Phenylketonuria Diagnosis by Massive Parallel Sequencing and Genotype-Phenotype Association in Brazilian Patients

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. DNA Extraction and Sequencing
2.3. Pathogenicity Determination and Prediction
2.4. Genotype–Phenotype Analysis
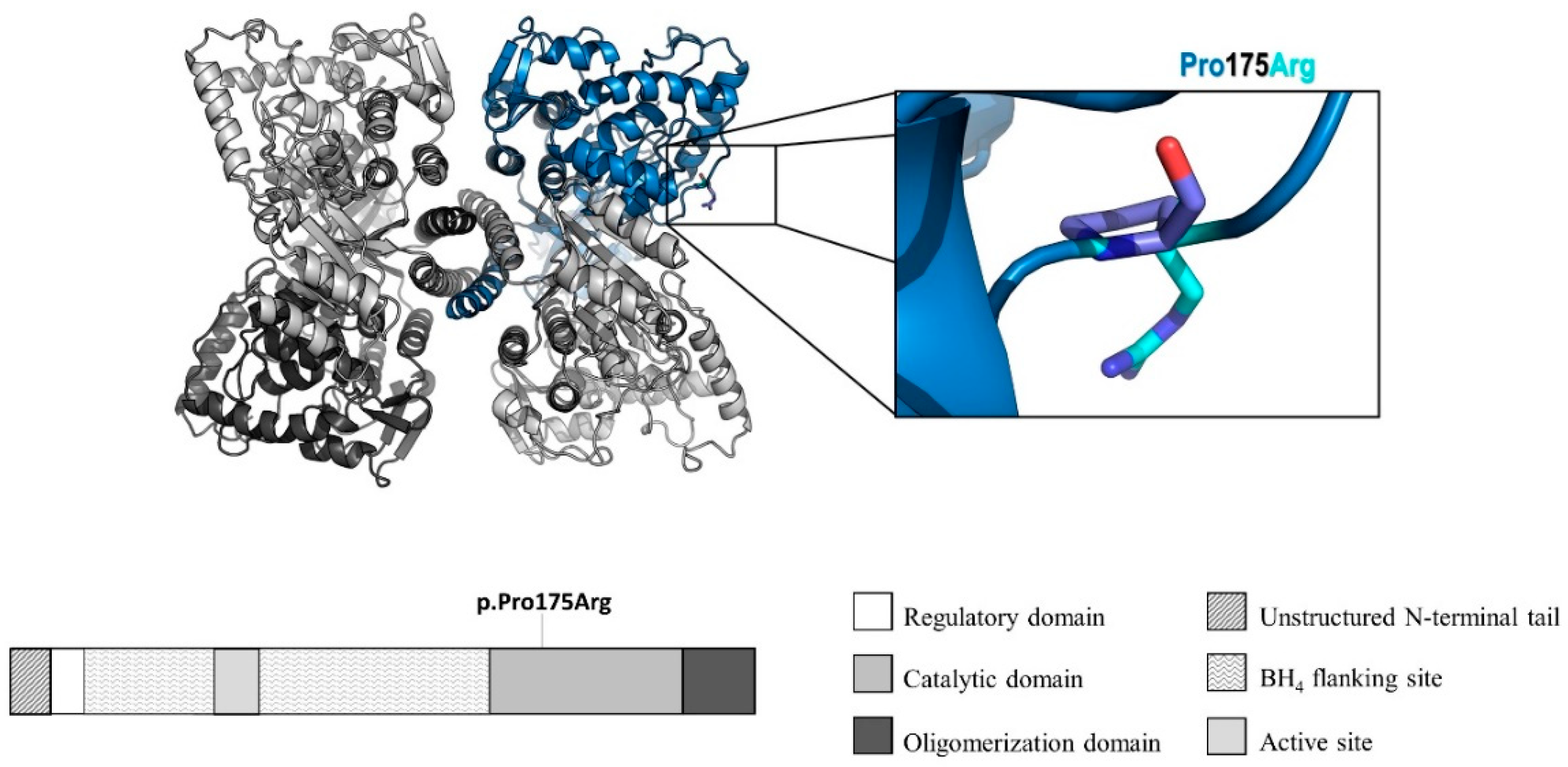
2.5. Molecular Modeling
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Blau, N.; Van Spronsen, F.J.; Levy, H.L. Phenylketonuria. Lancet 2010, 376, 1417–1427. [Google Scholar] [CrossRef]
- Kochhar, J.S.; Chan, S.Y.; Ong, P.S.; Kang, L. Clinical therapeutics for phenylketonuria. Drug Deliv. Transl. Res. 2012, 2, 223–237. [Google Scholar] [CrossRef]
- van Spronsen, F.J. Phenylketonuria: A 21st century perspective. Nat. Rev. Endocrinol. 2010, 6, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Al Hafid, N.; Christodoulou, J. Phenylketonuria: A review of current and future treatments. Transl. Pediatr. 2015, 4, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Muntau, A.C.; Röschinger, W.; Habich, M.; Demmelmair, H.; Hoffmann, B.; Sommerhoff, C.P.; Roscher, A.A. Tetrahydrobiopterin as an Alternative Treatment for Mild Phenylketonuria. N. Engl. J. Med. 2002, 347, 2122–2132. [Google Scholar] [CrossRef]
- Brasil Ministério da Saúde. Triagem Neonatal Biológica; Ministério da Saúde: Brasília, Brazil, 2016; ISBN 9788533424074.
