Identification of Potential Risk Genes and the Immune Landscape of Idiopathic Pulmonary Arterial Hypertension via Microarray Gene Expression Dataset Reanalysis


Abstract
:1. Introduction
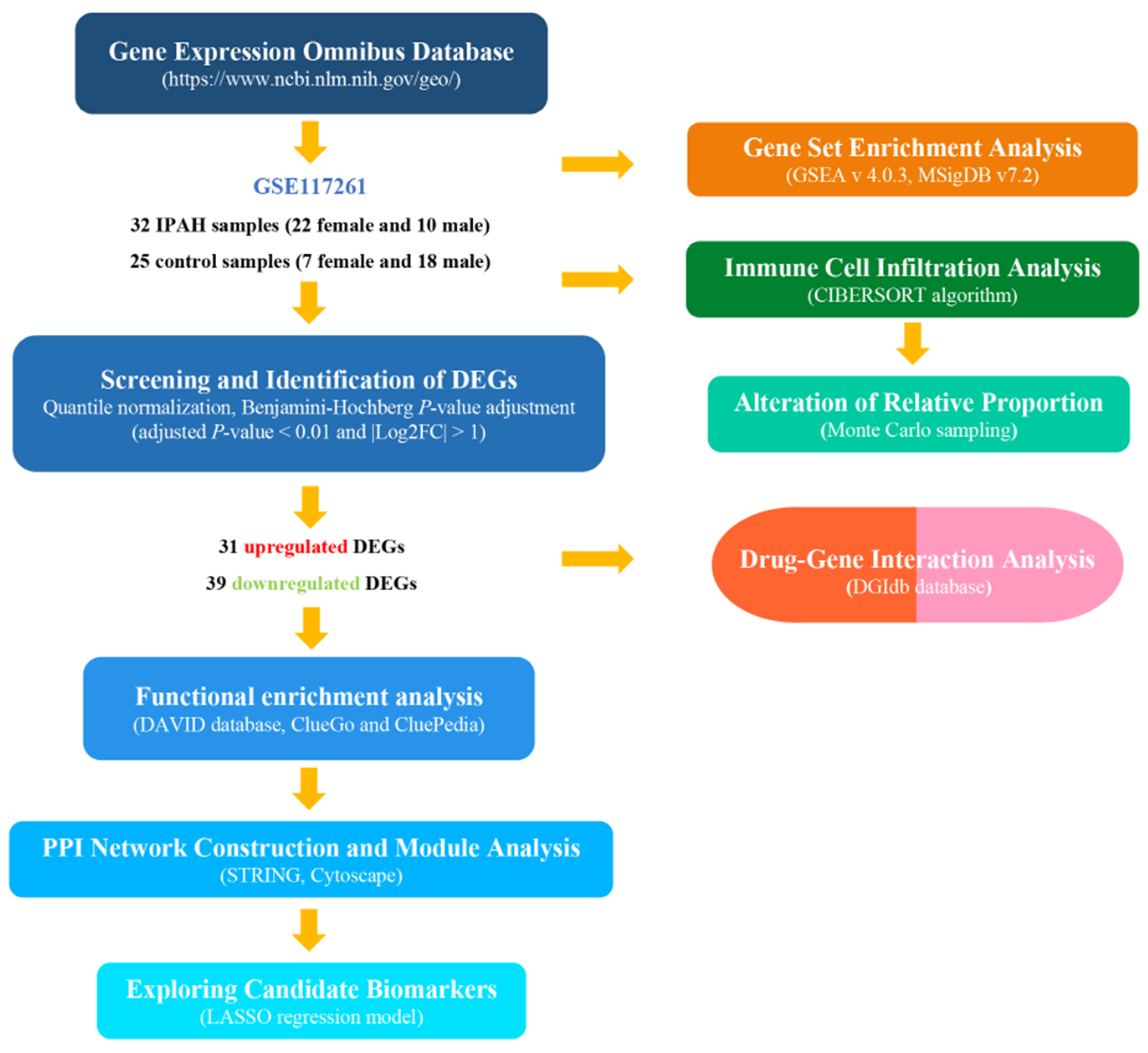
2. Materials and Methods
2.1. Data Resources
2.2. Screening and Identification of Differentially Expressed Genes
2.3. Functional Analysis of the Expression Profiles
2.4. Protein Interaction and Module Analysis
2.5. Evaluation of Immune Cell Infiltration
2.6. Prediction Model Analysis
2.7. Drug–Gene Interaction Analysis
3. Results
3.1. Screening and Identification of Differentially Expressed Genes between IPAH and Control Samples
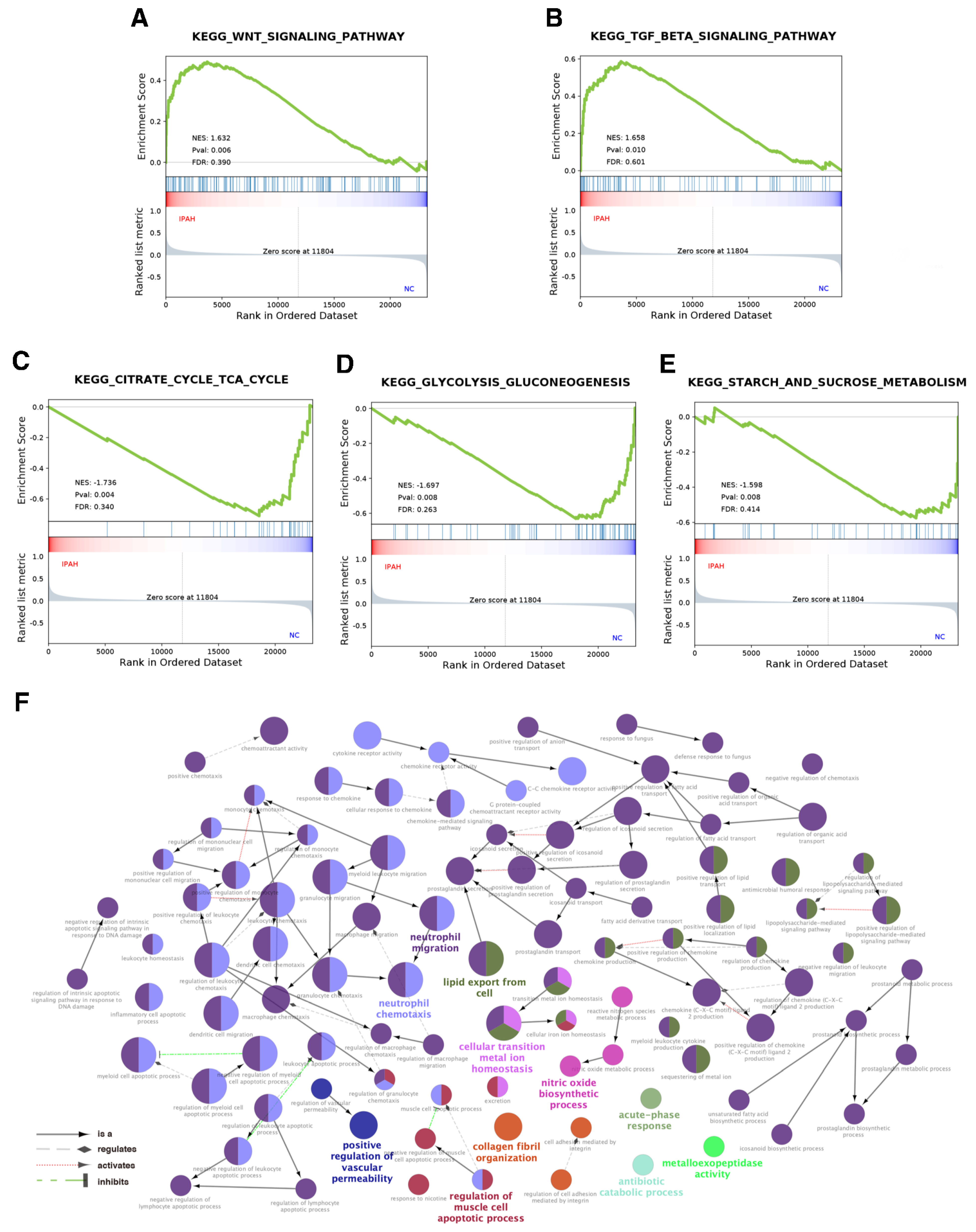
3.2. Functional Annotation and Enrichment of the Expression Profiles
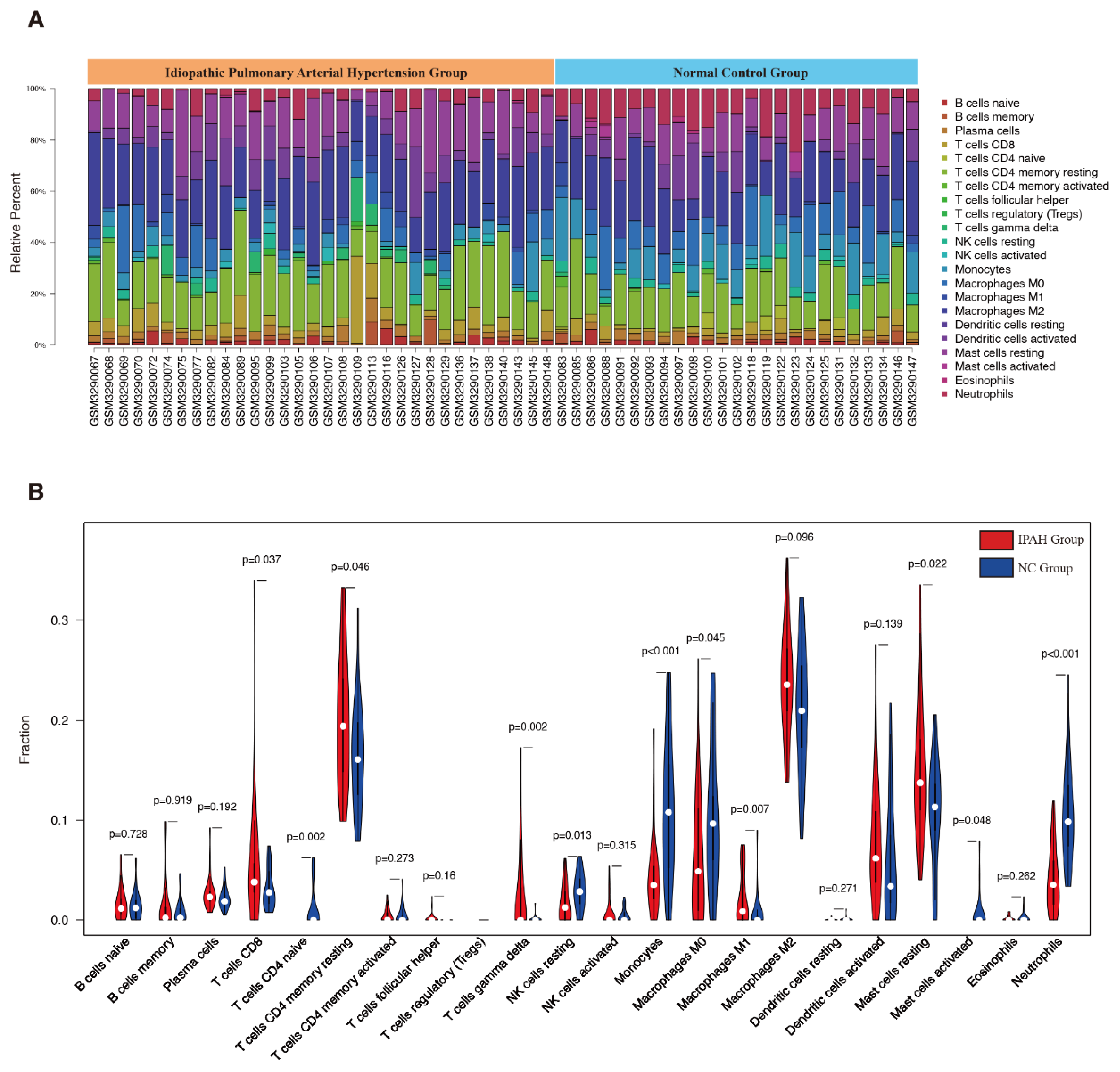
3.3. Evaluation of Immune Cell Infiltration
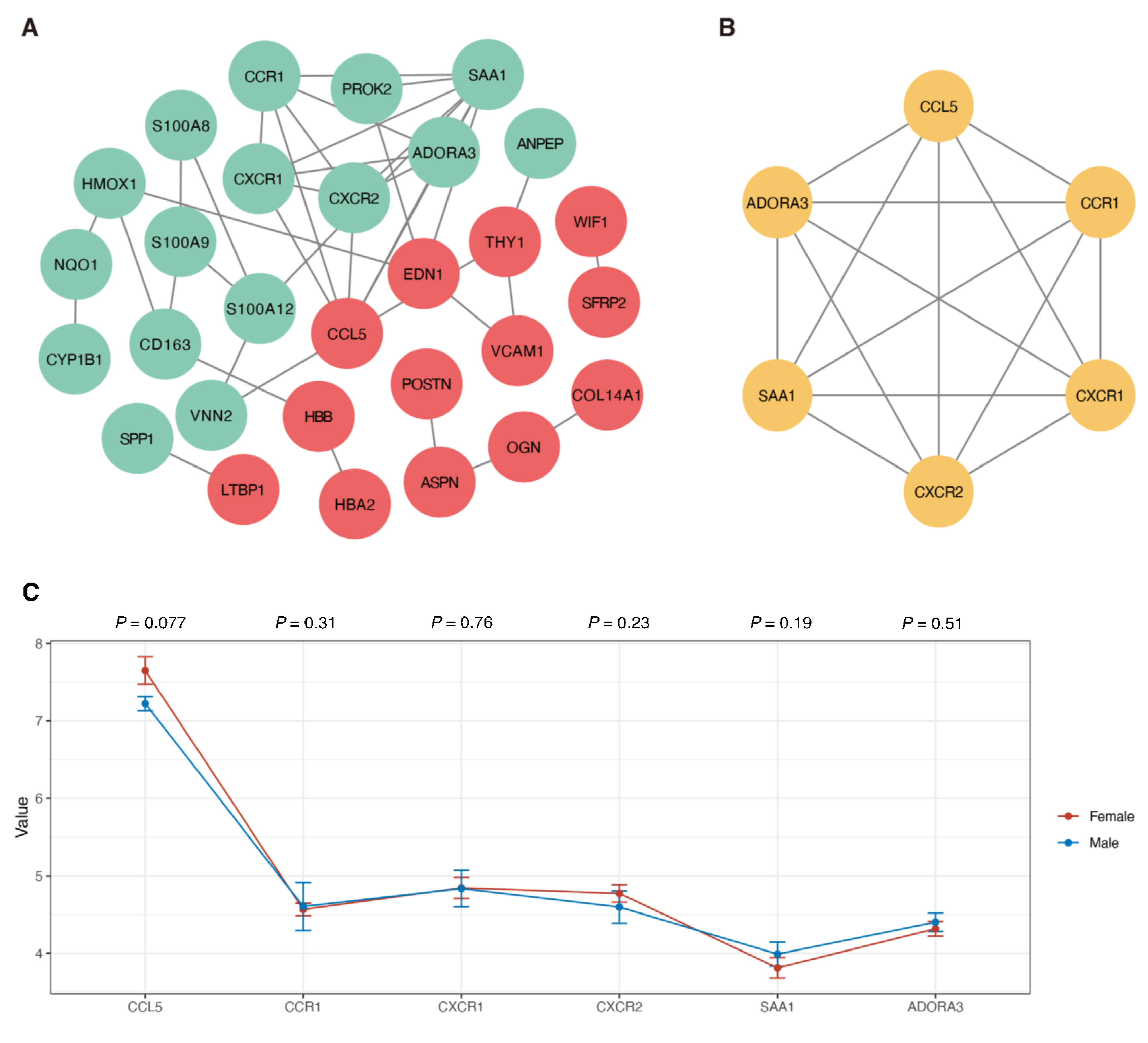
3.4. Protein Interaction and Module Analysis
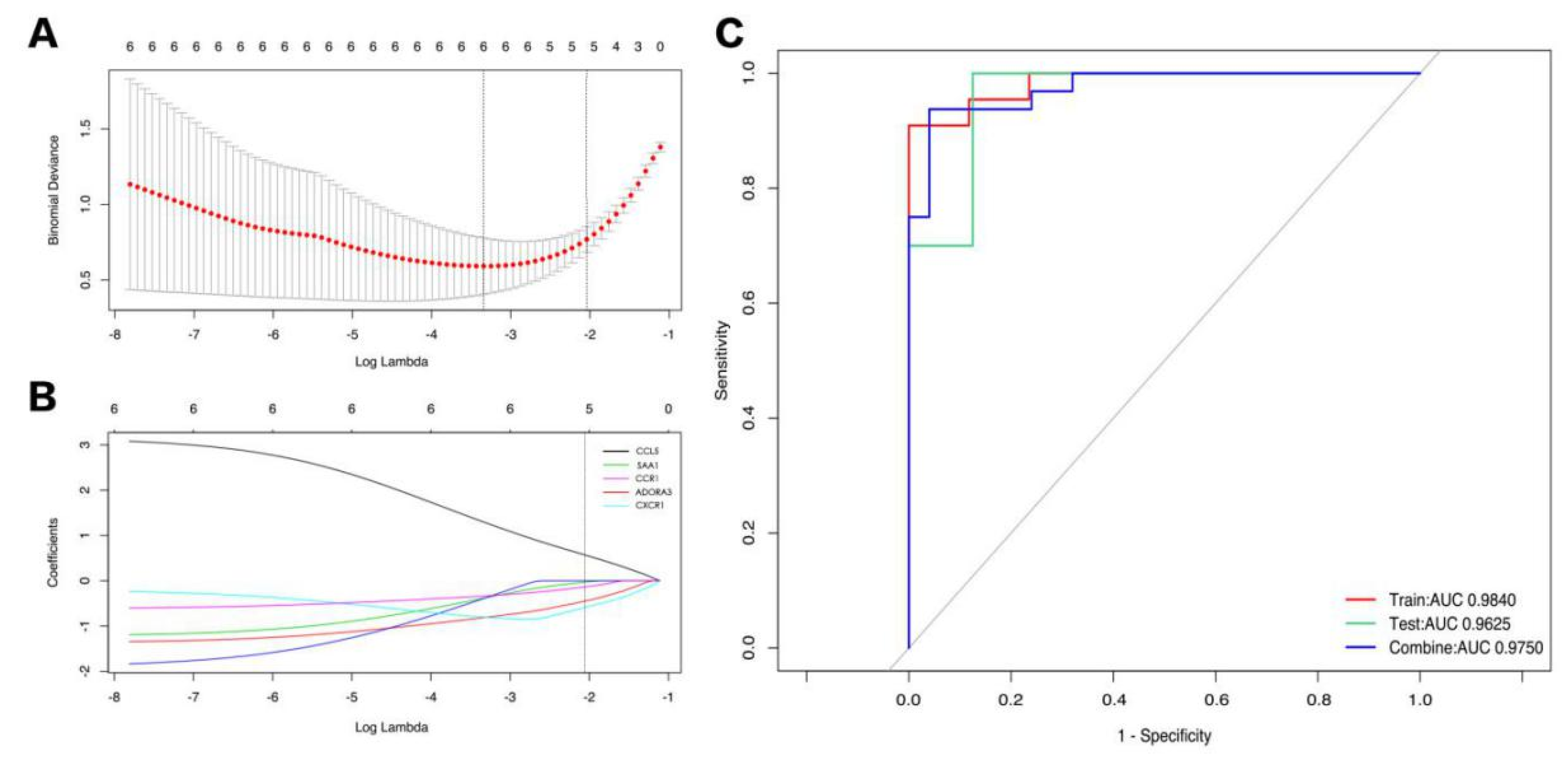
3.5. Exploring Candidate Biomarkers by Lasso Regression and Receiver Operating Characteristic Curves
3.6. Drug–Gene Interaction Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| IPAH | idiopathic pulmonary arterial hypertension; |
| PAH | pulmonary arterial hypertension; |
| DEGs | differentially expressed genes; |
| FC | fold change; |
| GO | gene ontology; |
| GEO | gene expression omnibus; |
| PPI | protein–protein interaction; |
| GSEA | gene set enrichment analysis; |
| MSigDB | molecular signatures database; |
| NES | normalized enrichment scores; |
| STRING | search tool for the retrieval of interacting genes/proteins; |
| MCODE | molecular complex detection; |
| DGIdb | drug–gene interaction database; |
| ROC | receiver operating characteristic; |
| AUC | area under the curve; |
| BP | biological process; |
| CC | cellular component; |
| MF | molecular function; |
| BMP | bone morphogenetic protein; |
| TGF-β | transforming growth factor-β. |
References
- Galie, N.; Humbert, M.; Vachiéry, J.-L.; Gibbs, S.; Lang, I.M.; Torbicki, A.; Simonneau, G.; Peacock, A.; Noordegraaf, A.V.; Beghetti, M.; et al. 2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension. Russ. J. Cardiol. 2016, 37, 5–64. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Lin, F.; Xiao, Z.; Sun, B.; Wei, Z.; Liu, B.; Xue, L.; Xiong, C. Investigational pharmacotherapy and immunotherapy of pulmonary arterial hypertension: An update. Biomed. Pharmacother. 2020, 129, 110355. [Google Scholar] [CrossRef] [PubMed]
- Thenappan, T.; Ormiston, M.L.; Ryan, J.J.; Archer, S.L. Pulmonary arterial hypertension: Pathogenesis and clinical management. BMJ 2018, 360, j5492. [Google Scholar] [CrossRef] [PubMed]
