An Overview of Duplicated Gene Detection Methods: Why the Duplication Mechanism Has to Be Accounted for in Their Choice

 ,
,
Abstract
:1. Introduction
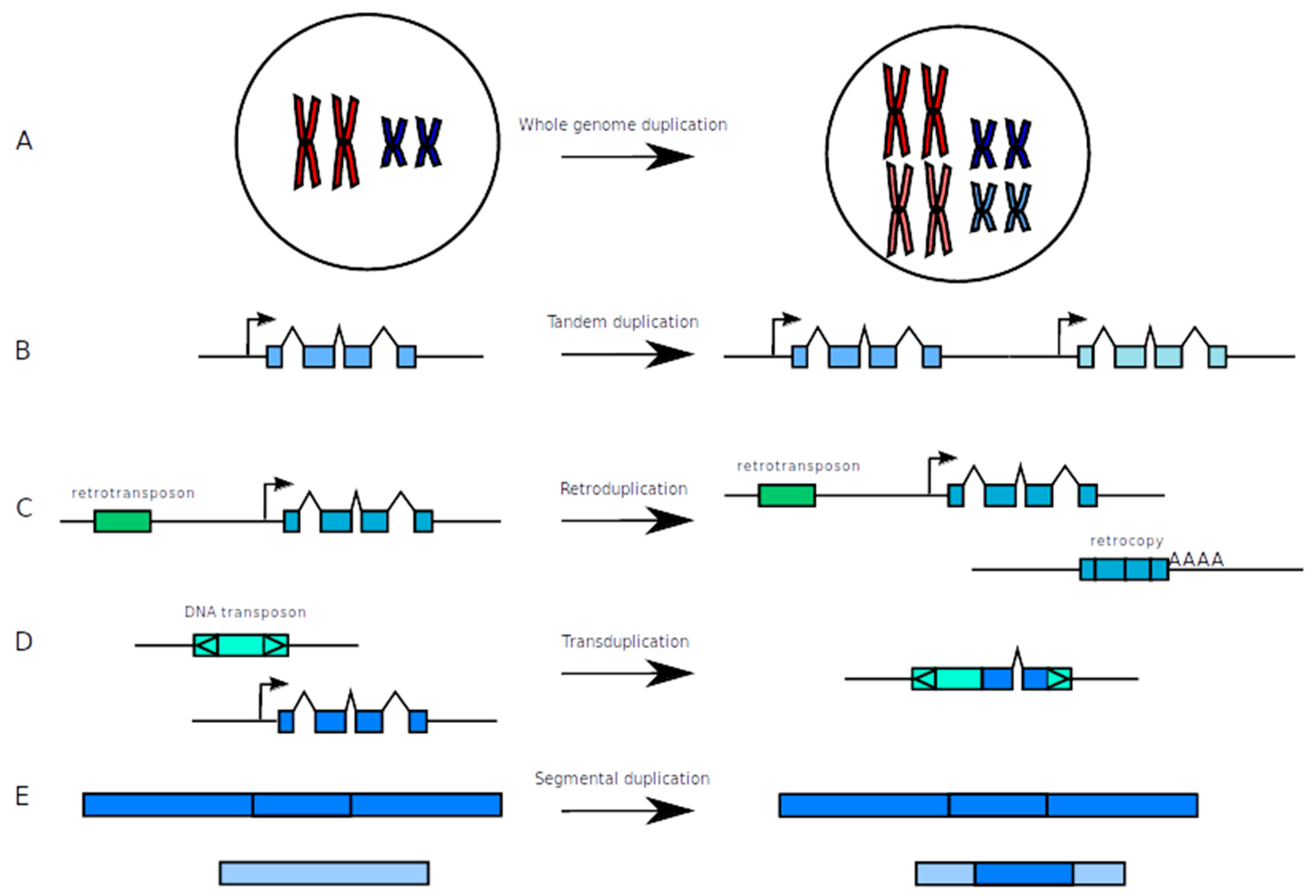
2. Evolutionary Processes Leading to the Formation and the Fate of Duplicated Genes
2.1. How to Make New from Old: Duplication Mechanisms
2.1.1. Whole Genome Duplication (WGD)
2.1.2. Tandem Duplications
2.1.3. Duplications Via the Action of Transposable Elements
2.1.4. Segmental Duplications
2.1.5. Differences among Duplication Types
2.2. Evolutionary Fates of Duplicated Genes
2.2.1. Pseudogenization and Neo-Functionalization
2.2.2. Sub-Functionalization and Functional Redundancy
2.2.3. The Fates of Duplicated Genes Depend on Different Factors
3. Bioinformatic Approaches to Identify Duplications in Genomes
3.1. Paralog Detection
3.1.1. Homology Assessment
3.1.2. Multispecies Graph-Based Methods
3.1.3. Multispecies Tree-Based Methods
3.2. Detection of Syntenic Blocks (WGD-Segmental Duplications)
3.2.1. Approaches Based on the Construction of Homologous Gene Matrices
i-ADHoRe (Iterative Automatic Detection of Homologous Regions) 3.0
- prob_cutoff, is used to store the maximum probability for a cluster to be generated by chance. The default value is 0.001.
- gap_size, indicating the maximum distance between genes in a cluster. The default value is 15.
- cluster_gap, indicating the maximum distance between individual base clusters in a cluster. The default value is 20.
- q_value, storing the minimum r2-value which measures the quality of the linear regression prediction.
- anchor_points, the minimum number of anchor points which is comprised between 3 and 6.
OrthoCluster
- l max defining the upper bound on the number of genes in each cluster.
- l min defining the lower bound on the number of genes in each cluster.
- op maximal percentage of out-map genes allowed.
- ip defining the maximal percentage of mismatched in-map genes allowed.
- op and ip can control the number of genes involved in transpositions in synteny block.
- i maximal number of mismatched in-map genes allowed.
- o maximal number of out-map genes allowed.
- r to find order-preserving clusters.
- s to find strandedness-preserving clusters.
3.2.2. Algorithms Using Dynamic Programming Optimizations
MCScanX and MultiSyn
- match_score, defines a threshold used to validate a synteny block. Default value is 50.
- gap_penalty, defines the penalty added when opening a gap. Default value is 21.
- match_size, defines the number of genes required to consider it as a collinear block. Default value is 5.
- e_value, defines the statistical significance of the synteny block alignment. Default value is 1e-10.
- max_gaps, maximum number of gaps allowed. Default value is 25.
- overlap_window stores the maximum number of genes to collapse BLAST matches. Default value is 5.
