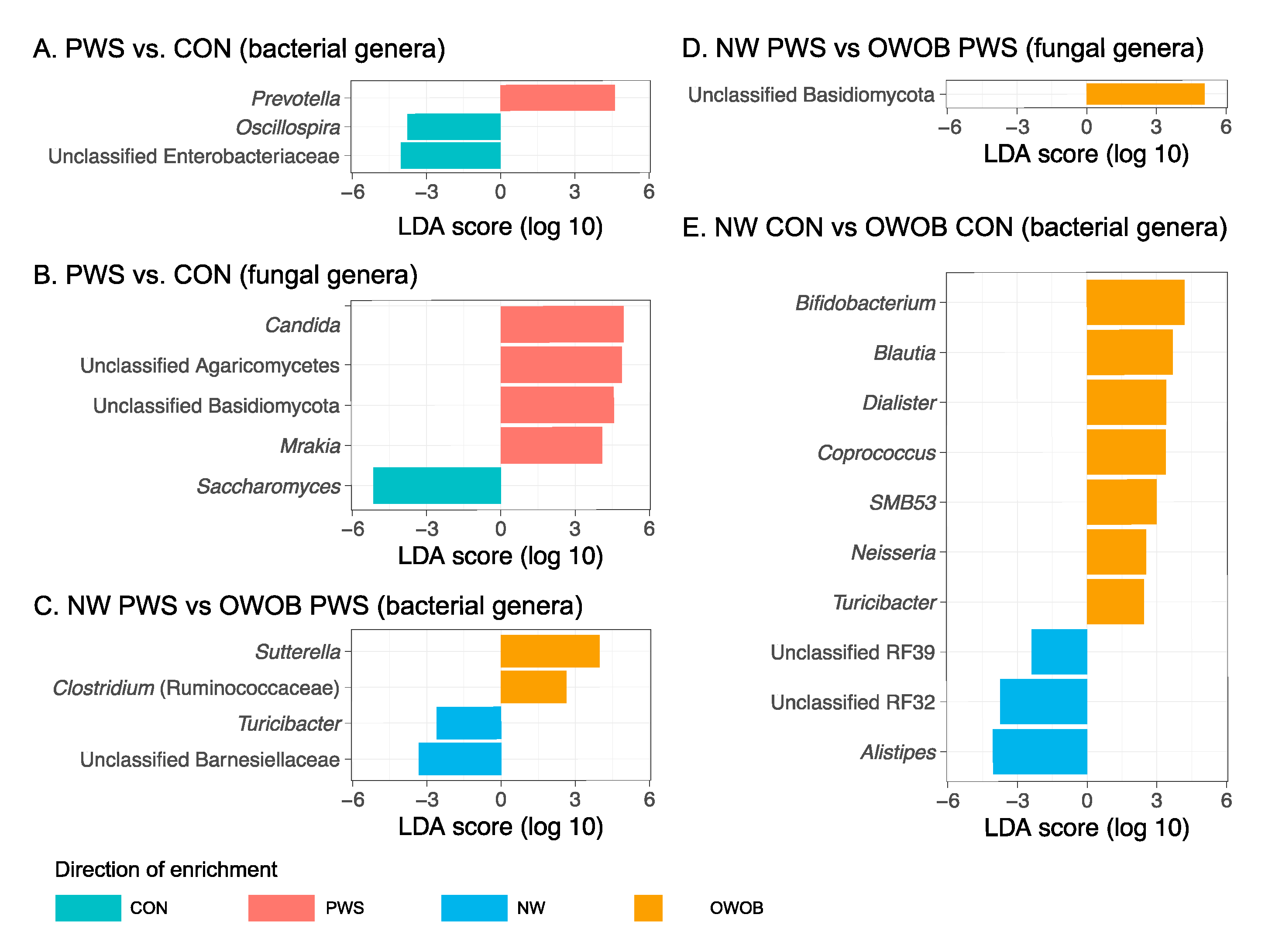
3.1. Gut Bacterial Communities in PWS
3.2. Gut Fungal Communities in PWS
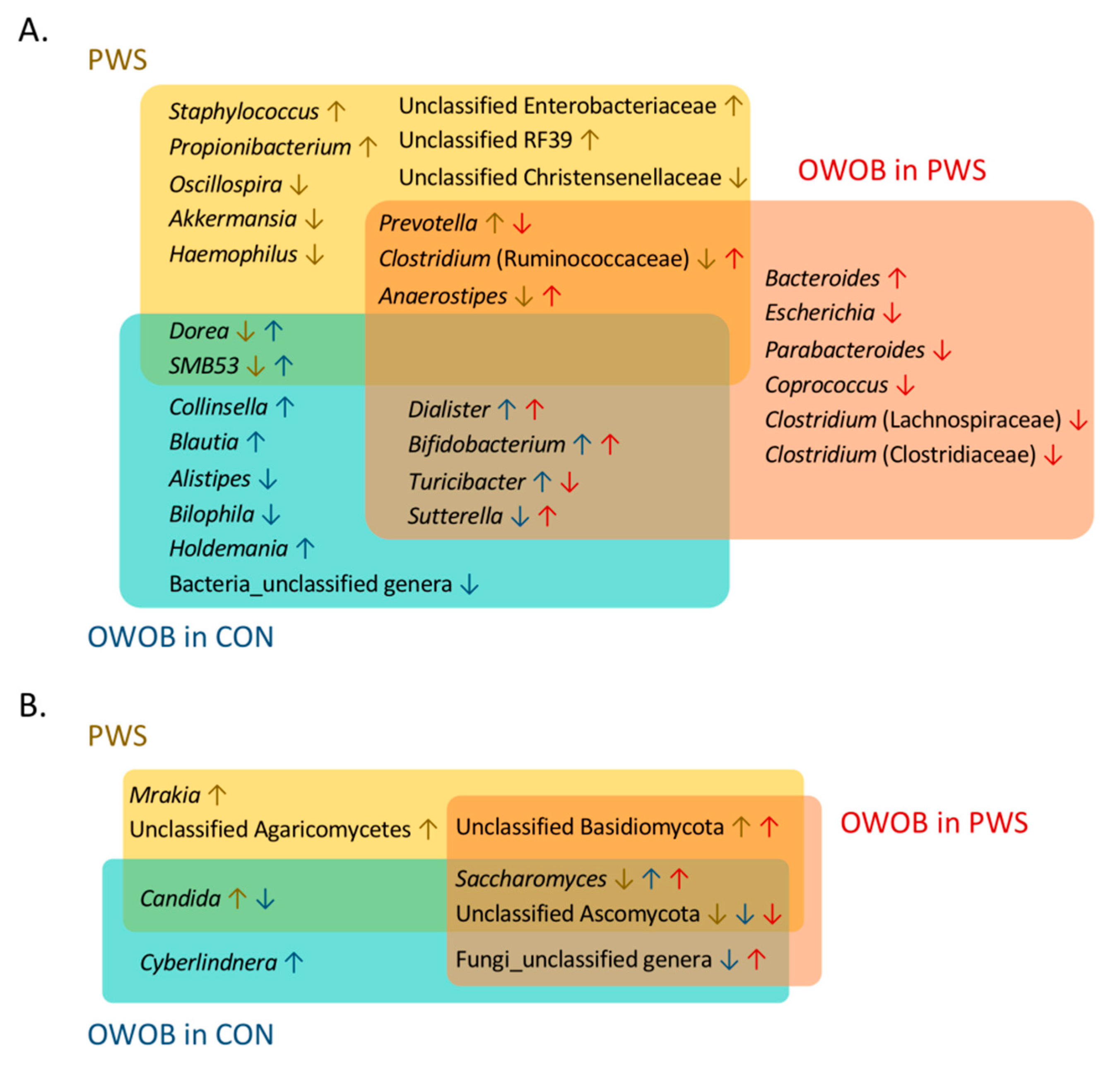
3.3. Abundance of Certain Taxa Is Associated with PWS
3.4. Gut Microbiota and Dietary Carbohydrate Intake in the Whole Dataset
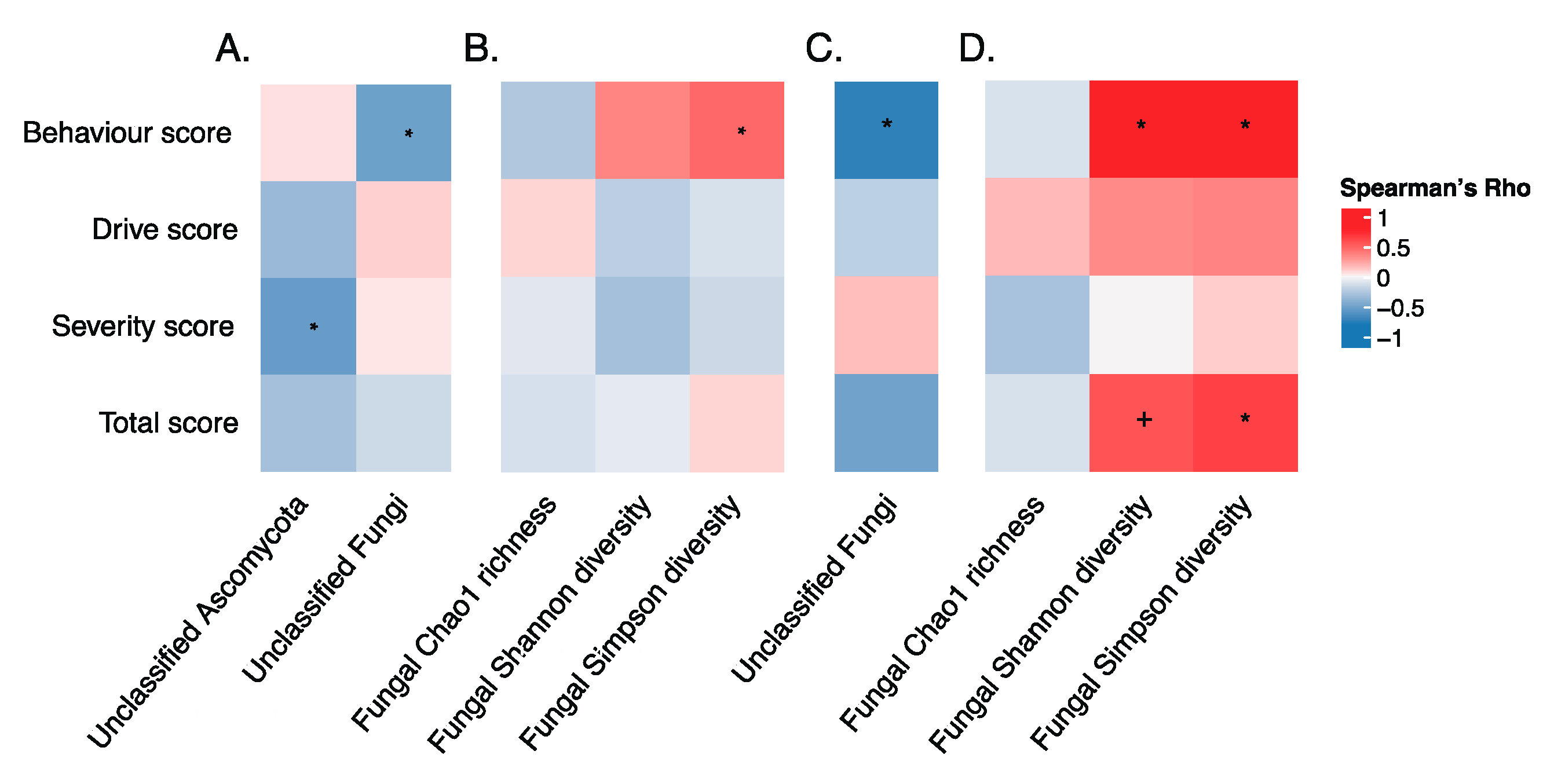
3.5. Gut Microbiota and Hyperphagia in PWS
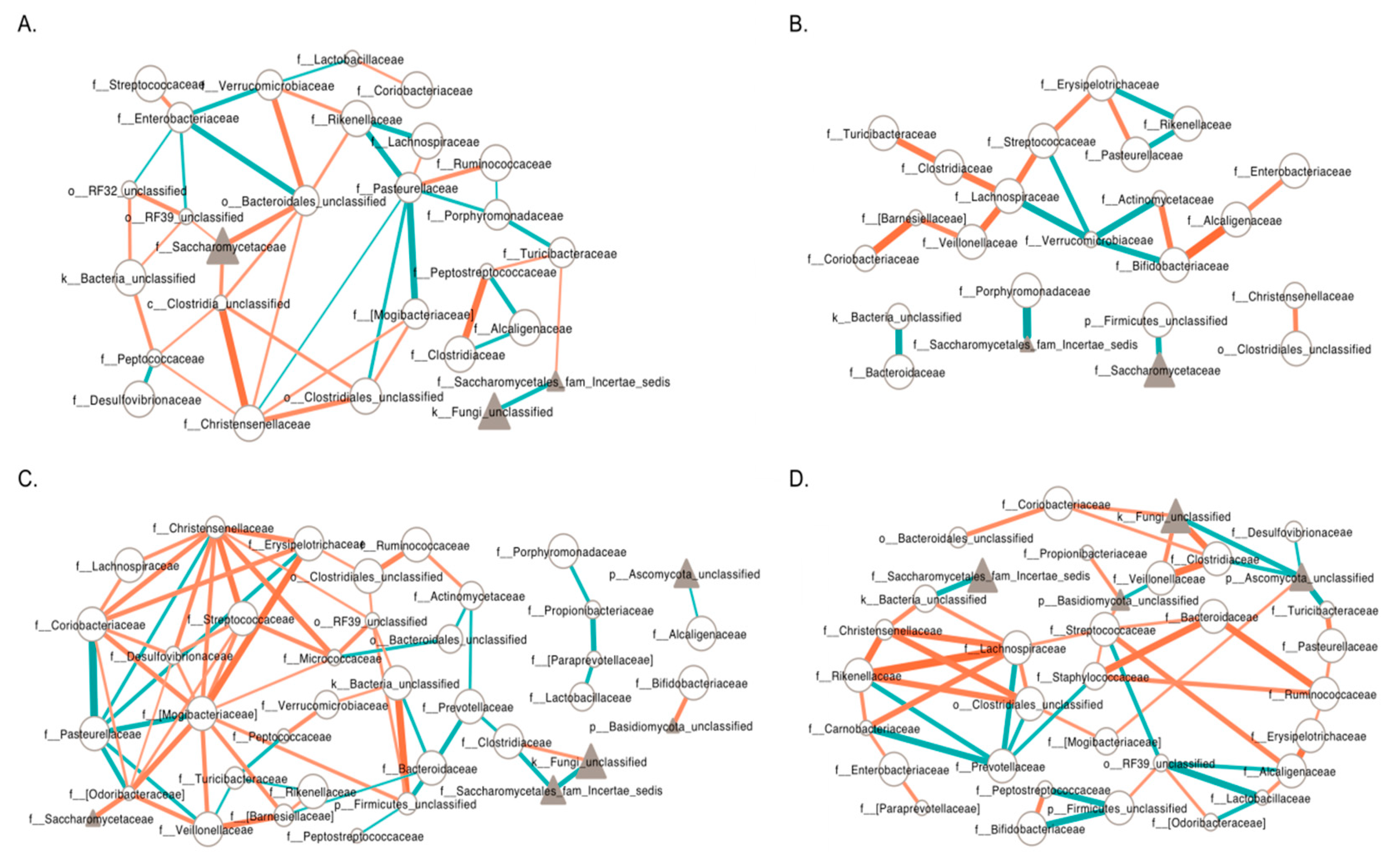
3.6. Inter-Kingdom Ecological Networks in PWS
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Appendix A



References
- Butler, M.G.; Miller, J.L.; Forster, J.L. Prader-Willi Syndrome-Clinical Genetics, Diagnosis and Treatment Approaches: An Update. Curr. Pediatr. Rev. 2019, 15, 207–244. [Google Scholar] [CrossRef] [PubMed]
- Irizarry, K.A.; Miller, M.; Freemark, M.; Haqq, A.M. Prader Willi Syndrome: Genetics, Metabolomics, Hormonal Function, and New Approaches to Therapy. Adv. Pediatr. 2016, 63, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.J.; Miller, J.L.; Kuipers, P.J.; German, J.R.; Beaudet, A.L.; Sahoo, T.; Driscoll, D.J. Unique and atypical deletions in Prader-Willi syndrome reveal distinct phenotypes. Eur. J. Hum. Genet. 2012, 20, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.L.; Lynn, C.H.; Driscoll, D.C.; Goldstone, A.P.; Gold, J.A.; Kimonis, V.; Dykens, E.; Butler, M.G.; Shuster, J.J.; Driscoll, D.J. Nutritional phases in Prader-Willi syndrome. Am. J. Med. Genet. Part A 2011, 155, 1040–1049. [Google Scholar] [CrossRef] [Green Version]