- Van Uden, D.; Boomars, K.; Kool, M. Dendritic cell subsets and effector function in idiopathic and connective tissue disease-associated pulmonary arterial hypertension. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Welch, C.L.; Chung, W.K. Genetics and other omics in pediatric pulmonary arterial hypertension. Chest 2020, 157, 1287–1295. [Google Scholar] [CrossRef]
- Wang, X.-J.; Xu, X.-Q.; Wu, Y.; Jiang, R.; Zhang, J.; Huang, W.; Wei, W.-B.; Yan, Y.; Li, J.-H.; Liu, Q.-Q.; et al. Association of rare PTGIS variants with susceptibility and pulmonary vascular response in patients with idiopathic pulmonary arterial hypertension. JAMA Cardiol. 2020, 5, 677–684. [Google Scholar] [CrossRef] [Green Version]
- Ping, P.; Hermjakob, H.; Polson, J.; Benos, P.; Wang, W. Biomedical informatics on the cloud: A treasure hunt for advancing cardiovascular medicine. Circ. Res. 2018, 122, 1290–1301. [Google Scholar] [CrossRef]
- Stearman, R.S.; Bui, Q.M.; Geraci, M.W.; Speyer, G.; Handen, A.; Cornelius, A.R.; Graham, B.; Kim, S.; Mickler, E.A.; Tuder, R.M.; et al. Systems analysis of the human pulmonary arterial hypertension lung transcriptome. Am. J. Respir. Cell Mol. Biol. 2019, 60, 637–649. [Google Scholar] [CrossRef]
- Jameson, J.; Havran, W. Skin gammadelta T-cell functions in homeostasis and wound healing. Immunol. Rev. 2007, 215, 114–122. [Google Scholar] [CrossRef]
- Barrett, T.; Wilhite, S.; Ledoux, P.; Evangelista, C.; Kim, I.; Tomashevsky, M.; Marshall, K.; Phillippy, K.; Sherman, P.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets--update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, M.; Cai, J.; Yuan, Y.; Shi, Y.; Wu, H.; Liu, Q.; Yao, Y.; Chen, L.; Dang, W.; Zhang, X.; et al. A four-gene signature-derived risk score for glioblastoma: Prospects for prognostic and response predictive analyses. Cancer Biol. Med. 2019, 16, 595–605. [Google Scholar] [PubMed]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.-H.; Pagès, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bindea, G.; Galon, J.; Mlecnik, B. CluePedia Cytoscape plugin: Pathway insights using integrated experimental and in silico data. Bioinformatics 2013, 29, 661–663. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Franceschini, A.; Szklarczyk, D.; Frankild, S.; Kuhn, M.; Simonovic, M.; Roth, A.; Lin, J.; Minguez, P.; Bork, P.; Von Mering, C.; et al. STRING v9.1: Protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2012, 41, D808–D815. [Google Scholar] [CrossRef] [Green Version]
- Benza, R.L.; Miller, D.P.; Barst, R.J.; Badesch, D.B.; Frost, A.E.; McGoon, M.D. An evaluation of long-term survival from time of diagnosis in pulmonary arterial hypertension from the reveal registry. Chest 2012, 142, 448–456. [Google Scholar] [CrossRef]
