Genetic Admixture in the Population of Wild Apple (Malus sieversii) from the Tien Shan Mountains, Kazakhstan



Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and DNA Isolation
2.2. Nuclear and Chloroplast Microsatellite Genotyping
2.3. Data Analysis
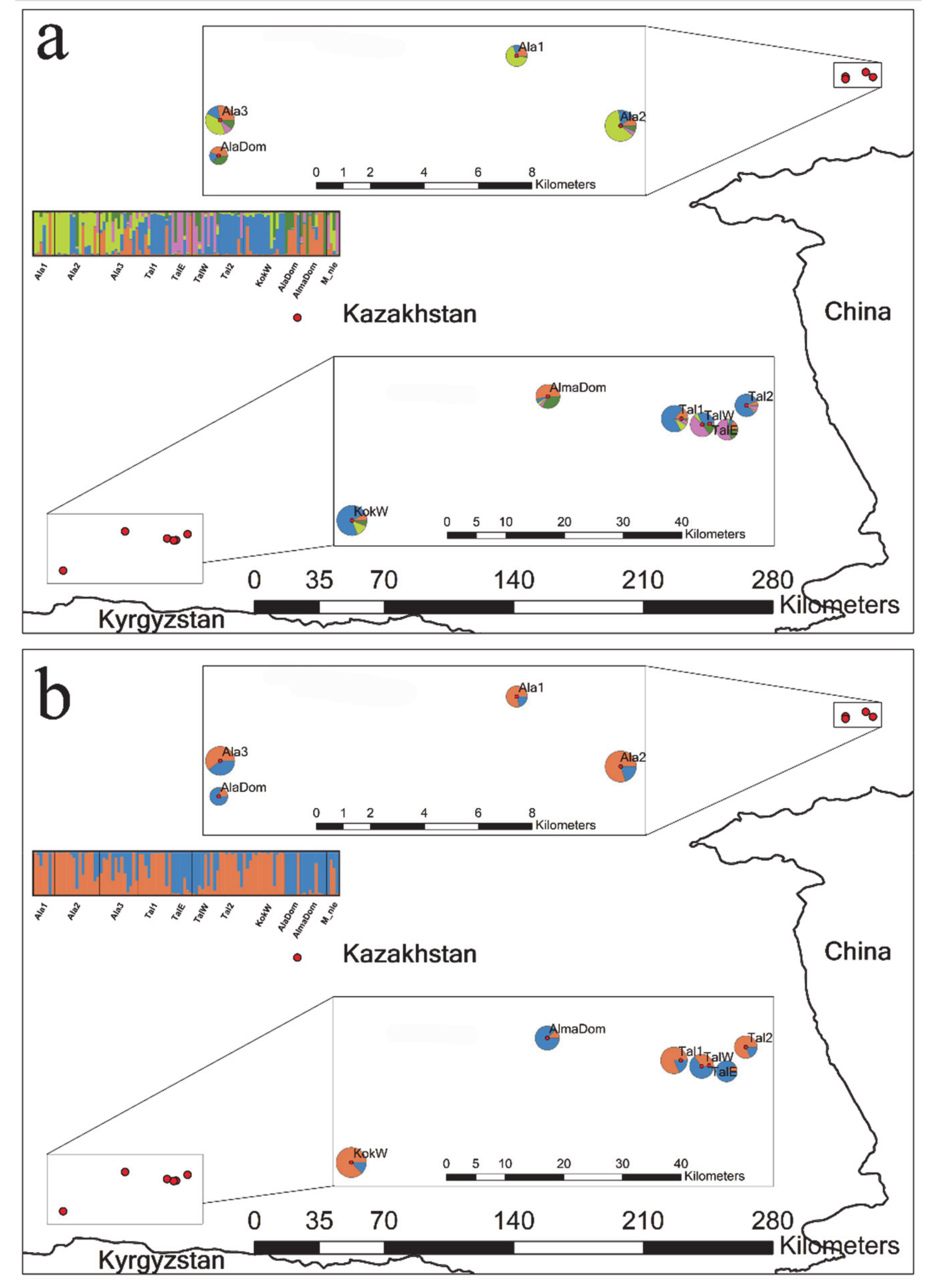
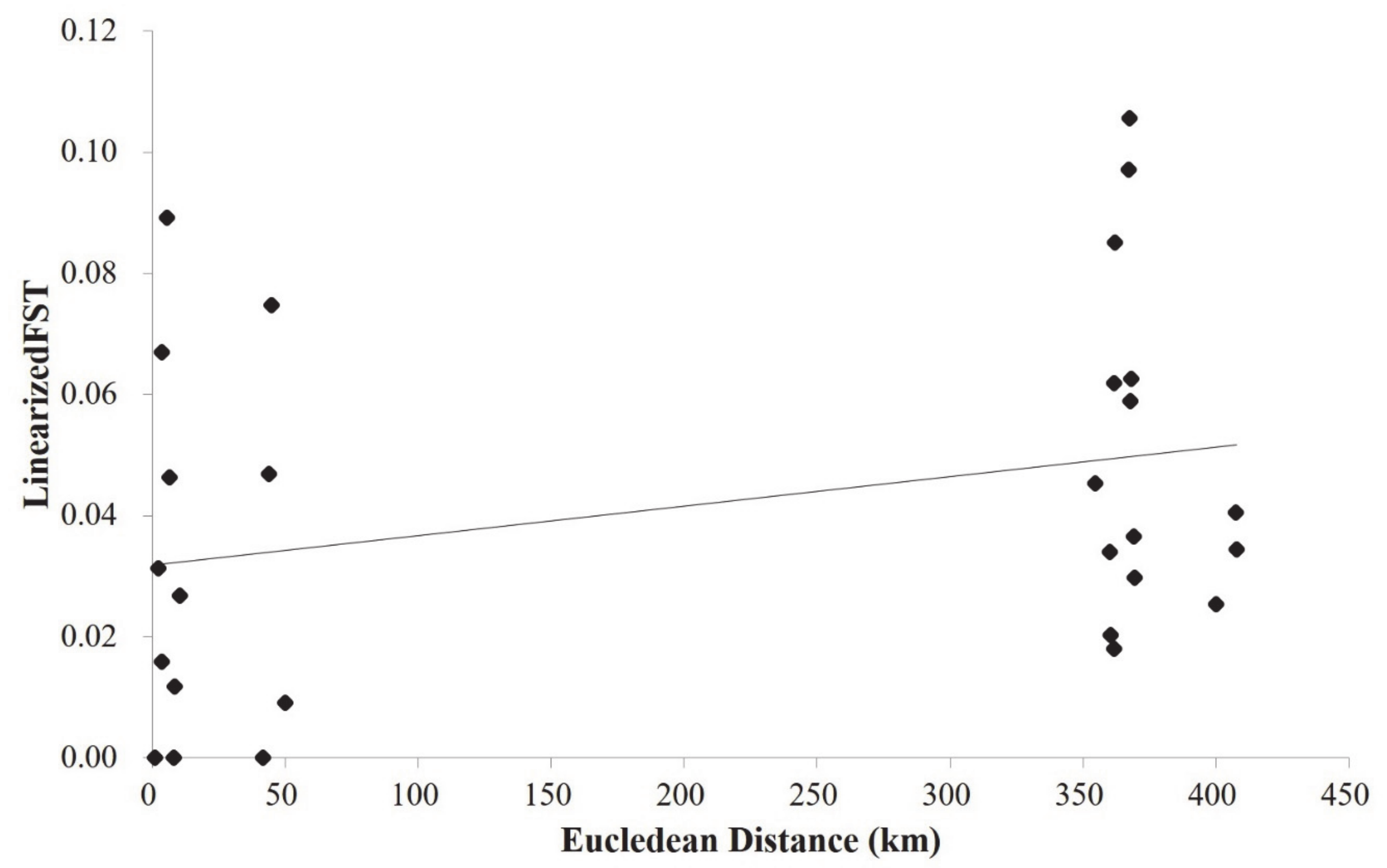
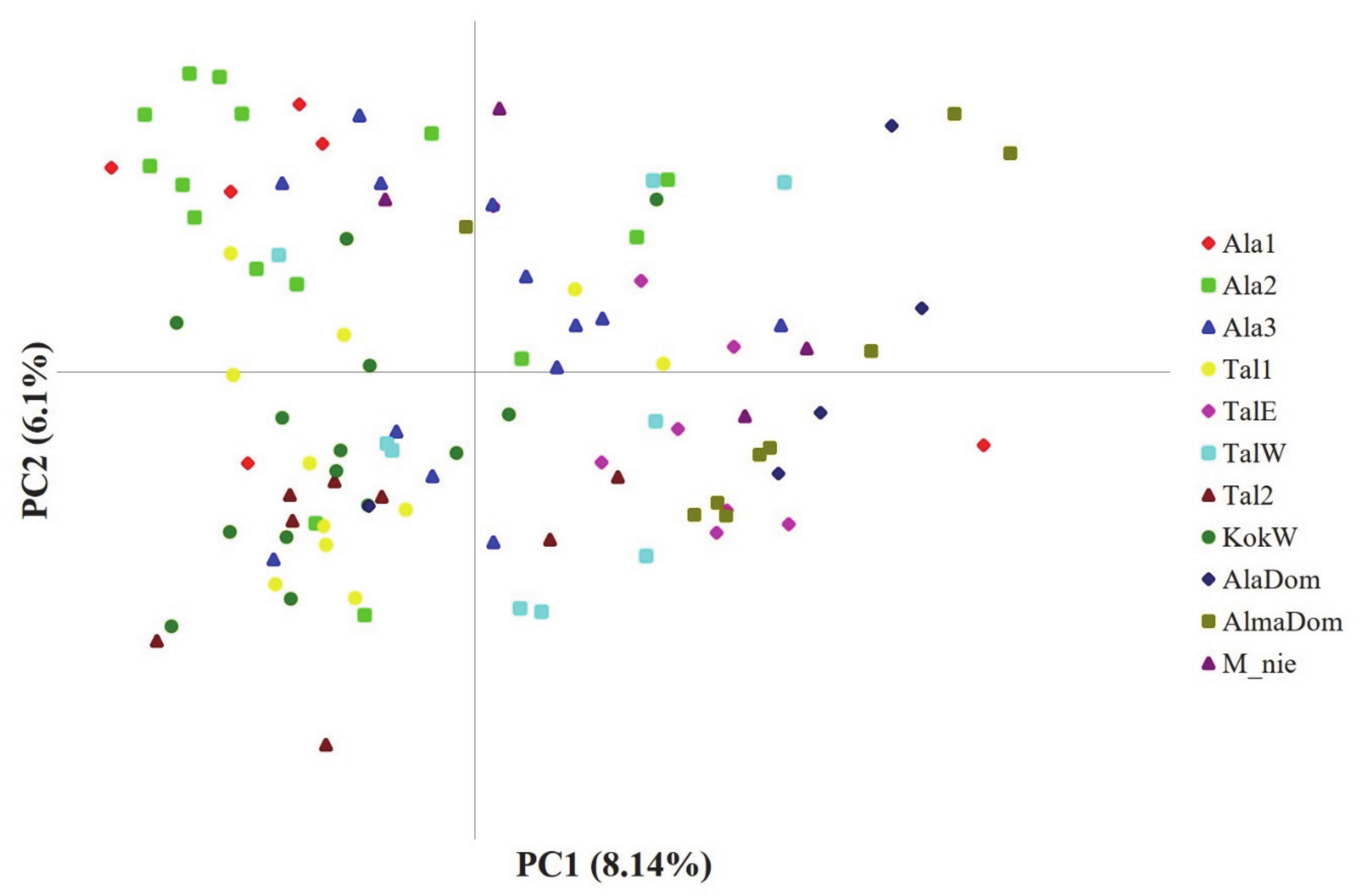
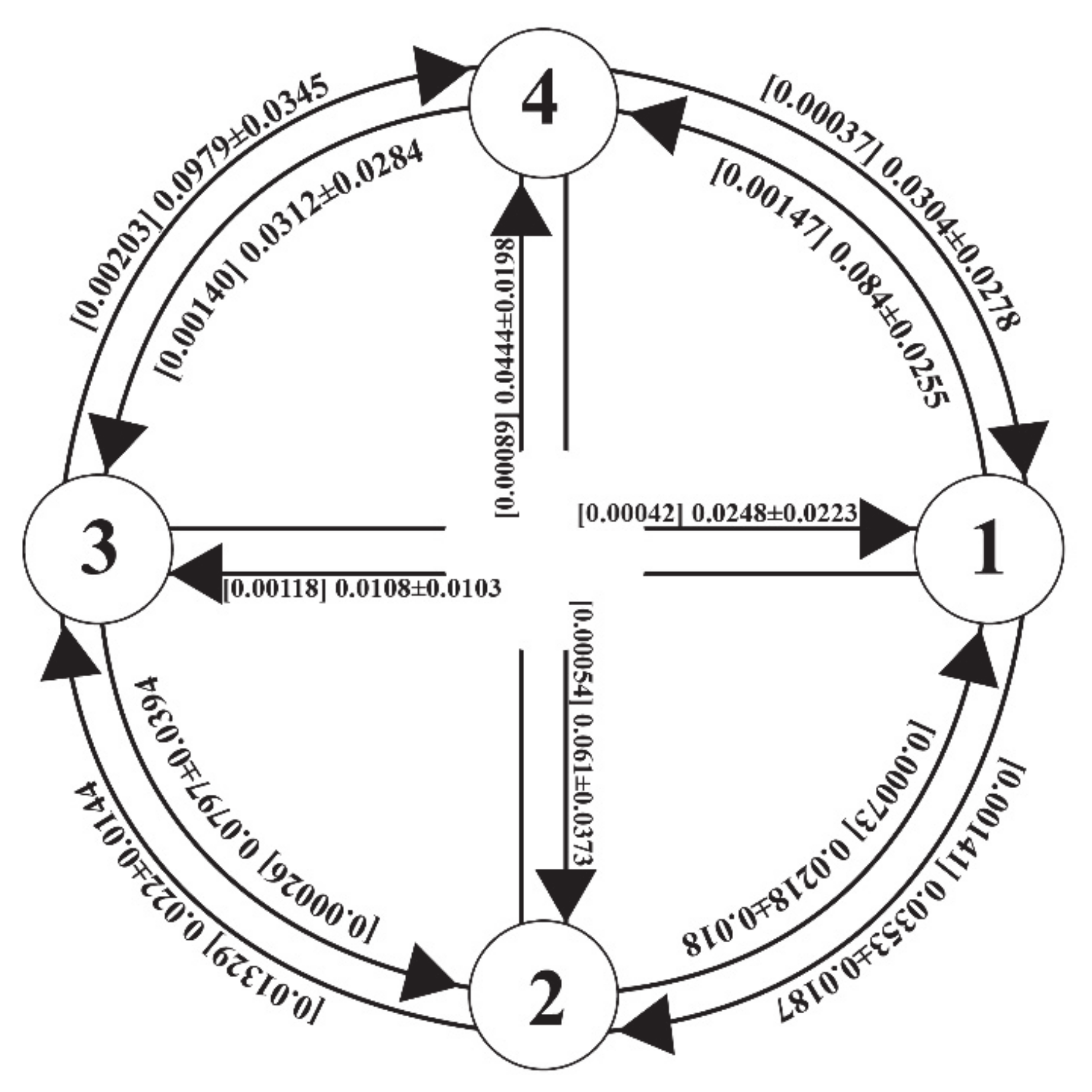
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rieseberg, L.H.; Zona, S.; Aberbom, L.; Martin, T.D. Hybridization in the Island Endemic, Catalina Mahogany. Conserv. Biol. 1989, 3, 52–58. [Google Scholar] [CrossRef]
- Rhymer, J.M.; Simberloff, D. Extinction by hybridization and introgression. Annu. Rev. Ecol. Syst. 1996, 27, 83–109. [Google Scholar] [CrossRef]
- Allendorf, F.W.; Leary, R.F.; Spruell, P.; Wenburg, J.K. The problems with hybrids: Setting conservation guidelines. Trends Ecol. Evol. 2001, 16, 613–622. [Google Scholar] [CrossRef]
- Taillebois, L.; Sabatino, S.; Manicki, A.; Daverat, F.; Nachón, D.J.; Lepais, O. Variable outcomes of hybridization between declining Alosa alosa and Alosa fallax. Evol. Appl. 2019, 13, 636–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todesco, M.; Pascual, M.A.; Owens, G.L.; Ostevik, K.L.; Moyers, B.T.; Hübner, S.; Heredia, S.M.; Hahn, M.A.; Caseys, C.; Bock, D.G.; et al. Hybridization and extinction. Evol. Appl. 2016, 9, 892–908. [Google Scholar] [CrossRef]
- Kareiva, P.; Watts, S.; McDonald, R.; Boucher, T. Domesticated nature: Shaping landscapes and ecosystems for human welfare. Science 2007, 316, 1866–1869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellstrand, N.C.; Prentice, H.C.; Hancock, J.F. Gene Flow and Introgression from Domesticated Plants into Their Wild Relatives. Annu. Rev. Ecol. Syst. 1999, 30, 539–563. [Google Scholar] [CrossRef]
- Cornille, A.; Gladieux, P.; Giraud, T. Crop-to-wild gene flow and spatial genetic structure in the closest wild relatives of the cultivated apple. Evol. Appl. 2013, 6, 737–748. [Google Scholar] [CrossRef]
- Macková, L.; Vít, P.; Ďurišová, Ľ.; Eliáš, P.; Urfus, T. Hybridization success is largely limited to homoploid Prunus hybrids: A multidisciplinary approach. Plant Syst. Evol. 2017, 303, 481–495. [Google Scholar] [CrossRef]
- Robinson, J.P.; Harris, S.A.; Juniper, B.E. Taxonomy of the genus Malus mill. (Rosaceae) with emphasis on the cultivated apple, Malus domestica Borkh. Plant Syst. Evol. 2001, 226, 35–58. [Google Scholar] [CrossRef]
