CRISPR/Cas9 in Cancer Immunotherapy: Animal Models and Human Clinical Trials

 ,
, 
 , , , , , , ,
, , , , , , ,  , ,
, ,  ,
, 
Abstract
:1. Introduction
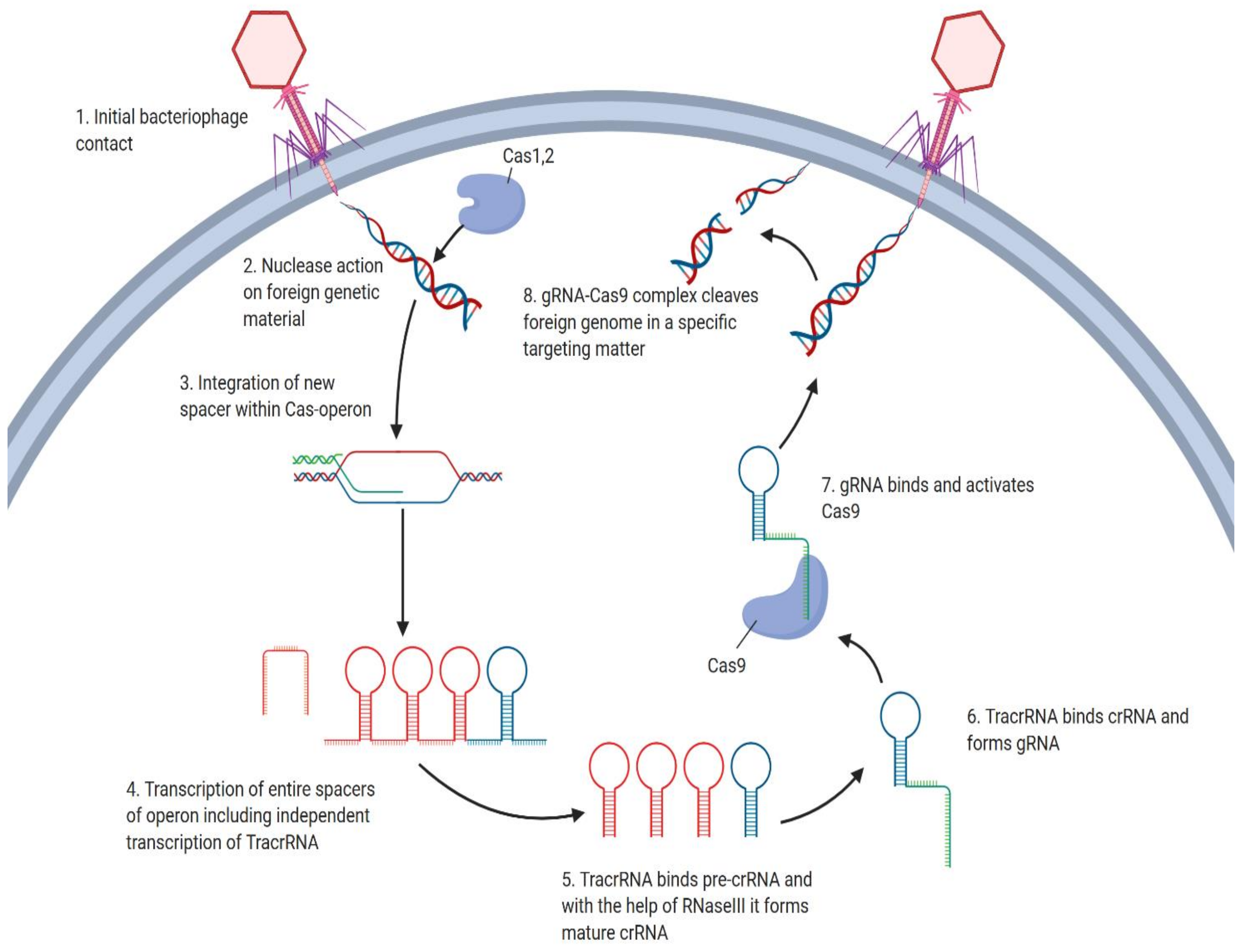
2. Overview of the CRISPR/Cas9 Mechanism
3. Applications and Advances of the CRISPR/Cas9 Technique in Animal Cancer Model and Human Clinical Trials
4. Precision Medicine and Immunotherapy in the Era of CRISPR/Cas9
5. Chimeric Antigen Receptors (CARs) for Cancer Immunotherapy
6. Limitations of the CRISPR/Cas9 System
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cooper, G.M.; Hausman, R.E. The Development and Causes of Cancer. Cell A Mol. Approach 2007, 743. [Google Scholar]
- Prager, G.W.; Braga, S.; Bystricky, B.; Qvortrup, C.; Criscitiello, C.; Esin, E.; Sonke, G.S.; Martínez, G.A.; Frenel, J.S.; Karamouzis, M.; et al. Global cancer control: Responding to the growing burden, rising costs and inequalities in access. ESMO Open 2018, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, K.; Takagi, Y.; Aoki, S.; Futamura, M.; Saji, S. Significant detection of circulating cancer cells in the blood by reverse transcriptase-polymerase chain reaction during colorectal cancer resection. Ann. Surg. 2000, 232, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Van Der Bij, G.J.; Oosterling, S.J.; Beelen, R.H.J.; Meijer, S.; Coffey, J.C.; Van Egmond, M. The perioperative period is an underutilized window of therapeutic opportunity in patients with colorectal cancer. Ann. Surg. 2009, 249, 727–734. [Google Scholar] [CrossRef] [Green Version]
- Oh, B.Y.; Kim, K.H.; Chung, S.S.; Hong, K.S.; Lee, R.A. Role of β1-integrin in colorectal cancer: Case-control study. Ann. Coloproctol. 2014, 30, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The different mechanisms of cancer drug resistance: A brief review. Adv. Pharm. Bull. 2017, 7, 339–348. [Google Scholar] [CrossRef]
- Martinez-Lage, M.; Puig-Serra, P.; Menendez, P.; Torres-Ruiz, R.; Rodriguez-Perales, S. CRISPR/Cas9 for cancer therapy: Hopes and challenges. Biomedicines 2018, 6. [Google Scholar] [CrossRef] [Green Version]
- Rath, D.; Amlinger, L.; Rath, A.; Lundgren, M. The CRISPR-Cas immune system: Biology, mechanisms and applications. Biochimie 2015, 117, 119–128. [Google Scholar] [CrossRef]
- Chylinski, K.; Le Rhun, A.; Charpentier, E. The tracrRNA and Cas9 families of type II CRISPR-Cas immunity systems. RNA Biol. 2013, 10, 726–737. [Google Scholar] [CrossRef] [Green Version]
- Modell, J.W.; Jiang, W.; Marraffini, L.A. CRISPR-Cas systems exploit viral DNA injection to establish and maintain adaptive immunity. Nature 2017, 544, 101–104. [Google Scholar] [CrossRef]