- Manes, A.; Palazzini, M.; Leci, E.; Reggiani, M.L.B.; Branzi, A.; Galie, N. Current era survival of patients with pulmonary arterial hypertension associated with congenital heart disease: A comparison between clinical subgroups. Eur. Heart J. 2014, 35, 716–724. [Google Scholar] [CrossRef] [Green Version]
- David, C.; Massagué, J. Contextual determinants of TGFβ action in development, immunity and cancer. Nature reviews. Mol. Cell. Biol. 2018, 19, 419–435. [Google Scholar]
- Ma, J.; Sanchez-Duffhues, G.; Goumans, M.-J.; Dijke, P.T. TGF-β;-induced endothelial to mesenchymal transition in disease and tissue engineering. Front. Cell Dev. Biol. 2020, 8, 260. [Google Scholar] [CrossRef]
- Soon, E.; Holmes, A.; Treacy, C.; Doughty, N.; Southgate, L.; Machado, R.; Trembath, R.; Jennings, S.; Barker, L.; Nicklin, P.; et al. Elevated levels of inflammatory cytokines predict survival in idiopathic and familial pulmonary arterial hypertension. Circulation 2010, 122, 920–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marsh, L.M.; Jandl, K.; Grünig, G.; Foris, V.; Bashir, M.; Ghanim, B.; Klepetko, W.; Olschewski, H.; Olschewski, A.; Kwapiszewska, G. The inflammatory cell landscape in the lungs of patients with idiopathic pulmonary arterial hypertension. Eur. Respir. J. 2018, 51, 1701214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guignabert, C.; Humbert, M. Targeting transforming growth factor β receptors in pulmonary hypertension. Eur. Respir. J. 2020, 2002341. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Wang, X.-J.; Li, S.-Q.; Yang, S.-H.; Lv, Z.-C.; Wang, L.-T.; He, Y.-Y.; Jiang, X.; Wang, Y.; Jing, Z.-C. Elevated levels of plasma transforming growth factor-β1 in idiopathic and heritable pulmonary arterial hypertension. Int. J. Cardiol. 2016, 222, 368–374. [Google Scholar] [CrossRef] [PubMed]
- Teo, J.-L.; Kahn, M. The Wnt signaling pathway in cellular proliferation and differentiation: A tale of two coactivators. Adv. Drug Deliv. Rev. 2010, 62, 1149–1155. [Google Scholar] [CrossRef]
- West, J.D.; Austin, E.D.; Gaskill, C.; Marriott, S.; Baskir, R.; Bilousova, G.; Jean, J.-C.; Hemnes, A.R.; Menon, S.; Bloodworth, N.C.; et al. Identification of a common Wnt-associated genetic signature across multiple cell types in pulmonary arterial hypertension. Am. J. Physiol. Physiol. 2014, 307, C415–C430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemnes, A.R.; Zhao, M.; Zhao, Z.; Gaskill, C.; Jetter, C.; Kropski, J.A.; Majka, S.M.; Austin, E.D.; West, J.; Newman, J.H.; et al. Critical genomic networks and vasoreactive variants in idiopathic pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2016, 194, 464–475. [Google Scholar] [CrossRef] [Green Version]