- Phipps, J.B.; Robertson, K.R.; Smith, P.G.; Rohrer, J.R. A checklist of the subfamily Maloideae (Rosaceae). Can. J. Bot. 1990, 68, 2209–2269. [Google Scholar] [CrossRef]
- Coart, E.; Vekemans, X.; Smulders, M.J.M.; Wagner, I.; Van Huylenbroeck, J.; Van Bockstaele, E.; Roldán-Ruiz, I. Genetic variation in the endangered wild apple (Malus sylvestris (L.) Mill.) in Belgium as revealed by amplified fragment length polymorphism and microsatellite markers. Mol. Ecol. 2003, 12, 845–857. [Google Scholar] [CrossRef] [PubMed]
- Coart, E.; Van Glabeke, S.; De Loose, M.; Larsen, A.S.; Roldán-Ruiz, I. Chloroplast diversity in the genus Malus: New insights into the relationship between the European wild apple (Malus sylvestris (L.) Mill.) and the domesticated apple (Malus domestica Borkh.). Mol. Ecol. 2006, 15, 2171–2182. [Google Scholar] [CrossRef] [PubMed]
- Cornille, A.; Gladieux, P.; Smulders, M.J.M.; Roldán-Ruiz, I.; Laurens, F.; Le Cam, B.; Nersesyan, A.; Clavel, J.; Olonova, M.; Feugey, L.; et al. New insight into the history of domesticated apple: Secondary contribution of the European wild apple to the genome of cultivated varieties. PLoS Genet. 2012, 8, e1002703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornille, A.; Giraud, T.; Smulders, M.J.M.; Roldán-Ruiz, I.; Gladieux, P. The domestication and evolutionary ecology of apples. Trends Genet. 2014, 30, 57–65. [Google Scholar] [CrossRef]
- Duan, N.; Bai, Y.; Sun, H.; Wang, N.; Ma, Y.; Li, M.; Wang, X.; Jiao, C.; Legall, N.; Mao, L.; et al. Genome re-sequencing reveals the history of apple and supports a two-stage model for fruit enlargement. Nat. Commun. 2017, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Vavilov, N.I.; Vavylov, M.I.; Vavílov, N.Í.; Dorofeev, V.F. Origin and Geography of Cultivated Plants; Cambridge University Press: Cambridge, UK, 1992. [Google Scholar]
- Harris, S.A.; Robinson, J.P.; Juniper, B.E. Genetic clues to the origin of the apple. Trends Genet. 2002, 18, 426–430. [Google Scholar] [CrossRef]
- Velasco, R.; Zharkikh, A.; Affourtit, J.; Dhingra, A.; Cestaro, A.; Kalyanaraman, A.; Fontana, P.; Bhatnagar, S.K.; Troggio, M.; Pruss, D.; et al. The genome of the domesticated apple (Malus × domestica Borkh.). Nat. Genet. 2010, 42, 833–839. [Google Scholar] [CrossRef]
- Janick, J. Horticultural Reviews: Wild Apple and Fruit Trees of Central Asia; Janick, J., Ed.; John Wiley & Sons: New York, NY, USA, 2003. [Google Scholar]