- Burmistrz, M.; Krakowski, K.; Krawczyk-Balska, A. RNA-targeting CRISPR–cas systems and their applications. Int. J. Mol. Sci. 2020, 21, 1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyda, F.; Hickman, A.B. Mechanism of spacer integration links the CRISPR/Cas system to transposition as a form of mobile DNA. Mob. DNA 2015, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lone, B.A.; Karna, S.K.L.; Ahmad, F.; Shahi, N.; Pokharel, Y.R. CRISPR/Cas9 System: A Bacterial Tailor for Genomic Engineering. Genet. Res. Int. 2018, 2018. [Google Scholar] [CrossRef] [PubMed]
- Vakulskas, C.A.; Behlke, M.A. Evaluation and reduction of crispr off-target cleavage events. Nucleic Acid Ther. 2019, 29, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, R.A.; Martin, C.; Nemudryi, A.A.; Wiedenheft, B. CRISPR RNA-guided autonomous delivery of Cas9. Nat. Struct. Mol. Biol. 2019, 26, 14–24. [Google Scholar] [CrossRef]
- Murovec, J.; Pirc, Ž.; Yang, B. New variants of CRISPR RNA-guided genome editing enzymes. Plant Biotechnol. J. 2017, 15, 917–926. [Google Scholar] [CrossRef] [Green Version]
- Jiang, F.; Doudna, J.A. CRISPR–Cas9 Structures and Mechanisms. Annu. Rev. Biophys. 2017, 46, 505–529. [Google Scholar] [CrossRef] [Green Version]
- Ye, L.; Wang, C.; Hong, L.; Sun, N.; Chen, D.; Chen, S.; Han, F. Programmable DNA repair with CRISPRa/i enhanced homology-directed repair efficiency with a single Cas9. Cell Discov. 2018, 4, 46. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Yang, Y.; Hong, W.; Huang, M.; Wu, M.; Zhao, X. Applications of genome editing technology in the targeted therapy of human diseases: Mechanisms, advances and prospects. Signal Transduct. Target. Ther. 2020, 5, 1–23. [Google Scholar] [CrossRef]
- Jo, N.; Sogabe, Y.; Yamada, Y.; Ukai, T.; Kagawa, H.; Mitsunaga, K.; Woltjen, K.; Yamada, Y. Platforms of in vivo genome editing with inducible Cas9 for advanced cancer modeling. Cancer Sci. 2019, 110, 926–938. [Google Scholar] [CrossRef] [Green Version]
- Jacinto, F.V.; Link, W.; Ferreira, B.I. CRISPR/Cas9-mediated genome editing: From basic research to translational medicine. J. Cell. Mol. Med 2020, 10.1111/jcmm.14916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayrhofer, M.; Mione, M. The toolbox for conditional zebrafish cancer models. In Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2016; Volume 916, pp. 21–59. [Google Scholar]
- Sayin, V.I.; Papagiannakopoulos, T. Application of CRISPR-mediated genome engineering in cancer research. Cancer Lett. 2017, 387, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Vijai, J.; Topka, S.; Villano, D.; Ravichandran, V.; Maxwell, K.N.; Maria, A.; Thomas, T.; Gaddam, P.; Lincoln, A.; Kazzaz, S.; et al. A Recurrent ERCC3 Truncating Mutation Confers Moderate Risk for Breast Cancer. Cancer Discov. 2016, 6, 1267–1275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lok, B.H.; Gardner, E.E.; Schneeberger, V.E.; Ni, A.; Desmeules, P.; Rekhtman, N.; De Stanchina, E.; Teicher, B.A.; Riaz, N.; Powell, S.N.; et al. PARP Inhibitor activity correlates with slfn11 expression and demonstrates synergy with temozolomide in small cell lung cancer. Clin. Cancer Res. 2017, 23, 523–535. [Google Scholar] [CrossRef] [Green Version]