SynChro
CYNTENATOR
SyMap
3.2.3. Approaches Based on Graphs
DRIMM-Synteny
3.3. Detection of Tandemly Arrayed Genes (TAGs)
3.3.1. Detection at Protein Level
3.3.2. Detection at DNA Level
3.4. Databases Storing Syntenic Block or Homology Information
3.4.1. Syntenic Information
3.4.2. Homology Relationships Databases
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ohno, S. Evolution by Gene Duplication; Springer: Berlin/Heidelberg, Germany, 1970; ISBN 978-3-642-86661-6. [Google Scholar]
- Kondrashov, F.A. Gene duplication as a mechanism of genomic adaptation to a changing environment. Proc. R. Soc. B Biol. Sci. 2012, 279, 5048–5057. [Google Scholar] [CrossRef] [Green Version]
- Van de Peer, Y.; Maere, S.; Meyer, A. The evolutionary significance of ancient genome duplications. Nat. Rev. Genet. 2009, 10, 725–732. [Google Scholar] [CrossRef] [Green Version]
- Vallejo-Marín, M.; Buggs, R.J.A.; Cooley, A.M.; Puzey, J.R. Speciation by genome duplication: Repeated origins and genomic composition of the recently formed allopolyploid species Mimulus peregrinus. Evolution 2015, 69, 1487–1500. [Google Scholar] [CrossRef]
- Ting, C.T.; Tsaur, S.C.; Sun, S.; Browne, W.E.; Chen, Y.C.; Patel, N.H.; Wu, C.I. Gene duplication and speciation in Drosophila: Evidence from the Odysseus locus. Proc. Natl. Acad. Sci. USA 2004, 101, 12232–12235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.; Gu, W.; Hurles, M.E.; Lupski, J.R. Copy number variation in human health, disease, and evolution. Annu. Rev. Genomics Hum. Genet. 2009, 10, 451–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dickerson, J.E.; Robertson, D.L. On the origins of Mendelian disease genes in man: The impact of gene duplication. Mol. Biol. Evol. 2012, 29, 61–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tollis, M.; Schneider-Utaka, A.K.; Maley, C.C. The Evolution of Human Cancer Gene Duplications across Mammals. Mol. Biol. Evol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Mendivil Ramos, O.; Ferrier, D.E.K. Mechanisms of Gene Duplication and Translocation and Progress towards Understanding Their Relative Contributions to Animal Genome Evolution. Int. J. Evol. Biol. 2012, 2012, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolfe, K. Robustness—it’s not where you think it is. Nat. Genet. 2000, 25, 3–4. [Google Scholar] [CrossRef]
- Sharman, A.C. Some new terms for duplicated genes. Semin. Cell Dev. Biol. 1999, 10, 561–563. [Google Scholar] [CrossRef]
- Sonnhammer, E.L.L.; Koonin, E.V. Orthology, paralogy and proposed classification for paralog subtypes. Trends Genet. 2002, 18, 619–620. [Google Scholar] [CrossRef]
- Koonin, E.V. Orthologs, Paralogs, and Evolutionary Genomics. Annu. Rev. Genet. 2005, 39, 309–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altenhoff, A.M.; Glover, N.M.; Dessimoz, C. Inferring Orthology and Paralogy. In Evolutionary Genomics; Anisimova, M., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2019; Volume 1910, pp. 149–175. ISBN 978-1-4939-9073-3. [Google Scholar]
- Van de Peer, Y.; Mizrachi, E.; Marchal, K. The evolutionary significance of polyploidy. Nat. Rev. Genet. 2017, 18, 411–424. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, J.; Schemske, D.W. Pathways, Mechanisms, and Rates of Polyploid Formation in Flowering Plants. Annu. Rev. Ecol. Syst. 1998, 29, 467–501. [Google Scholar] [CrossRef] [Green Version]
- Panchy, N.; Lehti-Shiu, M.; Shiu, S.-H. Evolution of Gene Duplication in Plants. Plant Physiol. 2016, 171, 2294–2316. [Google Scholar] [CrossRef] [Green Version]
- Jiao, Y.; Wickett, N.J.; Ayyampalayam, S.; Chanderbali, A.S.; Landherr, L.; Ralph, P.E.; Tomsho, L.P.; Hu, Y.; Liang, H.; Soltis, P.S.; et al. Ancestral polyploidy in seed plants and angiosperms. Nature 2011, 473, 97–100. [Google Scholar] [CrossRef]
- Feldman, M.; Levy, A.A. Genome Evolution Due to Allopolyploidization in Wheat. Genetics 2012, 192, 763–774. [Google Scholar] [CrossRef] [Green Version]
- Chalhoub, B.; Denoeud, F.; Liu, S.; Parkin, I.A.P.; Tang, H.; Wang, X.; Chiquet, J.; Belcram, H.; Tong, C.; Samans, B.; et al. Plant genetics. Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 2014, 345, 950–953. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Liu, D.; Wang, X.; Ji, C.; Cheng, F.; Liu, B.; Hu, Z.; Chen, S.; Pental, D.; Ju, Y.; et al. The genome sequence of allopolyploid Brassica juncea and analysis of differential homoeolog gene expression influencing selection. Nat. Genet. 2016, 48, 1225–1232. [Google Scholar] [CrossRef]
- Sun, F.; Fan, G.; Hu, Q.; Zhou, Y.; Guan, M.; Tong, C.; Li, J.; Du, D.; Qi, C.; Jiang, L.; et al. The high-quality genome of Brassica napus cultivar ‘ZS11′ reveals the introgression history in semi-winter morphotype. Plant J. 2017, 92, 452–468. [Google Scholar] [CrossRef] [Green Version]
- Lu, K.; Wei, L.; Li, X.; Wang, Y.; Wu, J.; Liu, M.; Zhang, C.; Chen, Z.; Xiao, Z.; Jian, H.; et al. Whole-genome resequencing reveals Brassica napus origin and genetic loci involved in its improvement. Nat. Commun. 2019, 10, 1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasahara, M. The 2R hypothesis: An update. Curr. Opin. Immunol. 2007, 19, 547–552. [Google Scholar] [CrossRef] [PubMed]
- Wendel, J.F.; Lisch, D.; Hu, G.; Mason, A.S. The long and short of doubling down: Polyploidy, epigenetics, and the temporal dynamics of genome fractionation. Curr. Opin. Genet. Dev. 2018, 49, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Freeling, M.; Scanlon, M.J.; Fowler, J.E. Fractionation and subfunctionalization following genome duplications: Mechanisms that drive gene content and their consequences. Curr. Opin. Genet. Dev. 2015, 35, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.E.; Johnson, K.; Hollister, A.; May, B. Meiotic models to explain classical linkage, pseudolinkage, and chromosome pairing in tetraploid derivative salmonid genomes. Isozymes 1983, 10, 239–260. [Google Scholar]
- Sacerdot, C.; Louis, A.; Bon, C.; Berthelot, C.; Roest Crollius, H. Chromosome evolution at the origin of the ancestral vertebrate genome. Genome Biol. 2018, 19, 166. [Google Scholar] [CrossRef]
- Pervaiz, N.; Shakeel, N.; Qasim, A.; Zehra, R.; Anwar, S.; Rana, N.; Xue, Y.; Zhang, Z.; Bao, Y.; Abbasi, A.A. Evolutionary history of the human multigene families reveals widespread gene duplications throughout the history of animals. BMC Evol. Biol. 2019, 19, 128. [Google Scholar] [CrossRef]
- Zhang, J. Evolution by gene duplication: An update. Trends Ecol. Evol. 2003, 18, 292–298. [Google Scholar] [CrossRef] [Green Version]
- Arguello, J.R.; Fan, C.; Wang, W.; Long, M. Origination of chimeric genes through DNA-level recombination. In Gene and Protein Evolution; Karger Publishers: Basel, Switzerland, 2007; Volume 3, pp. 131–146. [Google Scholar] [CrossRef] [Green Version]
- Reams, A.B.; Roth, J.R. Mechanisms of gene duplication and amplification. Cold Spring Harb. Perspect. Biol. 2015, 7, a016592. [Google Scholar] [CrossRef] [Green Version]
- Cook, D.E.; Lee, T.G.; Guo, X.; Melito, S.; Wang, K.; Bayless, A.M.; Wang, J.; Hughes, T.J.; Willis, D.K.; Clemente, T.E.; et al. Copy Number Variation of Multiple Genes at Rhg1 Mediates Nematode Resistance in Soybean. Science 2012, 338, 1206–1209. [Google Scholar] [CrossRef] [Green Version]
- Kono, T.J.Y.; Brohammer, A.B.; McGaugh, S.E.; Hirsch, C.N. Tandem Duplicate Genes in Maize Are Abundant and Date to Two Distinct Periods of Time. G3 Genes Genomes Genet. 2018, 8, 3049–3058. [Google Scholar] [CrossRef] [Green Version]
- Tan, B.C.; Guan, J.C.; Ding, S.; Wu, S.; Saunders, J.W.; Koch, K.E.; McCarty, D.R. Structure and Origin of the White Cap Locus and Its Role in Evolution of Grain Color in Maize. Genetics 2017, 206, 135–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.M.; Vanguri, S.; Boeke, J.D.; Gabriel, A.; Voytas, D.F. Transposable elements and genome organization: A comprehensive survey of retrotransposons revealed by the complete Saccharomyces cerevisiae genome sequence. Genome Res. 1998, 8, 464–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnable, P.S.; Ware, D.; Fulton, R.S.; Stein, J.C.; Wei, F.; Pasternak, S.; Liang, C.; Zhang, J.; Fulton, L.; Graves, T.A.; et al. The B73 maize genome: Complexity, diversity, and dynamics. Science 2009, 326, 1112–1115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brosius, J. Retroposons—Seeds of evolution. Science 1991, 251, 753. [Google Scholar] [CrossRef] [PubMed]