- Cornille, A.; Antolín, F.; Garcia, E.; Vernesi, C.; Fietta, A.; Brinkkemper, O.; Kirleis, W.; Schlumbaum, A.; Roldán-Ruiz, I. A Multifaceted Overview of Apple Tree Domestication. Trends Plant Sci. 2019, 24, 770–782. [Google Scholar] [CrossRef] [Green Version]
- Lu, L.; Gu, C.; Li, C.; Alexander, C.; Bartholomew, B.; Brach, A.R.; Boufford, D.E.; Ikeda, H.; Ohba, H.; Robertson, K.R.; et al. Rosaceae. In Flora of China; Wu, Z., Raven, P.H., Hong, D., Eds.; Science Press: Beijing, China, 2003; pp. 46–434. [Google Scholar]
- Ma, X.; Cai, Z.; Liu, W.; Ge, S.; Tang, L. Identification, genealogical structure and population genetics of S-alleles in Malus sieversii, the wild ancestor of domesticated apple. Heredity 2017, 119, 185–196. [Google Scholar] [CrossRef]
- Omasheva, M.Y.; Flachowsky, H.; Ryabushkina, N.A.; Pozharskiy, A.S.; Galiakparov, N.N.; Hanke, M.V. To what extent do wild apples in Kazakhstan retain their genetic integrity? Tree Genet. Genomes 2017, 13, 52. [Google Scholar] [CrossRef]
- Gross, B.L.; Henk, A.D.; Forsline, P.L.; Richards, C.M.; Volk, G.M. Identification of interspecific hybrids among domesticated apple and its wild relatives. Tree Genet. Genomes 2012, 8, 1223–1235. [Google Scholar] [CrossRef]
- Gompert, Z.; Buerkle, C.A. What, if anything, are hybrids: Enduring truths and challenges associated with population structure and gene flow. Evol. Appl. 2016, 9, 909–923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Payseur, B.A.; Rieseberg, L.H. A genomic perspective on hybridization and speciation. Mol. Ecol. 2016, 25, 2337–2360. [Google Scholar] [CrossRef] [Green Version]
- Volk, G.M.; Henk, A.D.; Richards, C.M.; Forsline, P.L.; Thomas Chao, C. Malus sieversii: A diverse central asian apple species in the USDA-ARS national plant germplasm system. HortScience 2013, 48, 1440–1444. [Google Scholar] [CrossRef] [Green Version]
- Richards, C.M.; Volk, G.M.; Reilley, A.A.; Henk, A.D.; Lockwood, D.R.; Reeves, P.A.; Forsline, P.L. Genetic diversity and population structure in Malus sieversii, a wild progenitor species of domesticated apple. Tree Genet. Genomes 2009, 5, 339–347. [Google Scholar] [CrossRef]
- Luby, J.; Forsline, P.; Aldwinckle, H.; North, H.; Zealand, N.; Geibel, M. Silk Road Apples—Collection, Evaluation, and Utilization of Malus sieversii from Central Asia. HortScience 2001, 36, 225–231. [Google Scholar] [CrossRef] [Green Version]
- Faramarzi, S.; Yadollahi, A.; Soltani, B.M. Preliminary evaluation of genetic diversity among Iranian red fleshed apples using microsatellite markers. J. Agric. Sci. Technol. 2014, 16, 373–384. [Google Scholar]