- Santoni-Rugiu, E.; Melchior, L.C.; Urbanska, E.M.; Jakobsen, J.N.; De Stricker, K.; Grauslund, M.; Sørensen, J.B. Intrinsic resistance to EGFR-tyrosine kinase inhibitors in EGFR-mutant non-small cell lung cancer: Differences and similarities with acquired resistance. Cancers (Basel) 2019, 11. [Google Scholar] [CrossRef] [Green Version]
- McFadden, D.G.; Papagiannakopoulos, T.; Taylor-Weiner, A.; Stewart, C.; Carter, S.L.; Cibulskis, K.; Bhutkar, A.; McKenna, A.; Dooley, A.; Vernon, A.; et al. Genetic and clonal dissection of murine small cell lung carcinoma progression by genome sequencing. Cell 2014, 156, 1298–1311. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Chen, M.; Xu, E.S.; Luo, L.; Ma, Y.; Huang, W.; Floyd, W.; Klann, T.S.; Kim, S.Y.; Gersbach, C.A.; et al. Genome-wide CRISPR Screen to Identify Genes that Suppress Transformation in the Presence of Endogenous Kras G12D. Sci. Rep. 2019, 9, 17220. [Google Scholar] [CrossRef]
- Ouyang, Q.; Liu, Y.; Tan, J.; Li, J.; Yang, D.; Zeng, F.; Huang, W.; Kong, Y.; Liu, Z.; Zhou, H.; et al. Loss of ZNF587B and SULF1 contributed to cisplatin resistance in ovarian cancer cell lines based on Genome-scale CRISPR/Cas9 screening. Am. J. Cancer Res. 2019, 9, 988–998. [Google Scholar]
- BeltCappellino, A.; Majerciak, V.; Lobanov, A.; Lack, J.; Cam, M.; Zheng, Z.-M. CRISPR/Cas9-Mediated Knockout and In Situ Inversion of the ORF57 Gene from All Copies of the Kaposi’s Sarcoma-Associated Herpesvirus Genome in BCBL-1 Cells. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Yang, Y.; Chai, L.; Bu, H.; Yang, Y.; Huang, H.; Ran, J.; Zhu, Y.; Li, L.; Chen, F.; et al. FRK plays an oncogenic role in non-small cell lung cancer by enhancing the stemness phenotype via induction of metabolic reprogramming. Int. J. Cancer 2020, 146, 208–222. [Google Scholar] [CrossRef]
- Eun, K.; Park, M.G.; Jeong, Y.W.; Jeong, Y.I.; Hyun, S.H.; Hwang, W.S.; Kim, S.H.; Kim, H. Establishment of TP53-knockout canine cells using optimized CRIPSR/Cas9 vector system for canine cancer research 06 Biological Sciences 0601 Biochemistry and Cell Biology 11 Medical and Health Sciences 1112 Oncology and Carcinogenesis. BMC Biotechnol. 2019, 19, 1. [Google Scholar]
- Jo, N.; Sogabe, Y.; Yamada, Y.; Ukai, T.; Kagawa, H.; Mitsunaga, K.; Woltjen, K.; Yamada, Y. Platforms of in vivo genome editing with inducible Cas9 for advanced cancer modeling. Cancer Sci. 2019, 110, 926–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Can Changes in the Structure of Chromosomes Affect Health and Development? - Genetics Home Reference – NIH. Available online: https://ghr.nlm.nih.gov/primer/mutationsanddisorders/structuralchanges.