- Moran, J.V.; DeBerardinis, R.J.; Kazazian, H.H. Exon shuffling by L1 retrotransposition. Science 1999, 283, 1530–1534. [Google Scholar] [CrossRef]
- Elrouby, N.; Bureau, T.E. A novel hybrid open reading frame formed by multiple cellular gene transductions by a plant long terminal repeat retroelement. J. Biol. Chem. 2001, 276, 41963–41968. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Harrison, P.M.; Liu, Y.; Gerstein, M. Millions of Years of Evolution Preserved: A Comprehensive Catalog of the Processed Pseudogenes in the Human Genome. Genome Res. 2003, 13, 2541–2558. [Google Scholar] [CrossRef]
- Casola, C.; Betrán, E. The Genomic Impact of Gene Retrocopies: What Have We Learned from Comparative Genomics, Population Genomics, and Transcriptomic Analyses? Genome Biol. Evol. 2017, 9, 1351–1373. [Google Scholar] [CrossRef]
- Betrán, E.; Thornton, K.; Long, M. Retroposed new genes out of the X in Drosophila. Genome Res. 2002, 12, 1854–1859. [Google Scholar] [CrossRef] [Green Version]
- Bai, Y.; Casola, C.; Feschotte, C.; Betrán, E. Comparative genomics reveals a constant rate of origination and convergent acquisition of functional retrogenes in Drosophila. Genome Biol. 2007, 8, R11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toups, M.A.; Hahn, M.W. Retrogenes reveal the direction of sex-chromosome evolution in mosquitoes. Genetics 2010, 186, 763–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emerson, J.J.; Kaessmann, H.; Betrán, E.; Long, M. Extensive gene traffic on the mammalian X chromosome. Science 2004, 303, 537–540. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zheng, H.; Fan, C.; Li, J.; Shi, J.; Cai, Z.; Zhang, G.; Liu, D.; Zhang, J.; Vang, S.; et al. High Rate of Chimeric Gene Origination by Retroposition in Plant Genomes. Plant Cell 2006, 18, 1791–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Wang, X.; Tang, H.; Tan, X.; Ficklin, S.P.; Feltus, F.A.; Paterson, A.H. Modes of gene duplication contribute differently to genetic novelty and redundancy, but show parallels across divergent angiosperms. PLoS ONE 2011, 6, e28150. [Google Scholar] [CrossRef] [Green Version]
- Juretic, N.; Hoen, D.R.; Huynh, M.L.; Harrison, P.M.; Bureau, T.E. The evolutionary fate of MULE-mediated duplications of host gene fragments in rice. Genome Res. 2005, 15, 1292–1297. [Google Scholar] [CrossRef] [Green Version]
- Le, Q.H.; Wright, S.; Yu, Z.; Bureau, T. Transposon diversity in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2000, 97, 7376–7381. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.; Wright, S.I.; Bureau, T.E. Mutator-like elements in Arabidopsis thaliana. Structure, diversity and evolution. Genetics 2000, 156, 2019–2031. [Google Scholar]
- Kawasaki, S.; Nitasaka, E. Characterization of Tpn1 family in the Japanese morning glory: En/Spm-related transposable elements capturing host genes. Plant Cell Physiol. 2004, 45, 933–944. [Google Scholar] [CrossRef]
- Zabala, G.; Vodkin, L.O. The wp mutation of Glycine max carries a gene-fragment-rich transposon of the CACTA superfamily. Plant Cell 2005, 17, 2619–2632. [Google Scholar] [CrossRef] [Green Version]
- Jiang, N.; Bao, Z.; Zhang, X.; Eddy, S.R.; Wessler, S.R. Pack-MULE transposable elements mediate gene evolution in plants. Nature 2004, 431, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Samonte, R.V.; Eichler, E.E. Segmental duplications and the evolution of the primate genome. Nat. Rev. Genet. 2002, 3, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, K.H.; Shields, D.C. Molecular evidence for an ancient duplication of the entire yeast genome. Nature 1997, 387, 708–713. [Google Scholar] [CrossRef] [PubMed]
- Bailey, J.A.; Gu, Z.; Clark, R.A.; Reinert, K.; Samonte, R.V.; Schwartz, S.; Adams, M.D.; Myers, E.W.; Li, P.W.; Eichler, E.E. Recent segmental duplications in the human genome. Science 2002, 297, 1003–1007. [Google Scholar] [CrossRef] [Green Version]
- Koszul, R.; Caburet, S.; Dujon, B.; Fischer, G. Eucaryotic genome evolution through the spontaneous duplication of large chromosomal segments. EMBO J. 2004, 23, 234–243. [Google Scholar] [CrossRef]
- Koszul, R.; Dujon, B.; Fischer, G. Stability of large segmental duplications in the yeast genome. Genetics 2006, 172, 2211–2222. [Google Scholar] [CrossRef] [Green Version]
- Fiston-Lavier, A.-S.; Anxolabehere, D.; Quesneville, H. A model of segmental duplication formation in Drosophila melanogaster. Genome Res. 2007, 17, 1458–1470. [Google Scholar] [CrossRef] [Green Version]
- Bailey, J.A.; Liu, G.; Eichler, E.E. An Alu transposition model for the origin and expansion of human segmental duplications. Am. J. Hum. Genet. 2003, 73, 823–834. [Google Scholar] [CrossRef] [Green Version]
- She, X.; Cheng, Z.; Zöllner, S.; Church, D.M.; Eichler, E.E. Mouse segmental duplication and copy number variation. Nat. Genet. 2008, 40, 909–914. [Google Scholar] [CrossRef] [Green Version]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [Green Version]
- Bailey, J.A.; Eichler, E.E. Primate segmental duplications: Crucibles of evolution, diversity and disease. Nat. Rev. Genet. 2006, 7, 552–564. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Ma, D.; Vasseur, L.; You, M. Segmental duplications: Evolution and impact among the current Lepidoptera genomes. BMC Evol. Biol. 2017, 17, 161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hakes, L.; Lovell, S.C.; Oliver, S.G.; Robertson, D.L. Specificity in protein interactions and its relationship with sequence diversity and coevolution. Proc. Natl. Acad. Sci. USA 2007, 104, 7999–8004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wapinski, I.; Pfeffer, A.; Friedman, N.; Regev, A. Natural history and evolutionary principles of gene duplication in fungi. Nature 2007, 449, 54–61. [Google Scholar] [CrossRef]
- Blanc, G.; Wolfe, K.H. Functional divergence of duplicated genes formed by polyploidy during Arabidopsis evolution. Plant Cell 2004, 16, 1679–1691. [Google Scholar] [CrossRef] [Green Version]
- Maere, S.; Bodt, S.D.; Raes, J.; Casneuf, T.; Montagu, M.V.; Kuiper, M.; De Peer, Y.V. Modeling gene and genome duplications in eukaryotes. Proc. Natl. Acad. Sci. USA 2005, 102, 5454–5459. [Google Scholar] [CrossRef] [Green Version]
- Hanada, K.; Zou, C.; Lehti-Shiu, M.D.; Shinozaki, K.; Shiu, S.-H. Importance of lineage-specific expansion of plant tandem duplicates in the adaptive response to environmental stimuli. Plant Physiol. 2008, 148, 993–1003. [Google Scholar] [CrossRef] [Green Version]
- Rodgers-Melnick, E.; Mane, S.P.; Dharmawardhana, P.; Slavov, G.T.; Crasta, O.R.; Strauss, S.H.; Brunner, A.M.; DiFazio, S.P. Contrasting patterns of evolution following whole genome versus tandem duplication events in Populus. Genome Res. 2012, 22, 95–105. [Google Scholar] [CrossRef] [Green Version]
- Freeling, M. Bias in plant gene content following different sorts of duplication: Tandem, whole-genome, segmental, or by transposition. Annu. Rev. Plant Biol. 2009, 60, 433–453. [Google Scholar] [CrossRef]
- Rizzon, C.; Ponger, L.; Gaut, B.S. Striking similarities in the genomic distribution of tandemly arrayed genes in Arabidopsis and rice. PLoS Comput. Biol. 2006, 2, e115. [Google Scholar] [CrossRef]
- Acharya, D.; Ghosh, T.C. Global analysis of human duplicated genes reveals the relative importance of whole-genome duplicates originated in the early vertebrate evolution. BMC Genom. 2016, 17, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casneuf, T.; De Bodt, S.; Raes, J.; Maere, S.; Van de Peer, Y. Nonrandom divergence of gene expression following gene and genome duplications in the flowering plant Arabidopsis thaliana. Genome Biol. 2006, 7, R13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Defoort, J.; Van de Peer, Y.; Carretero-Paulet, L. The Evolution of Gene Duplicates in Angiosperms and the Impact of Protein–Protein Interactions and the Mechanism of Duplication. Genome Biol. Evol. 2019, 11, 2292–2305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y. Locally duplicated ohnologs evolve faster than nonlocally duplicated ohnologs in Arabidopsis and rice. Genome Biol. Evol. 2013, 5, 362–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arabidopsis Interactome Mapping Consortium; Dreze, M.; Carvunis, A.R.; Charloteaux, B.; Galli, M.; Pevzner, S.J.; Tasan, M.; Ahn, Y.Y.; Balumuri, P.; Barabási, A.L.; et al. Evidence for network evolution in an Arabidopsis interactome map. Science 2011, 333, 601–607. [Google Scholar] [CrossRef] [Green Version]