- Rabinovitch, M.; Guignabert, C.; Humbert, M.; Nicolls, M.R. Inflammation and Immunity in the Pathogenesis of Pulmonary Arterial Hypertension. Circ. Res. 2014, 115, 165–175. [Google Scholar] [CrossRef]
- Dempsey, E.C.; Badesch, D.B.; Dobyns, E.L.; Stenmark, K.R. Enhanced growth capacity of neonatal pulmonary artery smooth muscle cells in vitro: Dependence on cell size, time from birth, insulin-like growth factor I, and auto-activation of protein Kinase, C.J. Cell. Physiol. 1994, 160, 469–481. [Google Scholar] [CrossRef]
- Chávez-Galán, L.; Olleros, M.; Vesin, D.; Garcia, I. Much more than M1 and M2 macrophages, there are also CD169(+) and TCR(+) macrophages. Front. Immunol. 2015, 6, 263. [Google Scholar]
- Hudalla, H.; Michael, Z.; Kourembanas, S.; Christou, H.; Christodoulou, N.; Willis, G.R.; Fernandez-Gonzalez, A.; Filatava, E.J.; Dieffenbach, P.; Fredenburgh, L.E.; et al. Carbonic anhydrase inhibition ameliorates inflammation and experimental pulmonary hypertension. Am. J. Respir. Cell Mol. Biol. 2019, 61, 512–524. [Google Scholar] [CrossRef]
- Voelkel, N.F.; Tamosiuniene, R.; Nicolls, M.R. Challenges and opportunities in treating inflammation associated with pulmonary hypertension. Expert Rev. Cardiovasc. Ther. 2016, 14, 939–951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Qu, S.; Zhu, J.; Chen, F.; Ma, L. Comprehensive analysis of lncRNA-associated competing endogenous RNA network and immune infiltration in idiopathic pulmonary fibrosis. J. Thorac. Dis. 2020, 12, 1856–1865. [Google Scholar] [CrossRef] [PubMed]
- Hanna, J.; Goldman-Wohl, D.; Hamani, Y.; Avraham, I.; Greenfield, C.; Natanson-Yaron, S.; Prus, D.; Cohen-Daniel, L.; Arnon, T.; Manaster, I.; et al. Decidual NK cells regulate key developmental processes at the human fetal-maternal interface. Nat. Med. 2006, 12, 1065–1074. [Google Scholar] [CrossRef] [PubMed]
- Morrell, N.W.; Chang, C.; Long, L.L.; Soon, E.; Jones, D.; Machado, R.; Treacy, C.; Toshner, M.; Campbell, K.; Riding, A.; et al. Impaired natural killer cell phenotype and function in idiopathic and heritable pulmonary arterial hypertension. Circulation 2012, 126, 1099–1109. [Google Scholar] [CrossRef] [Green Version]
- Edwards, A.L.; Gunningham, S.P.; Clare, G.C.; Hayman, M.W.; Smith, M.; Frampton, C.M.; Robinson, B.A.; Troughton, R.W.; Beckert, L. Professional killer cell deficiencies and decreased survival in pulmonary arterial hypertension. Respirology 2013, 18, 1271–1277. [Google Scholar] [CrossRef]