- Heymsfield, S.B.; Avena, N.M.; Baier, L.; Brantley, P.; Bray, G.A.; Burnett, L.C.; Butler, M.G.; Driscoll, D.J.; Egli, D.; Elmquist, J.; et al. Hyperphagia: Current concepts and future directions proceedings of the 2nd international conference on hyperphagia. Obesity 2014, 22, 1–35. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Yin, A.; Li, H.; Wang, R.; Wu, G.; Shen, J.; Zhang, M.; Wang, L.; Hou, Y.; Ouyang, H.; et al. Dietary Modulation of Gut Microbiota Contributes to Alleviation of Both Genetic and Simple Obesity in Children. EBioMedicine 2015, 2, 968–984. [Google Scholar] [CrossRef]
- Olsson, L.M.; Poitou, C.; Tremaroli, V.; Coupaye, M.; Aron-Wisnewsky, J.; Bäckhed, F.; Clément, K.; Caesar, R. Gut microbiota of obese subjects with Prader-Willi syndrome is linked to metabolic health. Gut 2019, 69, 1229–1238. [Google Scholar] [CrossRef] [Green Version]
- Limon, J.J.; Skalski, J.H.; Underhill, D.M. Commensal Fungi in Health and Disease. Cell Host Microbe 2017, 22, 156–165. [Google Scholar] [CrossRef]
- Dykens, E.M.; Maxwell, M.A.; Pantino, E.; Kossler, R.; Roof, E. Assessment of hyperphagia in prader-Willi syndrome. Obesity 2007, 15, 1816–1826. [Google Scholar] [CrossRef] [Green Version]
- Willett, W.C.; Howe, G.R.; Kushi, L.H. Adjustment for total energy intake in epidemiologic studies. Am. J. Clin. Nutr. 1997, 65, 1220S–1228S. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. Available online: https://www.cdc.gov/nchs/data/nhanes/nhanes_07_08/manual_an.pdf (accessed on 21 November 2016).
- de Onis, M.; Onyango, A.W.; Borghi, E.; Siyam, A.; Nishida, C.; Siekmann, J. Development of a WHO growth reference for school-aged children and adolescents. Bull World Health Organ. 2007, 85, 660–667. [Google Scholar] [CrossRef] [PubMed]