- Arsovski, A.A.; Pradinuk, J.; Guo, X.Q.; Wang, S.; Adams, K.L. Evolution of Cis-Regulatory Elements and Regulatory Networks in Duplicated Genes of Arabidopsis. Plant Physiol. 2015, 169, 2982–2991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prince, V.E.; Pickett, F.B. Splitting pairs: The diverging fates of duplicated genes. Nat. Rev. Genet. 2002, 3, 827–837. [Google Scholar] [CrossRef]
- Zou, C.; Lehti-Shiu, M.D.; Thibaud-Nissen, F.; Prakash, T.; Buell, C.R.; Shiu, S.-H. Evolutionary and expression signatures of pseudogenes in Arabidopsis and rice. Plant Physiol. 2009, 151, 3–15. [Google Scholar] [CrossRef] [Green Version]
- Rouquier, S.; Taviaux, S.; Trask, B.J.; Brand-Arpon, V.; Van den Engh, G.; Demaille, J.; Giorgi, D. Distribution of olfactory receptor genes in the human genome. Nat. Genet. 1998, 18, 243–250. [Google Scholar] [CrossRef]
- Quignon, P.; Kirkness, E.; Cadieu, E.; Touleimat, N.; Guyon, R.; Renier, C.; Hitte, C.; André, C.; Fraser, C.; Galibert, F. Comparison of the canine and human olfactory receptor gene repertoires. Genome Biol. 2003, 4, R80. [Google Scholar] [CrossRef] [Green Version]
- Hahn, M.W. Distinguishing among evolutionary models for the maintenance of gene duplicates. J. Hered. 2009, 100, 605–617. [Google Scholar] [CrossRef] [Green Version]
- Innan, H.; Kondrashov, F. The evolution of gene duplications: Classifying and distinguishing between models. Nat. Rev. Genet. 2010, 11, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M. The Neutral Theory of Molecular Evolution; Cambridge University Press: Cambridge, UK, 1983; ISBN 978-0-521-31793-1. [Google Scholar]
- Logeman, B.L.; Wood, L.K.; Lee, J.; Thiele, D.J. Gene duplication and neo-functionalization in the evolutionary and functional divergence of metazoan copper transporters Ctr1 and Ctr2. J. Biol. Chem. 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escriva, H.; Bertrand, S.; Germain, P.; Robinson-Rechavi, M.; Umbhauer, M.; Cartry, J.; Duffraisse, M.; Holland, L.; Gronemeyer, H.; Laudet, V. Neofunctionalization in vertebrates: The example of retinoic acid receptors. PLoS Genet. 2006, 2, e102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, T.E.; Langdale, J.A.; Kelly, S. The impact of widespread regulatory neofunctionalization on homeolog gene evolution following whole-genome duplication in maize. Genome Res. 2014, 24, 1348–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, C.; Chen, Y.; Long, M. Recurrent Tandem Gene Duplication Gave Rise to Functionally Divergent Genes in Drosophila. Mol. Biol. Evol. 2008, 25, 1451–1458. [Google Scholar] [CrossRef] [Green Version]
- Force, A.; Lynch, M.; Pickett, F.B.; Amores, A.; Yan, Y.L.; Postlethwait, J. Preservation of duplicate genes by complementary, degenerative mutations. Genetics 1999, 151, 1531–1545. [Google Scholar]
- Piatigorsky, J.; Wistow, G. The recruitment of crystallins: New functions precede gene duplication. Science 1991, 252, 1078–1079. [Google Scholar] [CrossRef]
- Hughes, A.L. The evolution of functionally novel proteins after gene duplication. Proc. R. Soc. Lond. B Biol. Sci. 1994, 256, 119–124. [Google Scholar] [CrossRef]
- Otto, S.P.; Yong, P. The evolution of gene duplicates. Adv. Genet. 2002, 46, 451–483. [Google Scholar] [CrossRef]
- Jackson, P.J.; Douglas, N.R.; Chai, B.; Binkley, J.; Sidow, A.; Barsh, G.S.; Millhauser, G.L. Structural and molecular evolutionary analysis of Agouti and Agouti-related proteins. Chem. Biol. 2006, 13, 1297–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlson, K.D.; Bhogale, S.; Anderson, D.; Zaragoza-Mendoza, A.; Madlung, A. Subfunctionalization of phytochrome B1/B2 leads to differential auxin and photosynthetic responses. Plant Direct 2020, 4, e00205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vavouri, T.; Semple, J.I.; Lehner, B. Widespread conservation of genetic redundancy during a billion years of eukaryotic evolution. Trends Genet. 2008, 24, 485–488. [Google Scholar] [CrossRef]
- Gout, J.F.; Lynch, M. Maintenance and Loss of Duplicated Genes by Dosage Subfunctionalization. Mol. Biol. Evol. 2015, 32, 2141–2148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, W.; Liao, B.Y.; Chang, A.Y.F.; Zhang, J. Maintenance of duplicate genes and their functional redundancy by reduced expression. Trends Genet. 2010, 26, 425–430. [Google Scholar] [CrossRef] [Green Version]
- Greer, J.M.; Puetz, J.; Thomas, K.R.; Capecchi, M.R. Maintenance of functional equivalence during paralogous HOX gene evolution. Nature 2000, 403, 661–665. [Google Scholar] [CrossRef]
- Dean, E.J.; Davis, J.C.; Davis, R.W.; Petrov, D.A. Pervasive and persistent redundancy among duplicated genes in yeast. PLoS Genet. 2008, 4, e1000113. [Google Scholar] [CrossRef] [Green Version]
- Averof, M.; Dawes, R.; Ferrier, D. Diversification of arthropod HOX genes as a paradigm for the evolution of gene functions. Semin. Cell Dev. Biol. 1996, 7, 539–551. [Google Scholar] [CrossRef]
- Wang, W.; Brunet, F.G.; Nevo, E.; Long, M. Origin of sphinx, a young chimeric RNA gene in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 2002, 99, 4448–4453. [Google Scholar] [CrossRef] [Green Version]
- Nisole, S.; Lynch, C.; Stoye, J.P.; Yap, M.W. A Trim5-cyclophilin A fusion protein found in owl monkey kidney cells can restrict HIV-1. Proc. Natl. Acad. Sci. USA 2004, 101, 13324–13328. [Google Scholar] [CrossRef] [Green Version]
- Sayah, D.M.; Sokolskaja, E.; Berthoux, L.; Luban, J. Cyclophilin A retrotransposition into TRIM5 explains owl monkey resistance to HIV-1. Nature 2004, 430, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J. Evolving protein functional diversity in new genes of Drosophila. Proc. Natl. Acad. Sci. USA 2004, 101, 16246–16250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machado, J.P.; Antunes, A. The genomic context of retrocopies increases their chance of functional relevancy in mammals. Genomics 2020, 112, 2410–2417. [Google Scholar] [CrossRef] [PubMed]
- Makino, T.; McLysaght, A. Positionally biased gene loss after whole genome duplication: Evidence from human, yeast, and plant. Genome Res. 2012, 22, 2427–2435. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.; Liu, Y.; Xia, E.; Gao, L. Prevalent role of gene features in determining evolutionary fates of whole-genome duplication duplicated genes in flowering plants. Plant Physiol. 2013, 161, 1844–1861. [Google Scholar] [CrossRef] [Green Version]
- Pan, D.; Zhang, L. Quantifying the major mechanisms of recent gene duplications in the human and mouse genomes: A novel strategy to estimate gene duplication rates. Genome Biol. 2007, 8, R158. [Google Scholar] [CrossRef] [Green Version]
- Marques-Bonet, T.; Girirajan, S.; Eichler, E.E. The origins and impact of primate segmental duplications. Trends Genet. 2009, 25, 443–454. [Google Scholar] [CrossRef] [Green Version]
- Assis, R.; Bachtrog, D. Neofunctionalization of young duplicate genes in Drosophila. Proc. Natl. Acad. Sci. USA 2013, 110, 17409–17414. [Google Scholar] [CrossRef] [Green Version]
- Pearson, W.R. An introduction to sequence similarity (“homology”) searching. Curr. Protoc. Bioinforma. 2013. [Google Scholar] [CrossRef]
- Shapiro, B.; Hofreiter, M. A paleogenomic perspective on evolution and gene function: New insights from ancient DNA. Science 2014, 343, 1236573. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, L.S.; Eddy, S.R.; Portugaly, E. Hidden Markov model speed heuristic and iterative HMM search procedure. BMC Bioinform. 2010, 11, 431. [Google Scholar] [CrossRef] [Green Version]
- Smith, T.F.; Waterman, M.S. Identification of common molecular subsequences. J. Mol. Biol. 1981, 147, 195–197. [Google Scholar] [CrossRef]
- Saebø, P.E.; Andersen, S.M.; Myrseth, J.; Laerdahl, J.K.; Rognes, T. PARALIGN: Rapid and sensitive sequence similarity searches powered by parallel computing technology. Nucleic Acids Res. 2005, 33, W535–W539. [Google Scholar] [CrossRef] [PubMed]