- Cheong, T.C.; Blasco, R.B.; Chiarle, R. The CRISPR/Cas9 system as a tool to engineer chromosomal translocation in vivo. In Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2018; Volume 1044, pp. 39–48. [Google Scholar]
- Wang, X.; Zhang, H.; Chen, X. Drug resistance and combating drug resistance in cancer. Cancer Drug Resist. 2019. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.-Y.; Ju, D.-T.; Chang, C.-F.; Reddy, P.M.; Velmurugan, B.K. A review on the effects of current chemotherapy drugs and natural agents in treating non–small cell lung cancer. BioMedicine 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Song, W.; Li, D.; Tao, L.; Luo, Q.; Chen, L. Solute carrier transporters: The metabolic gatekeepers of immune cells. Acta Pharm. Sin. B 2020, 10, 61–78. [Google Scholar] [CrossRef]
- Mohammad, I.S.; He, W.; Yin, L. Understanding of human ATP binding cassette superfamily and novel multidrug resistance modulators to overcome MDR. Biomed. Pharmacother. 2018, 100, 335–348. [Google Scholar] [CrossRef]
- Rodríguez-Rodríguez, D.R.; Ramírez-Solís, R.; Garza-Elizondo, M.A.; Garza-Rodríguez, M.D.L.; Barrera-Saldaña, H.A. Genome editing: A perspective on the application of CRISPR/Cas9 to study human diseases (Review). Int. J. Mol. Med. 2019, 43, 1559–1574. [Google Scholar]
- Chen, Y.; Zhang, Y. Application of the CRISPR/Cas9 System to Drug Resistance in Breast Cancer. Adv. Sci. 2018, 5. [Google Scholar] [CrossRef]
- Netz, U.; Carter, J.V.; Eichenberger, M.R.; Dryden, G.W.; Pan, J.; Rai, S.N.; Galandiuk, S. Genetic polymorphisms predict response to anti-tumor necrosis factor treatment in Crohn’s disease. World J. Gastroenterol. 2017, 23, 4958–4967. [Google Scholar] [CrossRef]
- Ko, B.; Paucar, D.; Halmos, B. EGFR T790M: Revealing the secrets of a gatekeeper. Lung Cancer Targets Ther. 2017, 8, 147–159. [Google Scholar] [CrossRef] [Green Version]
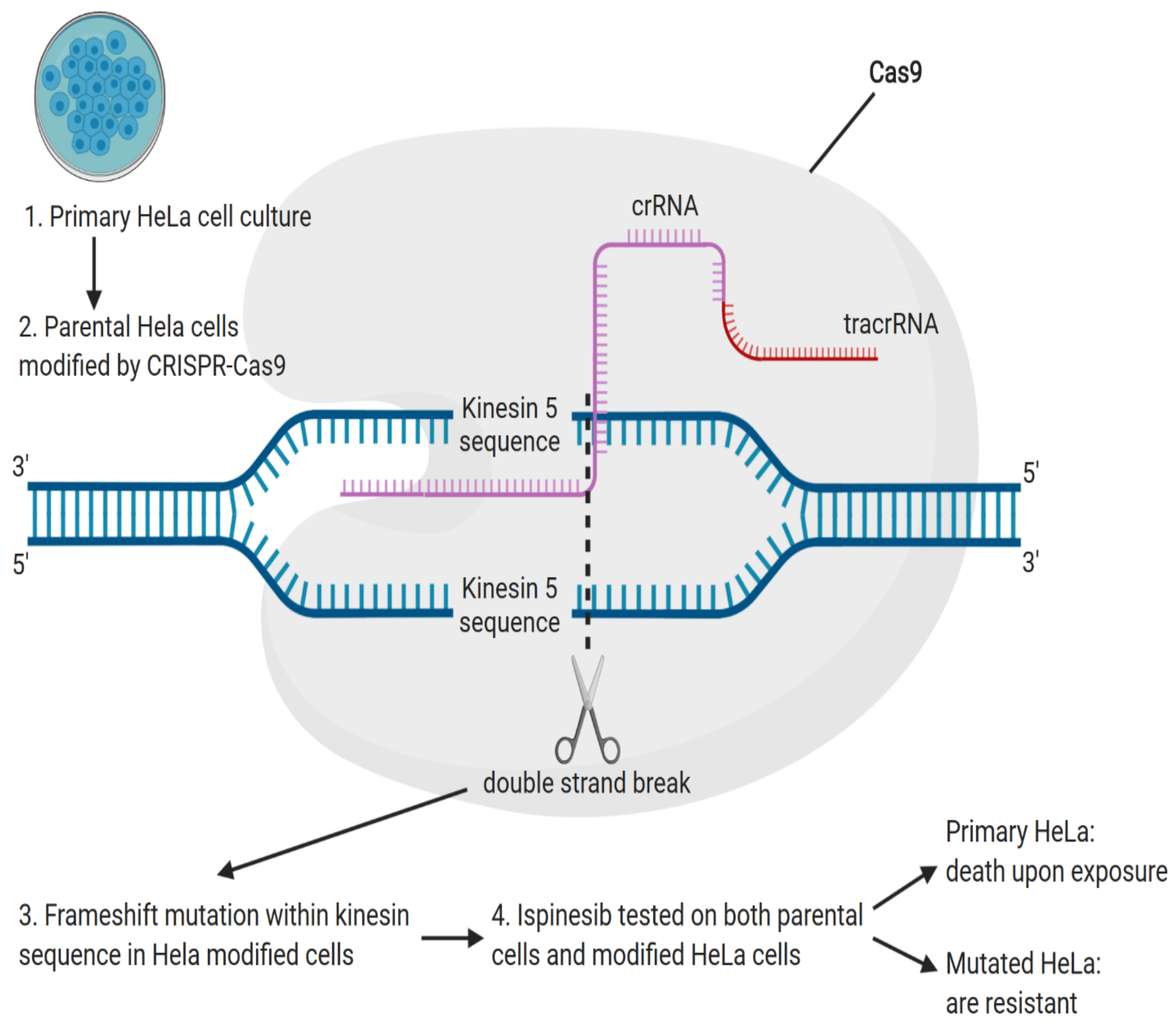
- Myers, S.M.; Collins, I. Recent findings and future directions for interpolar mitotic kinesin inhibitors in cancer therapy. Future Med. Chem. 2016, 8, 463–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasap, C.; Elemento, O.; Kapoor, T.M. DrugTargetSeqR: A genomics- and CRISPR-Cas9-based method to analyze drug targets. Nat. Chem. Biol. 2014, 10, 626–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sturgill, E.G.; Norris, S.R.; Guo, Y.; Ohi, R. Kinesin-5 inhibitor resistance is driven by kinesin-12. J. Cell Biol. 2016, 213, 213–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saeki, E.; Yasuhira, S.; Shibazaki, M.; Tada, H.; Doita, M.; Masuda, T.; Maesawa, C. Involvement of C-terminal truncation mutation of kinesin-5 in resistance to kinesin-5 inhibitor. PLoS ONE 2018, 13, e0209296. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Gu, T.; Patel, S.; Bode, A.M.; Lee, M.-H.; Dong, Z. CRISPR/Cas9 – An evolving biological tool kit for cancer biology and oncology. npj Precis. Oncol. 2019, 3. [Google Scholar] [CrossRef]
- Maeder, M.L.; Gersbach, C.A. Genome-editing technologies for gene and cell therapy. Mol. Ther. 2016, 24, 430–446. [Google Scholar] [CrossRef] [Green Version]
- Hong, A. CRISPR in personalized medicine: Industry perspectives in gene editing. Semin. Perinatol. 2018, 42, 501–507. [Google Scholar] [CrossRef]
- Alsibai, K.D.; Meseure, D. Significance of Tumor Microenvironment Scoring and Immune Biomarkers in Patient Stratification and Cancer Outcomes. In Histopathology - An Update; InTech: London, UK, 2018. [Google Scholar]
- Tran, E.; Robbins, P.F.; Rosenberg, S.A. Final common pathway’ of human cancer immunotherapy: Targeting random somatic mutations. Nat. Immunol. 2017, 18, 255–262. [Google Scholar] [CrossRef]