- Yan, D.; Jun, Z.; Ya-chao, R.; Zhi-xiao, H. Study on Genetic Diversitiy of Natural Population Malus Sieversii with Microsatellite. J. Plant Genet. Res. 2013, 14, 771–777. [Google Scholar]
- Silfverberg-Dilworth, E.; Matasci, C.L.; Van De Weg, W.E.; Van Kaauwen, M.P.W.; Walser, M.; Kodde, L.P.; Soglio, V.; Gianfranceschi, L.; Durel, C.E.; Costa, F.; et al. Microsatellite markers spanning the apple (Malus × domestica Borkh.) genome. Tree Genet. Genomes 2006, 2, 202–224. [Google Scholar] [CrossRef] [Green Version]
- Chybicki, I.J.; Burczyk, J. Simultaneous estimation of null alleles and inbreeding coefficients. J. Hered. 2009, 100, 106–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meirmans, P.G.; Van Tienderen, P.H. GENOTYPE and GENODIVE: Two programs for the analysis of genetic diversity of asexual organisms. Mol. Ecol. Notes 2004, 4, 792–794. [Google Scholar] [CrossRef]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 2006, 6, 288–295. [Google Scholar] [CrossRef]
- Kalinowski, S.T. hp-rare 1.0: A computer program for performing rarefaction on measures of allelic richness. Mol. Ecol. Notes 2005, 5, 187–189. [Google Scholar] [CrossRef]
- Guo, S.W.; Thompson, E.A. Performing the exact test of Hardy-Weinberg proportion for multiple alleles. Biometrics 1992, 361–372. [Google Scholar] [CrossRef]
- Rousset, F. Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics 1997, 145, 1219–1228. [Google Scholar] [CrossRef]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics 2003, 164, 1567–1587. [Google Scholar]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [Green Version]
- Earl, D.A. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Jakobsson, M.; Rosenberg, N.A. CLUMPP: A cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 2007, 23, 1801–1806. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, N.A. DISTRUCT: A program for the graphical display of population structure. Mol. Ecol. Notes 2004, 4, 137–138. [Google Scholar] [CrossRef]
- Beerli, P.; Palczewski, M. Unified framework to evaluate panmixia and migration direction among multiple sampling locations. Genetics 2010, 185, 313–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beerli, P.; Felsenstein, J. Maximum likelihood estimation of a migration matrix and effective population sizes in n subpopulations by using a coalescent approach. Proc. Natl. Acad. Sci. USA 2001, 98, 4563–4568. [Google Scholar] [CrossRef] [Green Version]