- Shinagawa, S.; Okazaki, T.; Ikeda, M.; Yudoh, K.; Kisanuki, Y.Y.; Yanagisawa, M.; Kawahata, K.; Ozaki, S. T cells upon activation promote endothelin 1 production in monocytes via IFN-γ; and TNF-α. Sci. Rep. 2017, 7, 14500. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.-R.; Malakhau, Y.; Yu, C.-H.; Phelan, S.-L.J.; Cumming, R.I.; Kan, M.J.; Mao, L.; Rajagopal, S.; Piantadosi, C.A.; Gunn, M.D. Nonclassical monocytes sense hypoxia, regulate pulmonary vascular remodeling, and promote pulmonary hypertension. J. Immunol. 2020, 204, 1474–1485. [Google Scholar] [CrossRef]
- Taylor, S.; Dirir, O.; Zamanian, R.T.; Rabinovitch, M.; Thompson, A.R. The role of neutrophils and neutrophil elastase in pulmonary arterial hypertension. Front. Med. 2018, 5, 217. [Google Scholar] [CrossRef]
- Aldabbous, L.; Abdul-Salam, V.; McKinnon, T.; Duluc, L.; Pepke-Zaba, J.; Southwood, M.; Ainscough, A.; Hadinnapola, C.; Wilkins, M.; Toshner, M.; et al. Neutrophil extracellular traps promote angiogenesis: Evidence from vascular pathology in pulmonary hypertension. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2078–2087. [Google Scholar] [CrossRef] [Green Version]
- Birukova, A.A.; Wu, T.; Tian, Y.; Meliton, A.; Sarich, N.; Tian, X.; Leff, A.; Birukov, K.G. Iloprost improves endothelial barrier function in lipopolysaccharide-induced lung injury. Eur. Respir. J. 2012, 41, 165–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peixoto, C.A.; Gomes, F.O.D.S. The role of phosphodiesterase-5 inhibitors in prostatic inflammation: A review. J. Inflamm. 2015, 12, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Ye, R.D. Serum amyloid A1: Structure, function and gene polymorphism. Gene 2016, 583, 48–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smole, U.; Gour, N.; Phelan, J.; Hofer, G.; Köhler, C.; Kratzer, B.; Tauber, P.A.; Xiao, X.; Yao, N.; Dvorak, J.; et al. Serum amyloid A is a soluble pattern recognition receptor that drives type 2 immunity. Nat. Immunol. 2020, 21, 756–765. [Google Scholar] [CrossRef]
- Gan, X.-W.; Wang, W.-S.; Lu, J.-W.; Ling, L.-J.; Zhou, Q.; Zhang, H.-J.; Ying, H.; Sun, K. De novo synthesis of SAA1 in the placenta participates in parturition. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Lee, J.; Hall, J.; Kroehling, L.; Wu, L.; Najar, T.; Nguyen, H.; Lin, W.; Yeung, S.; Silva, H.; Li, D.; et al. Serum amyloid a proteins induce pathogenic Th17 cells and promote inflammatory. Dis. Cell. 2020, 180, 79–91. [Google Scholar] [CrossRef]
- Lu, J.; Yu, Y.; Zhu, I.; Cheng, Y.; Sun, P.D. Structural mechanism of serum amyloid A-mediated inflammatory amyloidosis. Proc. Natl. Acad. Sci. USA 2014, 111, 5189–5194. [Google Scholar] [CrossRef] [Green Version]