- Rucci, E.; Garcia Sanchez, C.; Botella Juan, G.; Giusti, A.D.; Naiouf, M.; Prieto-Matias, M. SWIMM 2.0: Enhanced Smith-Waterman on Intel’s Multicore and Manycore Architectures Based on AVX-512 Vector Extensions. Int. J. Parallel Program 2019, 47, 296–316. [Google Scholar] [CrossRef]
- Koonin, E.V.; Galperin, M.Y. Sequence—Evolution—Function: Computational Approaches in Comparative Genomics; Kluwer Academic: Boston, MA, USA, 2003; ISBN 978-1-4020-7274-1. [Google Scholar]
- Sander, C.; Schneider, R. Database of homology-derived protein structures and the structural meaning of sequence alignment. Proteins 1991, 9, 56–68. [Google Scholar] [CrossRef]
- Rost, B. Twilight zone of protein sequence alignments. Protein Eng. 1999, 12, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Li, W.H.; Gu, Z.; Wang, H.; Nekrutenko, A. Evolutionary analyses of the human genome. Nature 2001, 409, 847–849. [Google Scholar] [CrossRef] [Green Version]
- Blanc, G.; Wolfe, K.H. Widespread Paleopolyploidy in Model Plant Species Inferred from Age Distributions of Duplicate Genes. Plant Cell 2004, 16, 1667–1678. [Google Scholar] [CrossRef] [Green Version]
- Wootton, J.C.; Federhen, S. Statistics of local complexity in amino acid sequences and sequence databases. Comput. Chem. 1993, 17, 149–163. [Google Scholar] [CrossRef]
- Shoja, V.; Zhang, L. A roadmap of tandemly arrayed genes in the genomes of human, mouse, and rat. Mol. Biol. Evol. 2006, 23, 2134–2141. [Google Scholar] [CrossRef] [PubMed]
- Britten, R.J. Almost all human genes resulted from ancient duplication. Proc. Natl. Acad. Sci. USA 2006, 103, 19027–19032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, D.; Zhang, L. Tandemly arrayed genes in vertebrate genomes. Comp. Funct. Genom. 2008, 545269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makino, T.; McLysaght, A. Ohnologs in the human genome are dosage balanced and frequently associated with disease. Proc. Natl. Acad. Sci. USA 2010, 107, 9270–9274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, P.P.; Arora, J.; Isambert, H. Identification of Ohnolog Genes Originating from Whole Genome Duplication in Early Vertebrates, Based on Synteny Comparison across Multiple Genomes. PLoS Comput. Biol. 2015, 11, e1004394. [Google Scholar] [CrossRef]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A.; et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2019, 47, D427–D432. [Google Scholar] [CrossRef]
- Mitchell, A.L.; Attwood, T.K.; Babbitt, P.C.; Blum, M.; Bork, P.; Bridge, A.; Brown, S.D.; Chang, H.Y.; El-Gebali, S.; Fraser, M.I.; et al. InterPro in 2019: Improving coverage, classification and access to protein sequence annotations. Nucleic Acids Res. 2019, 47, D351–D360. [Google Scholar] [CrossRef] [Green Version]
- Kuzniar, A.; Van Ham, R.C.H.J.; Pongor, S.; Leunissen, J.A.M. The quest for orthologs: Finding the corresponding gene across genomes. Trends Genet. 2008, 24, 539–551. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Koonin, E.V.; Lipman, D.J. A genomic perspective on protein families. Science 1997, 278, 631–637. [Google Scholar] [CrossRef] [Green Version]
- Remm, M.; Storm, C.E.; Sonnhammer, E.L. Automatic clustering of orthologs and in-paralogs from pairwise species comparisons. J. Mol. Biol. 2001, 314, 1041–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schreiber, F.; Sonnhammer, E.L.L. Hieranoid: Hierarchical orthology inference. J. Mol. Biol. 2013, 425, 2072–2081. [Google Scholar] [CrossRef] [PubMed]
- Jensen, L.J.; Julien, P.; Kuhn, M.; Von Mering, C.; Muller, J.; Doerks, T.; Bork, P. eggNOG: Automated construction and annotation of orthologous groups of genes. Nucleic Acids Res. 2008, 36, D250–D254. [Google Scholar] [CrossRef] [PubMed]
- Kriventseva, E.V.; Tegenfeldt, F.; Petty, T.J.; Waterhouse, R.M.; Simão, F.A.; Pozdnyakov, I.A.; Ioannidis, P.; Zdobnov, E.M. OrthoDB v8: Update of the hierarchical catalog of orthologs and the underlying free software. Nucleic Acids Res. 2015, 43, D250–D256. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Stoeckert, C.J.; Roos, D.S. OrthoMCL: Identification of Ortholog Groups for Eukaryotic Genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef] [Green Version]
- Linard, B.; Thompson, J.D.; Poch, O.; Lecompte, O. OrthoInspector: Comprehensive orthology analysis and visual exploration. BMC Bioinform. 2011, 12, 11. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. OrthoFinder: Solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 2015, 16, 157. [Google Scholar] [CrossRef] [Green Version]
- Train, C.-M.; Glover, N.M.; Gonnet, G.H.; Altenhoff, A.M.; Dessimoz, C. Orthologous Matrix (OMA) algorithm 2.0: More robust to asymmetric evolutionary rates and more scalable hierarchical orthologous group inference. Bioinformatics 2017, 33, i75–i82. [Google Scholar] [CrossRef] [Green Version]
- Dalquen, D.A.; Dessimoz, C. Bidirectional best hits miss many orthologs in duplication-rich clades such as plants and animals. Genome Biol. Evol. 2013, 5, 1800–1806. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Coghlan, A.; Ruan, J.; Coin, L.J.; Hériché, J.K.; Osmotherly, L.; Li, R.; Liu, T.; Zhang, Z.; Bolund, L.; et al. TreeFam: A curated database of phylogenetic trees of animal gene families. Nucleic Acids Res. 2006, 34, D572–D580. [Google Scholar] [CrossRef]
- Poptsova, M.S.; Gogarten, J.P. BranchClust: A phylogenetic algorithm for selecting gene families. BMC Bioinform. 2007, 8, 120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penel, S.; Arigon, A.M.; Dufayard, J.F.; Sertier, A.S.; Daubin, V.; Duret, L.; Gouy, M.; Perrière, G. Databases of homologous gene families for comparative genomics. BMC Bioinform. 2009, 10 (Suppl. 6), S3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huerta-Cepas, J.; Capella-Gutierrez, S.; Pryszcz, L.P.; Denisov, I.; Kormes, D.; Marcet-Houben, M.; Gabaldón, T. PhylomeDB v3.0: An expanding repository of genome-wide collections of trees, alignments and phylogeny-based orthology and paralogy predictions. Nucleic Acids Res. 2011, 39, D556–D560. [Google Scholar] [CrossRef] [PubMed]
- Storm, C.E.V.; Sonnhammer, E.L.L. Automated ortholog inference from phylogenetic trees and calculation of orthology reliability. Bioinformatics 2002, 18, 92–99. [Google Scholar] [CrossRef] [Green Version]
- Berglund-Sonnhammer, A.C.; Steffansson, P.; Betts, M.J.; Liberles, D.A. Optimal Gene Trees from Sequences and Species Trees Using a Soft Interpretation of Parsimony. J. Mol. Evol. 2006, 63, 240–250. [Google Scholar] [CrossRef]
- Van der Heijden, R.T.J.M.; Snel, B.; Van Noort, V.; Huynen, M.A. Orthology prediction at scalable resolution by phylogenetic tree analysis. BMC Bioinform. 2007, 8, 83. [Google Scholar] [CrossRef] [Green Version]
- Goodman, M.; Czelusniak, J.; Moore, G.W.; Romero-Herrera, A.E.; Matsuda, G. Fitting the Gene Lineage into its Species Lineage, a Parsimony Strategy Illustrated by Cladograms Constructed from Globin Sequences. Syst. Biol. 1979, 28, 132–163. [Google Scholar] [CrossRef]
- Åkerborg, Ö.; Sennblad, B.; Arvestad, L.; Lagergren, J. Simultaneous Bayesian gene tree reconstruction and reconciliation analysis. Proc. Natl. Acad. Sci. USA 2009, 106, 5714–5719. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Hunt, M.; Tsai, I.J. Inferring synteny between genome assemblies: A systematic evaluation. BMC Bioinform. 2018, 19, 26. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Haug-Baltzell, A.; Stephens, S.A.; Davey, S.; Scheidegger, C.E.; Lyons, E. SynMap2 and SynMap3D: Web-based whole-genome synteny browsers. Bioinformatics 2017, 33, 2197–2198. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tang, H.; DeBarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proost, S.; Fostier, J.; De Witte, D.; Dhoedt, B.; Demeester, P.; Van de Peer, Y.; Vandepoele, K. i-ADHoRe 3.0—Fast and sensitive detection of genomic homology in extremely large data sets. Nucleic Acids Res. 2012, 40, e11. [Google Scholar] [CrossRef] [PubMed]
- Rödelsperger, C.; Dieterich, C. CYNTENATOR: Progressive Gene Order Alignment of 17 Vertebrate Genomes. PLoS ONE 2010, 5, e8861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drillon, G.; Carbone, A.; Fischer, G. SynChro: A Fast and Easy Tool to Reconstruct and Visualize Synteny Blocks along Eukaryotic Chromosomes. PLoS ONE 2014, 9, e92621. [Google Scholar] [CrossRef] [PubMed]