- Wilson, G.A.; Rannala, B. Bayesian inference of recent migration rates using multilocus genotypes. Genetics 2003, 163, 1177–1191. [Google Scholar] [PubMed]
- Chiucchi, J.E.; Gibbs, H.L. Similarity of contemporary and historical gene flow among highly fragmented populations of an endangered rattlesnake. Mol. Ecol. 2010, 19, 5345–5358. [Google Scholar] [CrossRef]
- Marriage, T.N.; Hudman, S.; Mort, M.E.; Orive, M.E.; Shaw, R.G.; Kelly, J.K. Direct estimation of the mutation rate at dinucleotide microsatellite loci in Arabidopsis thaliana (Brassicaceae). Heredity 2009, 103, 310–317. [Google Scholar] [CrossRef] [Green Version]
- Team R. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2016; Available online: https://www.R-project.org (accessed on 28 December 2020).
- Cornuet, J.M.; Luikart, G. Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 1996, 144, 2001–2014. [Google Scholar] [CrossRef]
- Garza, J.C.; Williamson, E.G. Detection of reduction in population size using data from microsatellite loci. Mol. Ecol. 2001, 10, 305–318. [Google Scholar] [CrossRef]
- Williamson-Natesan, E.G. Comparison of methods for detecting bottlenecks from microsatellite loci. Conserv. Genet. 2005, 6, 551–562. [Google Scholar] [CrossRef]
- Piry, S.; Luikart, G.; Cornuet, J.M. BOTTLENECK: A computer program for detecting recent reductions in the effective population size using allele frequency data. J. Hered. 1999, 90, 502–503. [Google Scholar] [CrossRef]
- Hsieh, Y.C.; Chung, J.D.; Wang, C.N.; Chang, C.T.; Chen, C.Y.; Hwang, S.-Y. Historical connectivity, contemporary isolation and local adaptation in a widespread but discontinuously distributed species endemic to Taiwan, Rhododendron oldhamii (Ericaceae). Heredity 2013, 111, 147–156. [Google Scholar] [CrossRef] [Green Version]
- Sampson, J.F.; Byrne, M.; Yates, C.J.; Gibson, N.; Thavornkanlapachai, R.; Stankowski, S.; MacDonald, B.; Bennett, I. Contemporary pollen-mediated gene immigration reflects the historical isolation of a rare, animal-pollinated shrub in a fragmented landscape. Heredity 2014, 112, 172–181. [Google Scholar] [CrossRef] [Green Version]
- Fu, P.-C.; Gao, Q.-B.; Zhang, F.-Q.; Xing, R.; Wang, J.-L.; Liu, H.-R.; Chen, S.-L. Gene flow results in high genetic similarity between Sibiraea (Rosaceae) species in the Qinghai-Tibetan Plateau. Front. Plant Sci. 2016, 7, 1596. [Google Scholar] [CrossRef] [Green Version]