- Chang, Z.N.L.; Chen, Y.Y. CARs: Synthetic Immunoreceptors for Cancer Therapy and Beyond. Trends Mol. Med. 2017, 23, 430–450. [Google Scholar] [CrossRef] [Green Version]
- Pech, M.F.; Fong, L.E.; Villalta, J.E.; Chan, L.J.G.; Kharbanda, S.; O’brien, J.J.; McAllister, F.E.; Firestone, A.J.; Jan, C.H.; Settleman, J. Systematic identification of cancer cell vulnerabilities to natural killer cell-mediated immune surveillance. Elife 2019, 8. [Google Scholar] [CrossRef]
- Zhuang, X.; Veltri, D.P.; Long, E.O. Genome-Wide CRISPR Screen Reveals Cancer Cell Resistance to NK Cells Induced by NK-Derived IFN-γ. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castro, F.; Cardoso, A.P.; Gonçalves, R.M.; Serre, K.; Oliveira, M.J. Interferon-gamma at the crossroads of tumor immune surveillance or evasion. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonifant, C.L.; Jackson, H.J.; Brentjens, R.J.; Curran, K.J. Toxicity and management in CAR T-cell therapy. Mol. Ther. Oncolytics 2016, 3, 16011. [Google Scholar] [CrossRef] [PubMed]
- Legut, M.; Dolton, G.; Mian, A.A.; Ottmann, O.G.; Sewell, A.K. CRISPR-mediated TCR replacement generates superior anticancer transgenic t cells. Blood 2018, 131, 311–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stenger, D.; Stief, T.A.; Käuferle, T.; Willier, S.; Rataj, F.; Schober, K.; Vick, B.; Lotfi, R.; Wagner, B.; Grunewald, T.; et al. Endogenous TCR promotes in vivo persistence of CD19-CAR-T cells compared to a CRISPR/Cas9-mediated TCR knockout CAR. Blood 2020. [Google Scholar] [CrossRef] [PubMed]
- Rotolo, R.; Leuci, V.; Donini, C.; Cykowska, A.; Gammaitoni, L.; Medico, G.; Valabrega, G.; Aglietta, M.; Sangiolo, D. Car-based strategies beyond t lymphocytes: Integrative opportunities for cancer adoptive immunotherapy. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef] [Green Version]
- McGowan, E.; Lin, Q.; Ma, G.; Yin, H.; Chen, S.; Lin, Y. PD-1 disrupted CAR-T cells in the treatment of solid tumors: Promises and challenges. Biomed. Pharmacother. 2020, 121, 109625. [Google Scholar] [CrossRef]
- Su, S.; Hu, B.; Shao, J.; Shen, B.; Du, J.; Du, Y.; Zhou, J.; Yu, L.; Zhang, L.; Chen, F.; et al. CRISPR-Cas9 mediated efficient PD-1 disruption on human primary T cells from cancer patients. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [Green Version]
- Hu, W.; Zi, Z.; Jin, Y.; Li, G.; Shao, K.; Cai, Q.; Ma, X.; Wei, F. CRISPR/Cas9-mediated PD-1 disruption enhances human mesothelin-targeted CAR T cell effector functions. Cancer Immunol. Immunother. 2019, 68, 365–377. [Google Scholar] [CrossRef]
- Montaño, A.; Forero-Castro, M.; Hernández-Rivas, J.M.; García-Tuñón, I.; Benito, R. Targeted genome editing in acute lymphoblastic leukemia: A review. BMC Biotechnol. 2018, 18. [Google Scholar]
- Eyquem, J.; Mansilla-Soto, J.; Giavridis, T.; Van Der Stegen, S.J.C.; Hamieh, M.; Cunanan, K.M.; Odak, A.; Gönen, M.; Sadelain, M. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 2017, 543, 113–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoiber, S.; Cadilha, B.L.; Benmebarek, M.-R.; Lesch, S.; Endres, S.; Kobold, S. Limitations in the Design of Chimeric Antigen Receptors for Cancer Therapy. Cells 2019, 8, 472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viaud, S.; Ma, J.S.Y.; Hardy, I.R.; Hampton, E.N.; Benish, B.; Sherwood, L.; Nunez, V.; Ackerman, C.J.; Khialeeva, E.; Weglarz, M.; et al. Switchable control over in vivo CAR T expansion, B cell depletion, and induction of memory. Proc. Natl. Acad. Sci. USA 2018, 115, E10898–E10906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, M.; Moon, E.K. CAR T cells for solid tumors: New strategies for finding, infiltrating, and surviving in the tumor microenvironment. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomuleasa, C.; Fuji, S.; Berce, C.; Onaciu, A.; Chira, S.; Petrushev, B.; Micu, W.T.; Moisoiu, V.; Osan, C.; Constantinescu, C.; et al. Chimeric antigen receptor T-cells for the treatment of B-cell acute lymphoblastic leukemia. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Richards, R.M.; Sotillo, E.; Majzner, R.G. CAR T cell therapy for neuroblastoma. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Wang, Y.; Han, W.-D. Chimeric antigen receptor modified T-cells for cancer treatment. Chronic Dis. Transl. Med. 2018, 4, 225–243. [Google Scholar] [CrossRef]
- Zhao, L.; Cao, Y.J. Engineered T Cell Therapy for Cancer in the Clinic. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Springuel, L.; Lonez, C.; Alexandre, B.; Van Cutsem, E.; Machiels, J.P.H.; Van Den Eynde, M.; Prenen, H.; Hendlisz, A.; Shaza, L.; Carrasco, J.; et al. Chimeric Antigen Receptor-T Cells for Targeting Solid Tumors: Current Challenges and Existing Strategies. BioDrugs 2019, 33, 515–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravanpay, A.C.; Gust, J.; Johnson, A.J.; Rolczynski, L.S.; Cecchini, M.; Chang, C.A.; Hoglund, V.J.; Mukherjee, R.; Vitanza, N.A.; Orentas, R.J.; et al. EGFR806-CAR T cells selectively target a tumor-restricted EGFR epitope in glioblastoma. Oncotarget 2019, 10, 7080–7095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, S.R.; Maus, M.V. Gene editing for immune cell therapies. Nat. Biotechnol. 2019, 37, 1425–1434. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.D.; Yu, X.; Castano, A.P.; Darr, H.; Henderson, D.B.; Bouffard, A.A.; Larson, R.C.; Scarfò, I.; Bailey, S.R.; Gerhard, G.M.; et al. CRISPR-Cas9 disruption of PD-1 enhances activity of universal EGFRvIII CAR T cells in a preclinical model of human glioblastoma. J. Immunother. Cancer 2019, 7, 304. [Google Scholar] [CrossRef] [PubMed]
- Zgodzinski, W.; Grywalska, E.; Zinkiewicz, K.; Surdacka, A.; Majewski, M.; Zakoscielny, A.; Bury, P.; Rolinski, J.; Wallner, G.T. Peripheral blood T lymphocytes are downregulated by the PD-1/PD-L1 axis in advanced gastric cancer. Arch. Med. Sci. 2019, 15, 774–783. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Shimada, S.; Akiyama, Y.; Ishikawa, Y.; Ogura, T.; Ogawa, K.; Ono, H.; Mitsunori, Y.; Ban, D.; Kudo, A.; et al. Loss of KDM6A characterizes a poor prognostic subtype of human pancreatic cancer and potentiates HDAC inhibitor lethality. Int. J. Cancer 2019, 145, 192–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Treuren, T.; Vishwanatha, J.K. CRISPR deletion of MIEN1 in breast cancer cells. PLoS ONE 2018, 13, e0204976. [Google Scholar] [CrossRef] [Green Version]
- Ng, S.R. CRISPR-mediated interrogation of small cell lung cancer. Ph.D. Thesis, Department of Biology, Massachusetts Institute of Technology, Cambridge, MA, USA, 2018. [Google Scholar]
- Ye, R.; Pi, M.; Cox, J.V.; Nishimoto, S.K.; Quarles, L.D. CRISPR/Cas9 targeting of GPRC6A suppresses prostate cancer tumorigenesis in a human xenograft model. J. Exp. Clin. Cancer Res. 2017, 36, 90. [Google Scholar] [CrossRef]
- Engel, B.J.; Bowser, J.L.; Broaddus, R.R.; Carson, D.D. MUC1 stimulates EGFR expression and function in endometrial cancer. Oncotarget 2016, 7, 32796–32809. [Google Scholar] [CrossRef]
- Raza, U.; Saatci, Ö.; Uhlmann, S.; Ansari, S.A.; Eyüpoglu, E.; Yurdusev, E.; Mutlu, M.; Ersan, P.G.; Altundag, M.K.; Zhang, J.D.; et al. The miR-644a/CTBP1/p53 axis suppresses drug resistance by simultaneous inhibition of cell survival and epithelialmesenchymal transition in breast cancer. Oncotarget 2016, 7, 49859–49877. [Google Scholar] [CrossRef] [Green Version]