- Cannon, S.B.; Kozik, A.; Chan, B.; Michelmore, R.; Young, N.D. DiagHunter and GenoPix2D: Programs for genomic comparisons, large-scale homology discovery and visualization. Genome Biol. 2003, 4, R68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calabrese, P.P.; Chakravarty, S.; Vision, T.J. Fast identification and statistical evaluation of segmental homologies in comparative maps. Bioinformatics 2003, 19, i74–i80. [Google Scholar] [CrossRef] [Green Version]
- Soderlund, C.; Nelson, W.; Shoemaker, A.; Paterson, A. SyMAP: A system for discovering and viewing syntenic regions of FPC maps. Genome Res. 2006, 16, 1159–1168. [Google Scholar] [CrossRef] [Green Version]
- Sinha, A.U.; Meller, J. Cinteny: Flexible analysis and visualization of synteny and genome rearrangements in multiple organisms. BMC Bioinform. 2007, 8, 82. [Google Scholar] [CrossRef] [Green Version]
- Haas, B.J.; Delcher, A.L.; Wortman, J.R.; Salzberg, S.L. DAGchainer: A tool for mining segmental genome duplications and synteny. Bioinformatics 2004, 20, 3643–3646. [Google Scholar] [CrossRef] [Green Version]
- Hampson, S.; McLysaght, A.; Gaut, B.; Baldi, P. LineUp: Statistical detection of chromosomal homology with application to plant comparative genomics. Genome Res. 2003, 13, 999–1010. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Shi, X.; Li, Z.; Zhu, Q.; Kong, L.; Tang, W.; Ge, S.; Luo, J. Statistical inference of chromosomal homology based on gene colinearity and applications to Arabidopsis and rice. BMC Bioinform. 2006, 7, 447. [Google Scholar] [CrossRef] [Green Version]
- Pham, S.K.; Pevzner, P.A. DRIMM-Synteny: Decomposing genomes into evolutionary conserved segments. Bioinformatics 2010, 26, 2509–2516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paten, B.; Herrero, J.; Beal, K.; Fitzgerald, S.; Birney, E. Enredo and Pecan: Genome-wide mammalian consistency-based multiple alignment with paralogs. Genome Res. 2008, 18, 1814–1828. [Google Scholar] [CrossRef] [Green Version]
- Xu, A.W.; Moret, B.M.E. GASTS: Parsimony Scoring under Rearrangements. In Algorithms in Bioinformatics, Proceedings of the 11th International Workshop, WABI 2011, Saarbrücken, Germany, 5–7 September 2011; Przytycka, T.M., Sagot, M.F., Eds.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 351–363. [Google Scholar]
- Zhou, L.; Feng, B.; Yang, N.; Tang, J. Ancestral reconstruction with duplications using binary encoding and probabilistic model. In Proceedings of the 7th International conference on Bioinformatics and Computational Biology, Honolulu, HI, USA, 9–11 March 2015; pp. 97–104. [Google Scholar]
- Yang, N.; Hu, F.; Zhou, L.; Tang, J. Reconstruction of Ancestral Gene Orders Using Probabilistic and Gene Encoding Approaches. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [Green Version]
- Feng, B.; Zhou, L.; Tang, J. Ancestral Genome Reconstruction on Whole Genome Level. Curr. Genom. 2017, 18, 306–315. [Google Scholar] [CrossRef]
- Lucas, J.M.; Muffato, M.; Crollius, H.R. PhylDiag: Identifying complex synteny blocks that include tandem duplications using phylogenetic gene trees. BMC Bioinform. 2014, 15. [Google Scholar] [CrossRef] [Green Version]
- Hachiya, T.; Osana, Y.; Popendorf, K.; Sakakibara, Y. Accurate identification of orthologous segments among multiple genomes. Bioinformatics 2009, 25, 853–860. [Google Scholar] [CrossRef] [Green Version]
- Baek, J.H.; Kim, J.; Kim, C.K.; Sohn, S.H.; Choi, D.; Ratnaparkhe, M.B.; Kim, D.W.; Lee, T.H. MultiSyn: A Webtool for Multiple Synteny Detection and Visualization of User’s Sequence of Interest Compared to Public Plant Species. Evol. Bioinform. 2016. [Google Scholar] [CrossRef] [Green Version]
- Zeng, X.; Nesbitt, M.J.; Pei, J.; Wang, K.; Vergara, I.A.; Chen, N. OrthoCluster: A new tool for mining synteny blocks and applications in comparative genomics. In Advances in database technology, Proceedings of the 11th international conference on Extending database technology, Nantes, France, 25–29 March 2008; Association for Computing Machinery: New York, NY, USA, 2008; pp. 656–667. [Google Scholar]
- Fostier, J.; Proost, S.; Dhoedt, B.; Saeys, Y.; Demeester, P.; Van de Peer, Y.; Vandepoele, K. A greedy, graph-based algorithm for the alignment of multiple homologous gene lists. Bioinformatics 2011, 27, 749–756. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simillion, C.; Vandepoele, K.; Saeys, Y.; Van de Peer, Y. Building Genomic Profiles for Uncovering Segmental Homology in the Twilight Zone. Genome Res. 2004, 14, 1095–1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipman, D.J.; Pearson, W.R. Rapid and sensitive protein similarity searches. Science 1985, 227, 1435–1441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [Green Version]
- Pevzner, P.; Tesler, G. Genome rearrangements in mammalian evolution: Lessons from human and mouse genomes. Genome Res. 2003, 13, 37–45. [Google Scholar] [CrossRef] [Green Version]
- Swidan, F.; Rocha, E.P.C.; Shmoish, M.; Pinter, R.Y. An Integrative Method for Accurate Comparative Genome Mapping. PLoS Comput. Biol. 2006, 2. [Google Scholar] [CrossRef] [Green Version]
- Paten, B.; Earl, D.; Nguyen, N.; Diekhans, M.; Zerbino, D.; Haussler, D. Cactus: Algorithms for genome multiple sequence alignment. Genome Res. 2011, 21, 1512–1528. [Google Scholar] [CrossRef] [Green Version]
- Paten, B.; Diekhans, M.; Earl, D.; St. John, J.; Ma, J.; Suh, B.; Haussler, D. Cactus Graphs for Genome Comparisons. In Research in Computational Molecular Biology, Proceedings of the 14th Annual International Conference, RECOMB 2010, Lisbon, Portugal, 25–28 April 2010; Berger, B., Ed.; Springer: Berlin/Heidelberg, Germany, 2010; pp. 410–425. [Google Scholar]
- Zhang, L.; Gaut, B.S. Does Recombination Shape the Distribution and Evolution of Tandemly Arrayed Genes (TAGs) in the Arabidopsis thaliana Genome? Genome Res. 2003, 13, 2533–2540. [Google Scholar] [CrossRef] [Green Version]
- Morgulis, A.; Gertz, E.M.; Schäffer, A.A.; Agarwala, R. A fast and symmetric DUST implementation to mask low-complexity DNA sequences. J. Comput. Biol. 2006, 13, 1028–1040. [Google Scholar] [CrossRef]
- Wootton, J.C.; Federhen, S. Analysis of compositionally biased regions in sequence databases. Methods Enzymol. 1996, 266, 554–571. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frith, M.C. A new repeat-masking method enables specific detection of homologous sequences. Nucleic Acids Res. 2011, 39, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olson, D.; Wheeler, T. ULTRA: A Model Based Tool to Detect Tandem Repeats. In Proceedings of the 9th ACM International Conference on Bioinformatics, Computational Biology, and Health Informatics, Washington, DC, USA, 29 August–1 September 2018; Association for Computing Machinery: Washington, DC, USA, 2018; pp. 37–46. [Google Scholar]
- Soylev, A.; Le, T.M.; Amini, H.; Alkan, C.; Hormozdiari, F. Discovery of tandem and interspersed segmental duplications using high-throughput sequencing. Bioinformatics 2019, 35, 3923–3930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genovese, L.M.; Mosca, M.M.; Pellegrini, M.; Geraci, F. Dot2dot: Accurate whole-genome tandem repeats discovery. Bioinformatics 2019, 35, 914–922. [Google Scholar] [CrossRef] [PubMed]
- Audemard, E.; Schiex, T.; Faraut, T. Detecting long tandem duplications in genomic sequences. BMC Bioinform. 2012, 13, 83. [Google Scholar] [CrossRef] [Green Version]
- Zheng, D.; Gerstein, M.B. A computational approach for identifying pseudogenes in the ENCODE regions. Genome Biol. 2006, 7 (Suppl. 1), S13. [Google Scholar] [CrossRef] [Green Version]
- Despons, L.; Baret, P.V.; Frangeul, L.; Louis, V.L.; Durrens, P.; Souciet, J.-L. Genome-wide computational prediction of tandem gene arrays: Application in yeasts. BMC Genom. 2010, 11, 56. [Google Scholar] [CrossRef]
- Herrero, J.; Muffato, M.; Beal, K.; Fitzgerald, S.; Gordon, L.; Pignatelli, M.; Vilella, A.J.; Searle, S.M.J.; Amode, R.; Brent, S.; et al. Ensembl comparative genomics resources. Database 2016, 2016. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Hong, W.; Cho, M.; Sim, M.; Lee, D.; Ko, Y.; Kim, J. Synteny Portal: A web-based application portal for synteny block analysis. Nucleic Acids Res. 2016, 44, W35–W40. [Google Scholar] [CrossRef] [Green Version]