- Ellstrand, N.C. Gene Flow by Pollen: Implications for Plant Conservation Genetics. Oikos 1992, 63, 77. [Google Scholar] [CrossRef]
- Sagnard, F.; Deu, M.; Dembélé, D.; Leblois, R.; Touré, L.; Diakité, M.; Calatayud, C.; Vaksmann, M.; Bouchet, S.; Mallé, Y.; et al. Genetic diversity, structure, gene flow and evolutionary relationships within the Sorghum bicolor wild-weedy-crop complex in a western African region. Theor. Appl. Genet. 2011, 123, 1231–1246. [Google Scholar] [CrossRef] [Green Version]
- Jin, X.; Chen, Y.; Liu, P.; Li, C.; Cai, X.; Rong, J.; Lu, B.R. Introgression from cultivated rice alters genetic structures of wild relative populations: Implications for in situ conservation. AoB Plants 2018, 10, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Flowers, J.M.; Hazzouri, K.M.; Gros-Balthazard, M.; Mo, Z.; Koutroumpa, K.; Perrakis, A.; Ferrand, S.; Khierallah, H.S.M.; Fuller, D.Q.; Aberlenc, F.; et al. Cross-species hybridization and the origin of North African date palms. Proc. Natl. Acad. Sci. USA 2019, 116, 1651–1658. [Google Scholar] [CrossRef] [Green Version]
- Hamrick, J.L.; Godt, M.J.W. Effects of life history traits on genetic diversity in plant species. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 1996, 351, 1291–1298. [Google Scholar]
- Olson, M.S.; Hamrick, J.L.; Moore, R. Breeding systems, mating systems, and genomics of gender determination in angiosperm trees. In Comparative and Evolutionary Genomics of Angiosperm Trees; Springer: Cham, Switzerland, 2016; pp. 139–158. [Google Scholar]
- Jordano, P.; Garcia, C.; Godoy, J.A.; García-Castaño, J.L. Differential contribution of frugivores to complex seed dispersal patterns. Proc. Natl. Acad. Sci. USA 2007, 104, 3278–3282. [Google Scholar] [CrossRef] [Green Version]
- Ellstrand, N.C. Current knowledge of gene flow in plants: Implications for transgene flow. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2003, 358, 1163–1170. [Google Scholar] [CrossRef] [Green Version]
- Duputié, A.; David, P.; Debain, C.; McKey, D. Natural hybridization between a clonally propagated crop, cassava (Manihot esculenta Crantz) and a wild relative in French Guiana. Mol. Ecol. 2007, 16, 3025–3038. [Google Scholar] [CrossRef]
- Delplancke, M.; Alvarez, N.; Espíndola, A.; Joly, H.; Benoit, L.; Brouck, E.; Arrigo, N. Gene flow among wild and domesticated almond species: Insights from chloroplast and nuclear markers. Evol. Appl. 2012, 5, 317–329. [Google Scholar] [CrossRef] [Green Version]