- Anderson, E.L.; Li, W.; Klitgord, N.; Highlander, S.K.; Dayrit, M.; Seguritan, V.; Yooseph, S.; Biggs, W.; Venter, J.C.; Nelson, K.E.; et al. A robust ambient temperature collection and stabilization strategy: Enabling worldwide functional studies of the human microbiome. Sci. Rep. 2016, 6, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozich, J.J.; Westcott, S.L.; Baxter, N.T.; Highlander, S.K.; Schloss, P.D. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the miseq illumina sequencing platform. Appl. Environ. Microbiol. 2013, 79, 5112–5120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afgan, E.; Baker, D.; Batut, B.; Van Den Beek, M.; Bouvier, D.; Ech, M.; Chilton, J.; Clements, D.; Coraor, N.; Grüning, B.A.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef] [Green Version]
- Tun, H.M.; Bridgman, S.L.; Chari, R.; Field, C.J.; Guttman, D.S.; Becker, A.B.; Mandhane, P.J.; Turvey, S.E.; Subbarao, P.; Sears, M.R.; et al. Roles of birth mode and infant gut microbiota in intergenerational transmission of overweight and obesity from mother to offspring. JAMA Pediatr. 2018, 172, 368–377. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.R.; Shin, J.; Guevarra, R.B.; Lee, J.H.; Kim, D.W.; Seol, K.H.; Lee, J.H.; Kim, H.B.; Isaacson, R.E. Deciphering diversity indices for a better understanding of microbial communities. J. Microbiol. Biotechnol. 2017, 27, 2089–2093. [Google Scholar] [CrossRef] [Green Version]
- Mosca, A.; Leclerc, M.; Hugot, J.P. Gut microbiota diversity and human diseases: Should we reintroduce key predators in our ecosystem? Front. Microbiol. 2016, 7, 455. [Google Scholar] [CrossRef] [Green Version]
- McBurney, M.I.; Davis, C.; Fraser, C.M.; Schneeman, B.O.; Huttenhower, C.; Verbeke, K.; Walter, J.; Latulippe, M.E. Establishing What Constitutes a Healthy Human Gut Microbiome: State of the Science, Regulatory Considerations, and Future Directions. J. Nutr. 2019, 149, 1882–1895. [Google Scholar] [CrossRef]