- Muffato, M.; Louis, A.; Poisnel, C.E.; Crollius, H.R. Genomicus: A database and a browser to study gene synteny in modern and ancestral genomes. Bioinformatics 2010, 26, 1119–1121. [Google Scholar] [CrossRef]
- Ma, J.; Zhang, L.; Suh, B.B.; Raney, B.J.; Burhans, R.C.; Kent, W.J.; Blanchette, M.; Haussler, D.; Miller, W. Reconstructing contiguous regions of an ancestral genome. Genome Res. 2006, 16, 1557–1565. [Google Scholar] [CrossRef] [Green Version]
- Loots, G.; Ovcharenko, I. ECRbase: Database of evolutionary conserved regions, promoters, and transcription factor binding sites in vertebrate genomes. Bioinformatics 2007, 23, 122–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, M.P.; Vergara, I.A.; Frech, C.; Chen, Q.; Zeng, X.; Pei, J.; Chen, N. OrthoClusterDB: An online platform for synteny blocks. BMC Bioinform. 2009, 10, 192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonnhammer, E.L.L.; Östlund, G. InParanoid 8: Orthology analysis between 273 proteomes, mostly eukaryotic. Nucleic Acids Res. 2015, 43, D234–D239. [Google Scholar] [CrossRef] [PubMed]
- Miele, V.; Penel, S.; Duret, L. Ultra-fast sequence clustering from similarity networks with SiLiX. BMC Bioinform. 2011, 12, 116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Altenhoff, A.M.; Glover, N.M.; Train, C.-M.; Kaleb, K.; Warwick Vesztrocy, A.; Dylus, D.; De Farias, T.M.; Zile, K.; Stevenson, C.; Long, J.; et al. The OMA orthology database in 2018: Retrieving evolutionary relationships among all domains of life through richer web and programmatic interfaces. Nucleic Acids Res. 2018, 46, D477–D485. [Google Scholar] [CrossRef]
- Van Bel, M.; Diels, T.; Vancaester, E.; Kreft, L.; Botzki, A.; Van de Peer, Y.; Coppens, F.; Vandepoele, K. PLAZA 4.0: An integrative resource for functional, evolutionary and comparative plant genomics. Nucleic Acids Res. 2018, 46, D1190–D1196. [Google Scholar] [CrossRef]
- Conte, M.G.; Gaillard, S.; Lanau, N.; Rouard, M.; Périn, C. GreenPhylDB: A database for plant comparative genomics. Nucleic Acids Res. 2008, 36, D991–D998. [Google Scholar] [CrossRef] [Green Version]
- Goodstein, D.M.; Shu, S.; Howson, R.; Neupane, R.; Hayes, R.D.; Fazo, J.; Mitros, T.; Dirks, W.; Hellsten, U.; Putnam, N.; et al. Phytozome: A comparative platform for green plant genomics. Nucleic Acids Res. 2012, 40, D1178–D1186. [Google Scholar] [CrossRef]


{kind=link}
| Species | No. of Considered Genes | No. of Estimated Duplicated Genes | % Estimated Duplicated Genes | Methodology | Duplicated Gene Types | References |
|---|---|---|---|---|---|---|
| Arabidopsis thaliana | 25,557 | 11,937 | 46.7 | All-against-all nucleotide sequence similarity searches using BLASTN among the transcribed sequences. Sequences aligned over >300 bp and showing at least 40% identity were defined as pairs of paralogs. | Not specified, all paralogous pairs were searched | [125] |
| 27,558 | 12,761 | 46.3 * | All-against-all protein sequence similarity search using BLASTP (e-value cutoff of e-10). Sequences alignable over a length of 150 amino acids with an identity of 30% were defined as paralogs. Gene families were built through single-linkage clustering. | Not specified, genes families were obtained | [69] | |
| 25,972 | 10,483–17,406 | 40.4–67 | All-against-all protein sequence similarity search using BLASTP (e-value cutoff of 1.0). For each pair of genes, blast-hits were merged to compute the total length and the global similarity of the aligned regions. Two datasets were constructed with respectively 30 and 50% sequence identity over respectively 70 and 90% protein length. Gene families were built through single-linkage clustering. | Not specified, genes families were all obtained (gene families) | [73] | |
| 22,810 | 21,622 | 94.8 * | All-against-all protein sequence similarity search using BLASTP (top five non-self protein matches with e-value of 10e-10 were considered). Genes without hits that met a threshold of e-value 10e-10 were deemed singletons. Pairs of WGD duplicates were downloaded from published lists. Single gene duplications were derived by excluding pairs of WGD duplicates from the population of gene duplications. Tandem duplications were defined as being adjacent to each other on the same chromosome. Proximal duplications were defined as non-tandem genes on the same chromosome with no more than 20 annotated genes between each other. Single gene transposed-duplications were searched for from the remaining single gene duplications using syntenic blocks within and between 10 species to determine the ancestral locus. If the parental copy had more than two exons and the transposed copy was intronless, the pair of duplicates was classified as coming from a retrotransposition. Other cases of single gene-transposed duplications were classified as DNA based transpositions. Dispersed duplications corresponded to the remaining duplications not classified as WGD, tandem, proximal, or transposed duplications. | WGD, tandem, proximal, DNA based transposed, retrotransposed, and dispersed duplications | [48] | |
| Homo sapiens (human) | 33,869–>19,727 | 12,981 | 65.8 | All-against-all protein sequence similarity search using BLASTP with the BLOSUM62 matrix and the SEG filter [126], TribeMCL with the default parameters. Tandem duplications were then searched for among families. | Gene families (tandem duplications searched among families) | [127] |
| 13,298 | 11,386 | 85–97 | All-against-all protein sequence similarity search using BLASTP with cutoff expectation <2 and <10-e3. | Not specified, distant duplicates | [128] | |
| 31,126 | 14,473 | 46.5 * | Ensembl family database and genes >300 nt. Tandem duplications were then searched for among families. | Gene families (tandem duplications searched for among families) | [129] | |
| 20,415 | 15,569 | 76.3 | Pooling of different datasets from [130] and all-against-all protein sequence similarity search using BLASTP. | WGD and SSD | [131] | |
| 22,447 | 11,740 | 52.3 * | Ensembl version 77, >50% sequence identity, and high confidence for paralogy. | WGD and SSD | [74] | |
| Mus musculus (mouse) | 21,305 | 14,043 | 65.9 | All-against-all protein sequence similarity search using BLASTP with the BLOSUM62 matrix and the SEG filter [126], TribeMCL with the default parameters. Tandem duplications were then searched for among families. | Gene families (tandem duplications searched for among families) | [127] |
| 27,736 | 16,091 | 58.01 | Ensembl family database and genes >300 nt. Tandem duplications were then searched for among families. | Gene families (tandem duplications were searched for among families) | [129] | |
| Rattus norvegicus (rat) | 18,468 | 12,466 | 67.5 | All-against-all protein sequence similarity search using BLASTP with the BLOSUM62 matrix and the SEG filter [126], TribeMCL with the default parameters. Tandem duplications were then searched for. | Gene families (tandem duplications searched for among families) | [127] |
| 27,194 | 16,446 | 60.48 * | Ensembl family database and genes >300 nt. Tandem duplications were then searched for among families. | Gene families (tandem duplications searched for among families) | [129] | |
| Oryza sativa (rice) | 18,562 | 9149 | 49.3 | All-against-all nucleotide sequence similarity searches using BLASTN were done among the transcribed sequences. Sequences aligned over >300 bp and showing at least 40% identity were defined as pairs of paralogs. | Not specified, all paralogous pairs were searched | [125] |
| 42,534 | 8244–19,322 | 19.4–45.4 | All-against-all protein sequence similarity search using BLASTP (e-value cutoff of 1.0). For each pair of genes, blast-hits were merged to compute the total length and the global similarity of the aligned regions. Two datasets were constructed with respectively 30 and 50% sequence identity over respectively 70 and 90% protein length. Gene families were built through single-linkage clustering. | Not specified, genes families were all obtained (gene families) | [73] | |