- De Santo, C.; Arscott, R.; Booth, S.; Karydis, I.; Jones, M.; Asher, R.; Salio, M.; Middleton, M.; Cerundolo, V. Invariant NKT cells modulate the suppressive activity of IL-10-secreting neutrophils differentiated with serum amyloid A. Nat. Immunol. 2010, 11, 1039–1046. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Zhou, H.; Zhu, Z.; Ziyan, Z.; Wang, L.; Liang, Q.; Ye, R.D. Ex vivo and in vitro effect of serum amyloid a in the induction of macrophage M2 markers and efferocytosis of apoptotic neutrophils. J. Immunol. 2015, 194, 4891–4900. [Google Scholar] [CrossRef]
- Ye, R.D.; Sun, L. Emerging functions of serum amyloid A in inflammation. J. Leukoc. Biol. 2015, 98, 923–929. [Google Scholar] [CrossRef]
- Balabanian, K.; Foussat, A.; Simonneau, G.; Emilie, D.; Humbert, M.; Dorfmüller, P.; Durand-Gasselin, I.; Capel, F.; Bouchet-Delbos, L.; Portier, A.; et al. CX3C chemokine fractalkine in pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2002, 165, 1419–1425. [Google Scholar] [CrossRef] [PubMed]
- Dorfmüller, P.; Zarka, V.; Emilie, D.; Humbert, M.; Durand-Gasselin, I.; Monti, G.; Balabanian, K.; Garcia, G.; Capron, F.; Coulomb-Lherminé, A.; et al. Chemokine RANTES in severe pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2002, 165, 534–539. [Google Scholar] [CrossRef]
- Sweatt, A.J.; Hedlin, H.K.; Nicolls, M.R.; Rabinovitch, M.; Khatri, P.; Zamanian, R.T.; Balasubramanian, V.; Hsi, A.; Blum, L.K.; Robinson, W.H.; et al. Discovery of distinct immune phenotypes using machine learning in pulmonary arterial hypertension. Circ. Res. 2019, 124, 904–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zlotnik, A.; Yoshie, O. Chemokines: A new classification system and their role in immunity. Immunity 2000, 12, 121–127. [Google Scholar] [CrossRef] [Green Version]
- Nakano, M.; Fujii, T.; Hashimoto, M.; Yukawa, N.; Yoshifuji, H.; Ohmura, K.; Nakaizumi, A.; Mimori, T. Type I interferon induces CX3CL1 (fractalkine) and CCL5 (RANTES) production in human pulmonary vascular endothelial cells. Clin. Exp. Immunol. 2012, 170, 94–100. [Google Scholar] [CrossRef] [Green Version]
- Barnes, J.W.; Kucera, E.T.; Dweik, R.A.; Tian, L.; Mellor, N.E.; Dvorina, N.; Baldwin, W.W.; Aldred, M.A.; Farver, C.F.; Comhair, S.A.A.; et al. Bone morphogenic protein type 2 receptor mutation-independent mechanisms of disrupted bone morphogenetic protein signaling in idiopathic pulmonary arterial hypertension. Am. J. Respir. Cell Mol. Biol. 2016, 55, 564–575. [Google Scholar] [CrossRef] [Green Version]
- van der Bruggen, C.; Happé, C.; Dorfmüller, P.; Trip, P.; Spruijt, O.; Rol, N.; Hoevenaars, F.; Houweling, A.; Girerd, B.; Marcus, J.; et al. Bone morphogenetic protein receptor type 2 mutation in pulmonary arterial hypertension: A view on the right ventricle. Circulation 2016, 133, 1747–1760. [Google Scholar] [CrossRef] [Green Version]