- Garcia-Ribera, S.; Amat-Bou, M.; Climent, E.; Llobet, M.; Chenoll, E.; Corripio, R.; Ibáñez, L.; Ramon-Krauel, M.; Lerin, C. Specific dietary components and gut microbiota composition are associated with obesity in children and adolescents with prader–willi syndrome. Nutrients 2020, 12, 1063. [Google Scholar] [CrossRef]
- Cani, P.D.; Knauf, C. How gut microbes talk to organs: The role of endocrine and nervous routes. Mol. Metab. 2016, 5, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Queipo-Ortuño, M.I.; Seoane, L.M.; Murri, M.; Pardo, M.; Gomez-Zumaquero, J.M.; Cardona, F.; Casanueva, F.; Tinahones, F.J. Gut Microbiota Composition in Male Rat Models under Different Nutritional Status and Physical Activity and Its Association with Serum Leptin and Ghrelin Levels. PLoS ONE 2013, 8, e65465. [Google Scholar] [CrossRef] [PubMed]
- Fehlner-Peach, H.; Magnabosco, C.; Raghavan, V.; Scher, J.U.; Tett, A.; Cox, L.M.; Gottsegen, C.; Watters, A.; Wiltshire-Gordon, J.D.; Segata, N.; et al. Distinct Polysaccharide Utilization Profiles of Human Intestinal Prevotella copri Isolates. Cell Host Microbe 2019, 26, 680–690. [Google Scholar] [CrossRef] [PubMed]
- Goodrich, J.K.; Waters, J.L.; Poole, A.C.; Sutter, J.L.; Koren, O.; Blekhman, R.; Beaumont, M.; Van Treuren, W.; Knight, R.; Bell, J.T.; et al. Human genetics shape the gut microbiome. Cell 2014, 159, 789–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konikoff, T.; Gophna, U. Oscillospira: A Central, Enigmatic Component of the Human Gut Microbiota. Trends Microbiol. 2016, 24, 523–524. [Google Scholar] [CrossRef] [PubMed]