- Sekido, K.; Hayashi, Y.; Yamada, K.; Shiratake, K.; Matsumoto, S.; Maejima, T.; Komatsu, H. Efficient breeding system for red-fleshed apple based on linkage with S3-Rnase allele in “Pink Pearl”. HortScience 2010, 45, 534–537. [Google Scholar] [CrossRef] [Green Version]
- Wilson, B.; Mills, M.; Kulikov, M.; Clubbe, C. The future of walnut-fruit forests in Kyrgyzstan and the status of the iconic Endangered apple Malus niedzwetzkyana. Oryx 2019, 53, 415–423. [Google Scholar] [CrossRef] [Green Version]
- Whitlock, M.C.; McCauley, D.E. Some population genetic consequences of colony formation and extinction: Genetic correlations within founding groups. Evolution 1990, 44, 1717–1724. [Google Scholar] [CrossRef]
- Slatkin, M. Gene flow and the geographic structure of natural populations. Science 1987, 236, 787–792. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Region | Location | Abbreviation | Group Abbreviation | N | Lon | Lat | He (±sd) | Na (±se) |
|---|---|---|---|---|---|---|---|---|---|
| sieversii | East | Lepsi Rever side, Almaty, KAZ | Ala1 | Ala | 7 | 45.5-- | 80.6-- | 0.73 (0.14) | 4.12 (0.28) |
| East | Mt. Lepsy, Almaty, KAZ | Ala2 | Ala | 15 | 45.5-- | 80.7-- | 0.68 (0.23) | 3.99 (0.31) | |
| East | South-western Lepsy, Almaty, KAZ | Ala3 | Ala | 13 | 45.5-- | 80.5-- | 0.77 (0.16) | 4.42 (0.28) | |
| West | Mt. Lepsy (southern), KAZ | Tal1 | Tal1 | 11 | 45.5-- | 80.5-- | 0.75 (0.18) | 4.26 (0.28) | |
| West | Mt. Ryskulov (southern), Almaty, KAZ | TalE | TalEW | 7 | 43.2-- | 77.2-- | 0.75 (0.16) | 4.24 (0.29) | |
| West | South-western Orman (east Valley), Almaty, KAZ | TalW | TalEW | 9 | 43.2-- | 77.3-- | 0.74 (0.24) | 4.34 (0.34) | |
| West | South-western Orman (West valley), Almaty, KAZ | Tal2 | Tal2 | 8 | 43.2-- | 77.3-- | 0.71 (0.22) | 4.27 (0.34) | |
| West | Mt. Ryskulova (eastern), Almaty, KAZ | KokW | KokW | 14 | 43.2-- | 77.3-- | 0.76 (0.19) | 4.39 (0.30) | |
| domestica | - | Koklaisay, Almaty, KAZ | AlaDom | Dom | 5 | 43.1-- | 76.8-- | 0.78 (0.12) | 4.37 (0.27) |
| Belbulak, Almaty, KAZ | AlmaDom | Dom | 9 | 43.3-- | 77.0-- | 0.78 (0.09) | 4.33 (0.23) | ||
| niedzwetzkyana | - | Southern Bayzeren, Almaty, KAZ | M_nie | - | 4 | 45.6-- | 80.6-- | 0.77 (0.19) | 4.31 (0.38) |
| Ala1 | Ala2 | Ala3 | Tal1 | TalE | TalW | Tal2 | KokW | AlaDom | AlmaDom | M_nie | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Ala1 | 0.00 | ||||||||||
| Ala2 | 0.03 ns | 0.00 | |||||||||
| Ala3 | 0.01 ns | 0.04 | 0.00 | ||||||||
| Tal1 | 0.05 | 0.04 | 0.03 | 0.00 | |||||||
| TalE | 0.10 | 0.11 | 0.05 | 0.07 | 0.00 | ||||||