- Nie, X.; Tan, J.; Dai, Y.; Liu, Y.; Zou, J.; Sun, J.; Ye, S.; Shen, C.; Fan, L.; Chen, J.; et al. CCL5 deficiency rescues pulmonary vascular dysfunction, and reverses pulmonary hypertension via caveolin-1-dependent BMPR2 activation. J. Mol. Cell. Cardiol. 2018, 116, 41–56. [Google Scholar] [CrossRef]
- Jacobson, K.A.; Gao, Z.-G. Adenosine receptors as therapeutic targets. Nat. Rev. Drug Discov. 2006, 5, 247–264. [Google Scholar] [CrossRef] [Green Version]
- Van Der Putten, C.; Veth, J.; Sukurova, L.; Zuiderwijk-Sick, E.A.; Simonetti, E.; Koenen, H.J.P.M.; Burm, S.M.; Van Noort, J.M.; Ijzerman, A.P.; Van Hijum, S.A.F.T.; et al. TLR-Induced IL-12 and CCL2 Production by Myeloid Cells Is Dependent on Adenosine A3 Receptor–Mediated Signaling. J. Immunol. 2019, 202, 2421–2430. [Google Scholar] [CrossRef] [Green Version]
- D’Antongiovanni, V.; Fornai, M.; Pellegrini, C.; Benvenuti, L.; Blandizzi, C.; Antonioli, L. The Adenosine System at the Crossroads of Intestinal Inflammation and Neoplasia. Int. J. Mol. Sci. 2020, 21, 5089. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DEGs | |
|---|---|
| Upregulated DEGs | HBB, LTBP1, HBA2, PDE3A, CCL5, BMP6, MFAP4, ABCG2, RGS5, WIF1, SFRP2, EDN1, ASPN, COL14A1, OGN, DPT, RGS1, CD69, C10orf10, ESM1, GZMK, MXRA5, AGBL1, ENPP2, POSTN, VCAM1, CPA3, FABP4, IFI44L, ROBO2, THY1 |
| Downregulated DEGs | CSF3R, RNASE2, S100A9, MGAM, AQP9, SULT1B1, CR1, S100A8, NQO1, S100A12, LILRB3, CXCR1, IL1R2, NPL, PROK2, CXCR2, ADORA3, CD163, SIGLEC10, ANPEP, HMOX1, CCR1, VNN2, SAA1, LCN2, ELF5, GPR110, SPP1, SLC16A6, BPIFA1, CTSE, TIMP4, BPIFB1, SLC7A11, PLA2G7, CYP1B1, MT-TW, SERPINA3, SLCO4A1 |
| GOID | GO Term | p Value | Percentage of Term |
|---|---|---|---|
| GO:1990266 | Neutrophil migration | 1.61 × 10−9 | 52.35% |
| GO:0030593 | Neutrophil chemotaxis | 3.91 × 10−10 | 22.15% |
| GO:0140353 | Lipid export from cell | 1.08 × 10−6 | 10.74% |
| GO:0010660 | Regulation of muscle cell apoptotic process | 1.35 × 10−4 | 4.7% |
| GO:0046916 | Cellular transition metal ion homeostasis | 5.74 × 10−6 | 2.68% |
| GO:0030199 | Collagen fibril organization | 4.34 × 10−5 | 2.01% |
| GO:0006809 | Nitric oxide biosynthetic process | 2.29 × 10−4 | 2.01% |
| GO:0043117 | Positive regulation of vascular permeability | 2.93 × 10−5 | 1.34% |
| GO:0017001 | Antibiotic catabolic process | 2.67 × 10−3 | 0.67% |
| GO:0008235 | Metalloexopeptidase activity | 2.18 × 10−3 | 0.67% |
| GO:0006953 | Acute-phase response | 1.74 × 10−3 | 0.67% |
| Category | Term | Count | p-Value | Genes |
|---|---|---|---|---|
| GOTERM_BP_DIRECT | GO:0006935: Chemotaxis | 3 | 1.38 × 10−4 | CCR1, CXCR1, CXCR2 |