- Depommier, C.; Everard, A.; Druart, C.; Plovier, H.; Van Hul, M.; Vieira-Silva, S.; Falony, G.; Raes, J.; Maiter, D.; Delzenne, N.M.; et al. Supplementation with Akkermansia muciniphila in overweight and obese human volunteers: a proof-of-concept exploratory study. Nat. Med. 2019, 25, 1096–1103. [Google Scholar] [CrossRef]
- Plovier, H.; Everard, A.; Druart, C.; Depommier, C.; Van Hul, M.; Geurts, L.; Chilloux, J.; Ottman, N.; Duparc, T.; Lichtenstein, L.; et al. A purified membrane protein from Akkermansia muciniphila or the pasteurized bacterium improves metabolism in obese and diabetic mice. Nat. Med. 2017, 23, 107–113. [Google Scholar] [CrossRef] [Green Version]
- Dao, M.C.; Everard, A.; Aron-Wisnewsky, J.; Sokolovska, N.; Prifti, E.; Verger, E.O.; Kayser, B.D.; Levenez, F.; Chilloux, J.; Hoyles, L.; et al. Akkermansia muciniphila and improved metabolic health during a dietary intervention in obesity: Relationship with gut microbiome richness and ecology. Gut 2016, 64, 531–537. [Google Scholar] [CrossRef] [Green Version]
- Derrien, M.; Belzer, C.; de Vos, W.M. Akkermansia muciniphila and its role in regulating host functions. Microb. Pathog. 2017, 106, 171–181. [Google Scholar] [CrossRef]
- Caesar, R.; Tremaroli, V.; Kovatcheva-Datchary, P.; Cani, P.D.; Bäckhed, F. Crosstalk between gut microbiota and dietary lipids aggravates WAT inflammation through TLR signaling. Cell Metab. 2015, 22, 658–668. [Google Scholar] [CrossRef] [Green Version]
- Karlsson, C.L.J.; Önnerfält, J.; Xu, J.; Molin, G.; Ahrné, S.; Thorngren-Jerneck, K. The microbiota of the gut in preschool children with normal and excessive body weight. Obesity 2012, 20, 2257–2261. [Google Scholar] [CrossRef] [PubMed]