| TalW | 0.07 | 0.07 | 0.03 | 0.04 | 0.001 ns | 0.00 | |||||
| Tal2 | 0.07 | 0.09 | 0.05 | 0.02 ns | 0.09 | 0.05 | 0.00 | ||||
| KokW | 0.05 | 0.04 | 0.03 | 0.01 | 0.08 | 0.05 | 0.02 | 0.00 | |||
| AlaDom | 0.08 | 0.11 | 0.04 ns | 0.07 | 0.051 ns | 0.07 | 0.10 | 0.09 | 0.00 | ||
| AlmaDom | 0.10 | 0.12 | 0.07 | 0.09 | 0.05 | 0.09 | 0.12 | 0.09 | 0.02 ns | 0.00 | |
| M_nie | 0.08 | 0.07 | 0.05 ns | 0.07 | 0.03 ns | 0.05 ns | 0.13 | 0.07 | 0.06 ns | 0.07 | 0.00 |
| Source | Sum of Squares | Variance Components | Percentage of Variation | Fixation Index |
|---|---|---|---|---|
| Among groups (FCT) | 46.41 | 0.18 | 3.50 | 0.035 |
| Among populations within groups (FSC) | 56.74 | 0.12 | 2.29 | 0.024 |
| Among individuals within populations (FIS) | 527.85 | 0.95 | 18.31 | 0.194 |
| Within individuals (FIT) | 399.50 | 3.93 | 75.91 | 0.240 |
| Population | G-W Index (±sd) | P (Sign Test) | P (Wilcoxon Test) | Mode Shift | ||
|---|---|---|---|---|---|---|
| IAM | SMM | IAM | SMM | |||
| Ala1 | 0.22 (0.08) | 0.51 | 0.19 | 1.00 | 0.15 | no |
| Ala2 | 0.27 (0.10) | 0.10 | 0.01 | 0.74 | 0.01 | no |
| Ala3 | 0.25 (0.10) | 0.15 | 0.48 | 0.05 | 0.54 | no |
| Tal1 | 0.23 (0.08) | 0.05 | 0.43 | 0.09 | 1.00 | no |
| TalE | 0.19 (0.08) | 0.14 | 0.55 | 0.09 | 0.64 | no |
| TalW | 0.23 (0.11) | 0.31 | 0.12 | 0.24 | 0.24 | no |
| Tal2 | 0.21 (0.08) | 0.25 | 0.01 | 0.38 | 0.02 | no |
| KokW | 0.23 (0.08) | 0.01 | 0.04 | 0.02 | 0.15 | no |
| AlaDom | 0.20 (0.08) | 0.19 | 0.54 | 0.04 | 0.84 | yes |
| AlmaDom | 0.27 (0.11) | 0.06 | 0.26 | 0.00 | 0.95 | yes |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ha, Y.-H.; Oh, S.-H.; Lee, S.-R. Genetic Admixture in the Population of Wild Apple (Malus sieversii) from the Tien Shan Mountains, Kazakhstan. Genes 2021, 12, 104. https://doi.org/10.3390/genes12010104
Ha Y-H, Oh S-H, Lee S-R. Genetic Admixture in the Population of Wild Apple (Malus sieversii) from the Tien Shan Mountains, Kazakhstan. Genes. 2021; 12(1):104. https://doi.org/10.3390/genes12010104
Chicago/Turabian StyleHa, Young-Ho, Seung-Hwan Oh, and Soo-Rang Lee. 2021. "Genetic Admixture in the Population of Wild Apple (Malus sieversii) from the Tien Shan Mountains, Kazakhstan" Genes 12, no. 1: 104. https://doi.org/10.3390/genes12010104
APA StyleHa, Y. -H., Oh, S. -H., & Lee, S. -R. (2021). Genetic Admixture in the Population of Wild Apple (Malus sieversii) from the Tien Shan Mountains, Kazakhstan. Genes, 12(1), 104. https://doi.org/10.3390/genes12010104