- Kawamura, N.; Nimura, K.; Nagano, H.; Yamaguchi, S.; Nonomura, N.; Kaneda, Y. CRISPR/Cas9-mediated gene knockout of NANOG and NANOGP8 decreases the malignant potential of prostate cancer cells. Oncotarget 2015, 6, 22361–22374. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Zhang, C.; Wang, B.; Li, B.; Wang, Q.; Liu, D.; Wang, H.; Zhou, Y.; Shi, L.; Lan, F.; et al. Optimized CRISPR guide RNA design for two high-fidelity Cas9 variants by deep learning. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef]
- Lino, C.A.; Harper, J.C.; Carney, J.P.; Timlin, J.A. Delivering crispr: A review of the challenges and approaches. Drug Deliv. 2018, 25, 1234–1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soussi, T.; Wiman, K.G. TP53: An oncogene in disguise. Cell Death Differ. 2015, 22, 1239–1249. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.B.; Kim, D.Y.; Ko, J.H.; Kim, Y.S. Recent advances in the CRISPR genome editing tool set. Exp. Mol. Med. 2019, 51, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, H.A.; Pang, J.K.S.; Soh, B.-S. Mitigating off-target effects in CRISPR/Cas9-mediated in vivo gene editing. J. Mol. Med. 2020, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bin Moon, S.; Lee, J.M.; Kang, J.G.; Lee, N.E.; Ha, D.I.; Kim, D.Y.; Kim, S.H.; Yoo, K.; Kim, D.; Ko, J.H.; et al. Highly efficient genome editing by CRISPR-Cpf1 using CRISPR RNA with a uridinylate-rich 3′-overhang. Nat. Commun. 2018, 9, 1–11. [Google Scholar] [CrossRef]
- Thurtle-Schmidt, D.M.; Lo, T.W. Molecular biology at the cutting edge: A review on CRISPR/CAS9 gene editing for undergraduates. Biochem. Mol. Biol. Educ. 2018, 46, 195–205. [Google Scholar] [CrossRef] [Green Version]
- Matson, A.W.; Hosny, N.; Swanson, Z.A.; Hering, B.J.; Burlak, C. Optimizing sgRNA length to improve target specificity and efficiency for the GGTA1 gene using the CRISPR/Cas9 gene editing system. PLoS ONE 2019, 14, e0226107. [Google Scholar] [CrossRef]
- Araki, M.; Ishii, T. Providing appropriate risk information on genome editing for patients. Trends Biotechnol. 2016, 34, 86–90. [Google Scholar] [CrossRef]
- Joung, J.K. Standards needed for gene-editing errors. Nature 2015, 523, 158. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Ren, Q.; Yang, L.; Bao, Y.; Zhong, Z.; He, Y.; Liu, S.; Qi, C.; Liu, B.; Wang, Y.; et al. Single transcript unit CRISPR 2.0 systems for robust Cas9 and Cas12a mediated plant genome editing. Plant Biotechnol. J. 2019, 17, 1431–1445. [Google Scholar] [CrossRef] [Green Version]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of T to G C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Asami, M.; Perry, A.C.F. Asymmetric parental genome engineering by Cas9 during mouse meiotic exit. Sci. Rep. 2014, 4, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yen, S.T.; Zhang, M.; Deng, J.M.; Usman, S.J.; Smith, C.N.; Parker-Thornburg, J.; Swinton, P.G.; Martin, J.F.; Behringer, R.R. Somatic mosaicism and allele complexity induced by CRISPR/Cas9 RNA injections in mouse zygotes. Dev. Biol. 2014, 393, 3–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, D.; Xu, Z.; Zhang, Z.; Chen, X.; Zeng, X.; Zhang, Y.; Deng, T.; Ren, M.; Sun, Z.; Jiang, R.; et al. Engineer chimeric Cas9 to expand PAM recognition based on evolutionary information. Nat. Commun. 2019, 10, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raitskin, O.; Schudoma, C.; West, A.; Patron, N.J. Comparison of efficiency and specificity of CRISPR-associated (Cas) nucleases in plants: An expanded toolkit for precision genome engineering. PLoS ONE 2019, 14, e0211598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Safari, F.; Zare, K.; Negahdaripour, M.; Barekati-Mowahed, M.; Ghasemi, Y. CRISPR Cpf1 proteins: Structure, function and implications for genome editing. Cell Biosci. 2019, 9. [Google Scholar] [CrossRef] [Green Version]