- Bassetti, M.; Taramasso, L.; Nicco, E.; Molinari, M.P.; Mussap, M.; Viscoli, C. Epidemiology, species distribution, antifungal susceptibility and outcome of nosocomial candidemia in a tertiary care hospital in Italy. PLoS ONE 2011, 6, e24198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, L.; Yang, C.; Tang, J. Disruption of the intestinal mucosal barrier in Candida albicans infections. Microbiol. Res. 2013, 168, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Muñiz Pedrogo, D.A.; Jensen, M.D.; Van Dyke, C.T.; Murray, J.A.; Woods, J.A.; Chen, J.; Kashyap, P.C.; Nehra, V. Gut Microbial Carbohydrate Metabolism Hinders Weight Loss in Overweight Adults Undergoing Lifestyle Intervention With a Volumetric Diet. Mayo Clin. Proc. 2018, 93, 1104–1110. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.N.; Yu, Q.F.; Fu, N.; Liu, X.W.; Lu, F.G. Effects of four Bifidobacteria on obesity in high-fat diet induced rats. World J. Gastroenterol. 2010, 16, 3394. [Google Scholar] [CrossRef] [PubMed]
- Van Dijck, P.; Jabra-Rizk, M.A. Fungal-bacterial interactions: In health and disease. In Candida Albicans: Cellular and Molecular Biology, 2nd ed.; Springer: New York, NY, USA, 2017; pp. 115–143. [Google Scholar] [CrossRef]
- Coker, O.O.; Nakatsu, G.; Dai, R.Z.; Wu, W.K.K.; Wong, S.H.; Ng, S.C.; Chan, F.K.L.; Sung, J.J.Y.; Yu, J. Enteric fungal microbiota dysbiosis and ecological alterations in colorectal cancer. Gut 2019, 68, 654–662. [Google Scholar] [CrossRef]
- Sokol, H.; Leducq, V.; Aschard, H.; Pham, H.P.; Jegou, S.; Landman, C.; Cohen, D.; Liguori, G.; Bourrier, A.; Nion-Larmurier, I.; et al. Fungal microbiota dysbiosis in IBD. Gut 2017, 66, 1039–1048. [Google Scholar] [CrossRef] [Green Version]