| 27,910 | 21,461 | 76.9 * | All-against-all protein sequence similarity search using BLASTP (top five non-self protein matches with e-value of 10e-10 were considered). Genes without hits that met a threshold of e-value 10e-10 were deemed singletons. Pairs of WGD duplicates were downloaded from published lists. Single gene duplications were derived by excluding pairs of WGD duplicates from the population of gene duplications. Tandem duplications were defined as being adjacent to each other on the same chromosome. Proximal duplications were defined as non-tandem genes on the same chromosome with no more than 20 annotated genes between each other. Single gene transposed-duplications were searched for from the remaining single gene duplications using syntenic blocks within and between 10 species to determine the ancestral locus. If the parental copy had more than two exons and the transposed copy was intronless, the pair of duplicates was classified as coming from a retrotransposition. Other cases of single gene-transposed duplications were classified as DNA based transpositions. Dispersed duplications corresponded to the remaining duplications not classified as WGD, tandem, proximal, or transposed duplications. | WGD, tandem, proximal, DNA based transposed, retrotransposed, and dispersed duplications | [48] |
| Name | Input | Output Text | Output Plots | Main Algorithm | Specificities | Other Information | Documentation | Programming Language | Interface | References | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene Orientation | Genome Number | ||||||||||
| i-ADHoRe 3.0 | BLASTP output or gene families and list of genes in a gff like format | Tabulated text | Graphical visualization | Custom Greedy Graph | Typical implementation of the collinearity strategy | Yes | N | Complete | C++ Wrapper in Python | Command line interface | [158] |
| MCScanX-Tranposed | BLASTP output and a list of genes on chromosomes | Tabulated text | Graphical visualization | DAGChainer equivalent | Able to detect transposed gene duplications, detection of the type of duplicates | No | N | Incomplete and with errors | C++ | Command line interface | [157] |
| PhylDiag | Species gene list and gene tree | Tabulated text | Graphical visualization | DAGChainer equivalent | Uses gene trees to define gene homologies. Takes into account gene orientations, and tandem duplication blocks | Yes | 2 | Complete | Python | Command line interface | [174] |
| SynChro | List of protein-coding genes and their associated amino-acid sequences | Text files containing homology relationships (RBH and non-RBH) and syntenic blocks description | Chromosomal painting representation, genome-wide dotplot | Computes Reciprocal Best-Hits (RBH) to reconstruct the backbones of the synteny blocks and complete with non-RBH syntenic homologs | Only one parameter: the synteny block stringency. Use OPSCAN instead of BLAST due to its optimization to detect RBH | only in visualizations | N | Complete | Python, bash | Command line interface | [160] |
| Satsuma | Nucleic sequences | Tabulated text | Multiple interactive plots | Cross-correlation, implemented as a fast Fourier transform | Based on a search strategy at a global level and cross-correlation at the local level | Yes | 2 | Short | C++, on linux | Command line interface | [155] |
| DAGchainer | Homologous genes and associated E-value | Tabulated text | Dot plot | Identification of chains of ordered gene pairs by searching paths in directed acyclic graph | Use of dynamic programming making it fast and highly reliable. Many softwares are based on this algorithm | No | 2 | Short | C++, Perl | Command line interface, Graphical user interface | [165] |
| ColinearScan | Any type of genetic markers (physical or genetic distance between markers, gene numbers) | Tabulated text with syntenic blocks and associated p-value | None | Dynamic programming algorithm based on the Smith-Waterman algorithm | Statistical inference, high computational efficiency, and flexibility of input data types | No | 2 | Not available | C++, Perl | Command line interface | [167] |
| CYNTENATOR | Sequences or alignments and an annotation file | Text file gathering alignments | None | Profile-profile alignment setting, which is an extension of the Waterman-Eggert algorithm | Implementing a phylogenetic scoring function | - | N | Complete | C++ | Command line interface | [159] |
| FISH | List of the linear order and orientation of features on each contig andlist of the pairwise homologies between features | Text file results | Dot Plot | Dynamic programming algorithm based on the Smith-Waterman algorithm | Modeling of the probability of observing segmental homologies assumed by chance and taking this model into account to parameterize the algorithm and the statistical evaluation of its output | Yes | 2 | Not available | C++ | Command line interface | [162] |
| DRIMM-Synteny | Set of anchors (e.g., local alignments or pairs of similar genes) | Text file where each genome is represented as a shuffled sequence of the syntenic blocks | Dot Plot | Construction of A-Bruijn graph | Graph-based algorithm allowing to identify non-overlapping syntenic blocks | No | N | Not available | C# | Command line interface | [168] |
| DiagHunter | BLAST output | Two text files containing gene names and/or coordinates | Dot Plot | Homology matrix based algorithm | Typical implementation of the colinearity strategy. Identifies large-scale syntenic blocks despite high levels of background noise | No | 2 | Short | Perl, and requires the BioPerl and GD.pm modules | Command line interface | [161] |
| OSfinder | Genomic locations of anchor or BLASTP results | Genomic locations of chains and orthologous segments | Dot Plot and a synteny map | Machine Learning and Markov Chains | Use Markov chain models and machine learning techniques. Automatically optimizes the parameters used in the Markov chain models. Scoring scheme based on stochastic models | Yes | N | Complete | C++ | Command line interface | [175] |
| SyMap | Genome sequences in FASTA format and associated GFF files | Homologous genes, diagonals, and identified syntenic blocks. | Visualization available and interactive | DAGChainer | Interactive visualizations. Calculates synonymous and nonsynonymous mutation rates for syntenic gene pairs using CodeML of the PAML package | No | N | Complete | No requirements | Web user interface | [163] |
| Cinteny | Information about markers and the homologous groups. | Tabulated text | Three interactive visualizations Whole Genome Synteny, Chromosome Level Synteny, Synteny Around a Marker | Ternary search trees (TST) | On-the-fly computations allowing fast parameters adjustments | Yes | N | Complete | No requirements | Web user interface | [164] |
| MultiSyn | Protein sequences in FASTA format and genome annotation in BED | Output files from MCScanX | Multiple synteny plots | MCScanX | Efficient tool for non-programming skilled users. Precomputed data for 18 plant genomes | No | N | Not available | No requirement | Web user interface | [176] |
| OrthoCluster | Genome file and a file storing orthologous relationships among genes in all input genomes | Cluster file, with all the syntenic blocks detected, Stat file with information related to the size distribution of the syntenic blocks | One associated plot | Depth-first search method, can also use Cinteny or SyMap | Fast and easy to use. Can be applied using any types of markers as an input as long as their relationships can be established | Yes | N | Complete | C++ | Web user interface, Command Line | [177] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lallemand, T.; Leduc, M.; Landès, C.; Rizzon, C.; Lerat, E. An Overview of Duplicated Gene Detection Methods: Why the Duplication Mechanism Has to Be Accounted for in Their Choice. Genes 2020, 11, 1046. https://doi.org/10.3390/genes11091046
Lallemand T, Leduc M, Landès C, Rizzon C, Lerat E. An Overview of Duplicated Gene Detection Methods: Why the Duplication Mechanism Has to Be Accounted for in Their Choice. Genes. 2020; 11(9):1046. https://doi.org/10.3390/genes11091046
Chicago/Turabian StyleLallemand, Tanguy, Martin Leduc, Claudine Landès, Carène Rizzon, and Emmanuelle Lerat. 2020. "An Overview of Duplicated Gene Detection Methods: Why the Duplication Mechanism Has to Be Accounted for in Their Choice" Genes 11, no. 9: 1046. https://doi.org/10.3390/genes11091046
APA StyleLallemand, T., Leduc, M., Landès, C., Rizzon, C., & Lerat, E. (2020). An Overview of Duplicated Gene Detection Methods: Why the Duplication Mechanism Has to Be Accounted for in Their Choice. Genes, 11(9), 1046. https://doi.org/10.3390/genes11091046