- Wan, T.; Chen, Y.; Pan, Q.; Xu, X.; Kang, Y.; Gao, X.; Huang, F.; Wu, C.; Ping, Y. Genome editing of mutant KRAS through supramolecular polymer-mediated delivery of Cas9 ribonucleoprotein for colorectal cancer therapy. J. Control. Release 2020, 322, 236–247. [Google Scholar] [CrossRef]
- Kimmelman, J. The ethics of human gene transfer. Nat. Rev. Genet. 2008, 9, 239–244. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | Country | Phase | Cell Type | Target | Intervention | ID |
|---|---|---|---|---|---|---|
| Gastrointestinal Epithelial Cancer | USA | I/II | Tumor Infiltrating Lymphocytes (TIL) | CISH (Cytokine-induced SH2 protein) | Drug: Cyclophosphamide Drug: Fludarabine Biological: Tumor-Infiltrating Lymphocytes (TIL) Drug: Aldesleukin | NCT04426669 |
| Gastrointestinal Neoplasms | ||||||
| Gastrointestinal Cancer | ||||||
| Colorectal Cancer | ||||||
| Pancreatic Cancer | ||||||
| Gall Bladder Cancer | ||||||
| Colon Cancer | ||||||
| Esophageal Cancer | ||||||
| Stomach Cancer | ||||||
| B Cell Leukemia | China | I/II | B-cells | CD19, CD20, or CD22 Knockout | Biological: Universal Dual Specificity CD19 and CD20 or CD22 CAR-T Cells | NCT03398967 |
| B Cell Lymphoma | ||||||
| B Cell Leukemia | China | I/II | B-cell | CD19 | Biological: UCART019 | NCT03166878 |
| B Cell Lymphoma | ||||||
| Refractory B-cell malignancies | USA | I/II | bVCB-cel B-cell | Creation of a CD19-directed T cell | CD19-directed T-cell immunotherapy | NCT04035434 |
| Cancer Type | CRISPR/Cas9 Modifications | Therapeutic Contributions | Reference |
|---|---|---|---|
| Pancreatic cancer | KDM6A knockout | Increase in the aggressiveness of pancreatic ductal adenocarcinoma | [78] |
| Breast cancer | MIEN-1 knockout | Increase of progression and metastatic potential | [79] |
| SCLC | P107 knockout | Inhibition of tumor suppressor activity | [80] |
| Prostate cancer | GPRC6A knockout | Inhibition of cell proliferation | [81] |
| Endometrial cancer | MUC1 knockout | Inhibition of EGFR expression | [82] |
| Breast cancer | miR-644a knockout | Inhibition of growth, metastasis, and treatment resistance | [83] |
| Prostate cancer | NANOG and NANOGP8 knockout | The decrease in malignant potential | [84] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khalaf, K.; Janowicz, K.; Dyszkiewicz-Konwińska, M.; Hutchings, G.; Dompe, C.; Moncrieff, L.; Jankowski, M.; Machnik, M.; Oleksiewicz, U.; Kocherova, I.; et al. CRISPR/Cas9 in Cancer Immunotherapy: Animal Models and Human Clinical Trials. Genes 2020, 11, 921. https://doi.org/10.3390/genes11080921
Khalaf K, Janowicz K, Dyszkiewicz-Konwińska M, Hutchings G, Dompe C, Moncrieff L, Jankowski M, Machnik M, Oleksiewicz U, Kocherova I, et al. CRISPR/Cas9 in Cancer Immunotherapy: Animal Models and Human Clinical Trials. Genes. 2020; 11(8):921. https://doi.org/10.3390/genes11080921
Chicago/Turabian StyleKhalaf, Khalil, Krzysztof Janowicz, Marta Dyszkiewicz-Konwińska, Greg Hutchings, Claudia Dompe, Lisa Moncrieff, Maurycy Jankowski, Marta Machnik, Urszula Oleksiewicz, Ievgeniia Kocherova, and et al. 2020. "CRISPR/Cas9 in Cancer Immunotherapy: Animal Models and Human Clinical Trials" Genes 11, no. 8: 921. https://doi.org/10.3390/genes11080921
APA StyleKhalaf, K., Janowicz, K., Dyszkiewicz-Konwińska, M., Hutchings, G., Dompe, C., Moncrieff, L., Jankowski, M., Machnik, M., Oleksiewicz, U., Kocherova, I., Petitte, J., Mozdziak, P., Shibli, J. A., Iżycki, D., Józkowiak, M., Piotrowska-Kempisty, H., Skowroński, M. T., Antosik, P., & Kempisty, B. (2020). CRISPR/Cas9 in Cancer Immunotherapy: Animal Models and Human Clinical Trials. Genes, 11(8), 921. https://doi.org/10.3390/genes11080921