- Hoarau, G.; Mukherjee, P.K.; Gower-Rousseau, C.; Hager, C.; Chandra, J.; Retuerto, M.A.; Neut, C.; Vermeire, S.; Clemente, J.; Colombel, J.F.; et al. Bacteriome and mycobiome interactions underscore microbial dysbiosis in familial Crohn’s disease. MBio 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Dai, B.; Tang, Y.; Lei, L.; Li, N.; Liu, C.; Ge, T.; Zhang, L.; Xu, Y.; Hu, Y.; et al. Altered Bacterial-Fungal Interkingdom Networks in the Guts of Ankylosing Spondylitis Patients. mSystems 2019, 4. [Google Scholar] [CrossRef] [Green Version]
- Rothschild, D.; Weissbrod, O.; Barkan, E.; Kurilshikov, A.; Korem, T.; Zeevi, D.; Costea, P.I.; Godneva, A.; Kalka, I.N.; Bar, N.; et al. Environment dominates over host genetics in shaping human gut microbiota. Nature 2018, 555, 210–215. [Google Scholar] [CrossRef]
- Wang, J.; Thingholm, L.B.; Skiecevičie, J.; Rausch, P.; Kummen, M.; Hov, J.R.; Degenhardt, F.; Heinsen, F.A.; Rühlemann, M.C.; Szymczak, S.; et al. Genome-wide association analysis identifies variation in Vitamin D receptor and other host factors influencing the gut microbiota. Nat. Genet. 2016, 48, 1396–1406. [Google Scholar] [CrossRef] [PubMed]
- Walter, J.; Armet, A.M.; Finlay, B.B.; Shanahan, F. Establishing or Exaggerating Causality for the Gut Microbiome: Lessons from Human Microbiota-Associated Rodents. Cell 2020, 180, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.L.A.; Vieira-Silva, S.; Liston, A.; Raes, J. How informative is the mouse for human gut microbiota research? Dis. Model. Mech. 2015, 8, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PWS (n = 25) | CON (n = 25) | p-Values | |
|---|---|---|---|
| Sex (F/M) | 14/11 | 9/16 | 0.256 |
| Age (years) | 6.2 (5.2, 12.9) | 8.8 (6.3, 10.5) | 0.455 |
| BMI %ile | 79.3 (65.5, 94.1) | 76.6 (51.2, 91.5) | 0.655 |