| GOTERM_BP_DIRECT | GO:0090026: Positive regulation of monocyte chemotaxis | 2 | 6.32 × 10−3 | CCR1, CCL5 |
| GOTERM_BP_DIRECT | GO:0006953: Acute-phase response | 1 | 6.32 × 10−3 | SAA1 |
| GOTERM_BP_DIRECT | GO:0070098: Chemokine-mediated signaling pathway | 2 | 1.789 × 10−2 | CXCR2, CCL5 |
| GOTERM_BP_DIRECT | GO:0060326: Cell chemotaxis | 1 | 1.98 × 10−2 | SAA1 |
| GOTERM_BP_DIRECT | GO:0034364: High-density lipoprotein particle | 1 | 5.59 × 10−3 | SAA1 |
| GOTERM_BP_DIRECT | GO:0005615: Extracellular space | 2 | 4.37 × 10−2 | SAA1, CCL5 |
| GOTERM_BP_DIRECT | GO:0042056: Chemoattractant activity | 2 | 4.65 × 10−5 | SAA1, CCL5 |
| GOTERM_BP_DIRECT | GO:0016494: C-X-C Chemokine receptor activity | 2 | 3.61 × 10−3 | CXCR1, CXCR2 |
| KEGG_PATHWAY | cfa04062: Chemokine signaling pathway | 4 | 6.06 × 10−5 | CCR1, CXCR1, CXCR2, CCL5 |
| KEGG_PATHWAY | cfa04060: Cytokine–cytokine receptor interaction | 4 | 9.44 × 10−5 | CCR1, CXCR1, CXCR2, CCL5 |
| Gene | Drug | Interaction Type |
|---|---|---|
| CCL5 | FLUTICASONE PROPIONATE | anti-inflammatory agent |
| CXCR1 | CHEMBL411250 | agonist |
| CXCR1 | PROPOFOL | agonist |
| CXCR1 | CHOLINE ALFOSCERATE | agonist |
| CXCR2 | PROPOFOL | agonist |
| CXCR2 | BENZPIPERYLON | agonist |
| CXCR2 | CHEMBL411250 | agonist |
| CXCR2 | MEPHENTERMINE | agonist |
| CCR1 | ENOXAPARIN | agonist |
| CCR1 | GLYCERIN | agonist |
| CCR1 | GUANIDINO ACETATE | agonist |
| CCR1 | GUANINE | agonist |
| ADORA3 | IB-MECA | agonist |
| ADORA3 | ADENOSINE | agonist |
| ADORA3 | CF102 | agonist |
| ADORA3 | CHEMBL175543 | agonist |
| ADORA3 | CHEMBL472925 | agonist |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, J.; Yang, Y.; Yang, Y.; Xiong, C. Identification of Potential Risk Genes and the Immune Landscape of Idiopathic Pulmonary Arterial Hypertension via Microarray Gene Expression Dataset Reanalysis. Genes 2021, 12, 125. https://doi.org/10.3390/genes12010125
Xu J, Yang Y, Yang Y, Xiong C. Identification of Potential Risk Genes and the Immune Landscape of Idiopathic Pulmonary Arterial Hypertension via Microarray Gene Expression Dataset Reanalysis. Genes. 2021; 12(1):125. https://doi.org/10.3390/genes12010125
Chicago/Turabian StyleXu, Jing, Yicheng Yang, Yuejin Yang, and Changming Xiong. 2021. "Identification of Potential Risk Genes and the Immune Landscape of Idiopathic Pulmonary Arterial Hypertension via Microarray Gene Expression Dataset Reanalysis" Genes 12, no. 1: 125. https://doi.org/10.3390/genes12010125
APA StyleXu, J., Yang, Y., Yang, Y., & Xiong, C. (2021). Identification of Potential Risk Genes and the Immune Landscape of Idiopathic Pulmonary Arterial Hypertension via Microarray Gene Expression Dataset Reanalysis. Genes, 12(1), 125. https://doi.org/10.3390/genes12010125