- Stenson, P.D.; Mort, M.; Ball, E.V.; Evans, K.; Hayden, M.; Heywood, S.; Hussain, M.; Phillips, A.D.; Cooper, D.N. The Human Gene Mutation Database: Towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum. Genet. 2017, 136, 665–677. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.Y.; Qu, Y.J.; Song, F.; Zhang, T.; Bai, J.L.; Jin, Y.W.; Wang, H. Fast clinical molecular diagnosis of hyperphenylalaninemia using next-generation sequencing-based on a custom AmpliSeqTM panel and Ion Torrent PGM sequencing. Mol. Genet. Metab. 2014, 113, 261–266. [Google Scholar] [CrossRef]
- Blau, N.; Shen, N.; Carducci, C. Molecular genetics and diagnosis of phenylketonuria: State of the art. Expert Rev. Mol. Diagn. 2014, 14, 655–671. [Google Scholar] [CrossRef]
- Giugliani, L.; Sitta, A.; Vargas, C.R.; Santana-da-Silva, L.C.; Nalin, T.; Saraiva-Pereira, M.L.; Giugliani, R.; Schwartz, I.V.D. Responsividade à tetrahidrobiopterina em pacientes com deficiência de fenilalanina hidroxilase. J. Pediatr. 2011, 87, 245–251. [Google Scholar] [CrossRef] [Green Version]
- Nalin, T.; Perry, I.D.S.; Sitta, A.; Vargas, C.R.; Saraiva-Pereira, M.L.; Giugliani, R.; Blau, N.; Schwartz, I.V.D. Optimized loading test to evaluate responsiveness to tetrahydrobiopterin (BH 4) in Brazilian patients with phenylalanine hydroxylase deficiency. Mol. Genet. Metab. 2011, 104, S80–S85. [Google Scholar] [CrossRef] [Green Version]
- Robinson, J.T.; Thorvaldsdóttir, H.; Wenger, A.M.; Zehir, A.; Mesirov, J.P. Variant review with the integrative genomics viewer. Cancer Res. 2017, 77, e31–e34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, P.; Gauthier, L.D.; et al. The Mutational Constraint Spectrum Quantified from Variation in 141, 456 Humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Naslavsky, M.S.; Yamamoto, G.L.; de Almeida, T.F.; Ezquina, S.A.M.; Sunaga, D.Y.; Pho, N.; Bozoklian, D.; Sandberg, T.O.M.; Brito, L.A.; Lazar, M.; et al. Exomic variants of an elderly cohort of Brazilians in the ABraOM database. Hum. Mutat. 2017, 38, 751–763. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [PubMed]
- Garbade, S.F.; Shen, N.; Himmelreich, N.; Haas, D.; Trefz, F.K.; Hoffmann, G.F.; Burgard, P.; Blau, N. Allelic phenotype values: A model for genotype-based phenotype prediction in phenylketonuria. Genet. Med. 2019, 21, 580–590. [Google Scholar] [CrossRef]
- Nalin, T.; Perry, I.D.S.; Refosco, L.F.; Oliveira Netto, C.B.; de Souza, C.F.M.; Vieira, T.A.; Picon, P.D.; Schwartz, I.V.D. Phenylketonuria in the Public Health System: Assessment of Adherence. Clin. Biomed. Res. 2010, 30, 225–232. [Google Scholar]
- Flydal, M.I.; Alcorlo-Pagés, M.; Johannessen, F.G.; Martínez-Caballero, S.; Skjærven, L.; Fernandez-Leiro, R.; Martinez, A.; Hermoso, J.A. Structure of full-length human phenylalanine hydroxylase in complex with tetrahydrobiopterin. Proc. Natl. Acad. Sci. USA 2019, 166, 11229–11234. [Google Scholar] [CrossRef] [Green Version]
- Johansson, M.U.; Zoete, V.; Michielin, O.; Guex, N. Defining and searching for structural motifs using DeepView/Swiss-PdbViewer. BMC Bioinform. 2012, 13. [Google Scholar] [CrossRef] [Green Version]
- Zheng, W.; Zhang, C.; Bell, E.W.; Zhang, Y. I-TASSER gateway: A protein structure and function prediction server powered by XSEDE. Futur. Gener. Comput. Syst. 2019, 99, 73–85. [Google Scholar] [CrossRef]
- Guerois, R.; Nielsen, J.E.; Serrano, L. Predicting changes in the stability of proteins and protein complexes: A study of more than 1000 mutations. J. Mol. Biol. 2002, 320, 369–387. [Google Scholar] [CrossRef]