| Weight status (OWOB/NW) | 10/15 | 8/17 | 0.769 |
| Hyperphagia scores ** | 19 (16, 26) | 15 (14, 18) | 0.014 * |
| Protein (g) | 71 (65, 76) | 64 (56, 73) | 0.071 |
| Carbohydrate (g) | 189 (149, 206.3) | 225 (193, 240) | 0.002 * |
| Sugar (g) | 198 (179, 212) | 203 (184, 224) | 0.441 |
| Dietary fiber (g) | 19 (16, 23) | 17 (14, 21) | 0.168 |
| Fat (g) | 204 (184, 211) | 202 (196, 209) | 0.848 |
| SatFat (g) | 195 (192, 203) | 198 (195, 204) | 0.147 |
| UnSatFat (g) | 376 (368, 390) | 373 (362, 388) | 0.386 |
| Cholesterol (mg) | 179 (107, 306) | 163 (109, 267) | 0.848 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peng, Y.; Tan, Q.; Afhami, S.; Deehan, E.C.; Liang, S.; Gantz, M.; Triador, L.; Madsen, K.L.; Walter, J.; Tun, H.M.; et al. The Gut Microbiota Profile in Children with Prader–Willi Syndrome. Genes 2020, 11, 904. https://doi.org/10.3390/genes11080904
Peng Y, Tan Q, Afhami S, Deehan EC, Liang S, Gantz M, Triador L, Madsen KL, Walter J, Tun HM, et al. The Gut Microbiota Profile in Children with Prader–Willi Syndrome. Genes. 2020; 11(8):904. https://doi.org/10.3390/genes11080904
Chicago/Turabian StylePeng, Ye, Qiming Tan, Shima Afhami, Edward C. Deehan, Suisha Liang, Marie Gantz, Lucila Triador, Karen L. Madsen, Jens Walter, Hein M. Tun, and et al. 2020. "The Gut Microbiota Profile in Children with Prader–Willi Syndrome" Genes 11, no. 8: 904. https://doi.org/10.3390/genes11080904
APA StylePeng, Y., Tan, Q., Afhami, S., Deehan, E. C., Liang, S., Gantz, M., Triador, L., Madsen, K. L., Walter, J., Tun, H. M., & Haqq, A. M. (2020). The Gut Microbiota Profile in Children with Prader–Willi Syndrome. Genes, 11(8), 904. https://doi.org/10.3390/genes11080904