- Ferreira, P.; Sant’Anna, O.; Varejão, N.; Lima, C.; Novis, S.; Barbosa, R.V.; Caldeira, C.M.; Rumjanek, F.D.; Ventura, S.; Cruz, M.W.; et al. Structure-based analysis of A19D, a variant of transthyretin involved in familial amyloid cardiomyopathy. PLoS ONE 2013, 8, e82484. [Google Scholar] [CrossRef]
- Da Santana Silva, L.C.; Santos Carvalho, T.; Da Britto Silva, F.; Morari, L.; Aguirres Fachel, Â.; Pires, R.; Farret Refosco, L.; Desnick, R.J.; Giugliani, R.; Saraiva Pereira, M.L.; et al. Molecular characterization of phenylketonuria in South Brazil. Mol. Genet. Metab. 2003, 79, 17–24. [Google Scholar] [CrossRef]
- Zekanowski, C.; Nowacka, M.; Cabalska, B.; Bal, J. Molecular basis of mild hyperphenylalaninaemia in Poland. J. Med. Genet. 1997, 34, 1035–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daniele, A.; Cardillo, G.; Pennino, C.; Carbone, M.T.; Scognamiglio, D.; Correra, A.; Pignero, A.; Castaldo, G.; Salvatore, F. Molecular Epidemiology of Phenylalanine Hydroxylase Deficiency in Southern Italy: A 96% Detection Rate with Ten Novel Mutations. Ann. Hum. Genet. 2007, 71, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Eisensmith, R.C.; Woo, S.L.C. Molecular basis of phenylketonuria and related hyperphenylalaninemias: Mutations and polymorphisms in the human phenylalanine hydroxylase gene. Hum. Mutat. 1992, 1, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, K.; Takarada, Y.; Otsuka, N.; Kagawa, S.; Matsuoka, A.; Kalanin, J. Genetic diagnosis of phenylketonuria: Identification of the mutations of phenylalanine hydroxylase gene by PCR direct sequencing. Rinsho Byori. 1992, 40, 1060–1066. [Google Scholar]
- Dworniczak, B.; Aulehla-Scholz, C.; Horst, J. Phenylketonuria: Detection of a frequent haplotype 4 allele mutation. Hum. Genet. 1989, 84, 95–96. [Google Scholar] [CrossRef]
- Dworniczak, B.; Kalaydjieva, L.; Pankoke, S.; Aulehla-Scholz, C.; Allen, G.; Horst, J. Analysis of exon 7 of the human phenylalanine hydroxylase gene: A mutation hot spot? Hum. Mutat. 1992, 1, 138–146. [Google Scholar] [CrossRef]
- Guldberg, P.; Romano, V.; Ceratto, N.; Bosco, P.; Ciuna, M.; Indelicato, A.; Mollica, F.; Meli, C.; Giovannini, M.; Riva, E. Mutational spectrum of phenylalanine hydroxylase deficiency in Sicily: Implications for diagnosis of hyperphenylalaninemia in southern Europe. Hum. Mol. Genet. 1993, 2, 1703–1707. [Google Scholar] [CrossRef]
- Wang, T.; Okano, Y.; Eisensmith, R.C.; Lo, W.H.Y.; Huang, S.-Z.; Zeng, Y.-T.; Yuan, L.-F.; Liu, S.-R.; Woo, S.L.C. Missense mutations prevalent in orientals with phenylketonuria: Molecular characterization and clinical implications. Genomics 1991, 10, 449–456. [Google Scholar] [CrossRef]
- Zschocke, J.; Graham, C.A.; Carson, D.J.; Nevin, N.C. Phenylketonuria mutation analysis in Northern Ireland: A rapid stepwise approach. Am. J. Hum. Genet. 1995, 57, 1311–1317. [Google Scholar] [PubMed]
- Abadie, V.; Lyonnet, S.; Maurin, N.; Berthelon, M.; Caillaud, C.; Giraud, F.; Mattei, J.F.; Rey, J.; Rey, F.; Munnich, A. CpG dinucleotides are mutation hot spots in phenylketonuria. Genomics 1989, 5, 936–939. [Google Scholar] [CrossRef]
- Svensson, E.; Andersson, B.; Hagenfeldt, L. Two mutations within the coding sequence of the phenylalanine hydroxylase gene. Hum. Genet. 1990, 85, 300–304. [Google Scholar] [CrossRef] [PubMed]
- Lyonnet, S.; Caillaud, C.; Rey, F.; Berthelon, M.; Frézal, J.; Rey, J.; Munnich, A. Molecular genetics of phenylketonuria in Mediterranean countries: A mutation associated with partial phenylalanine hydroxylase deficiency. Am. J. Hum. Genet. 1989, 44, 511–517. [Google Scholar]
- Dianzani, I.; Forrest, S.M.; Camaschella, C.; Saglio, G.; Ponzone, A.; Cotton, R.G. Screening for mutations in the phenylalanine hydroxylase gene from Italian patients with phenylketonuria by using the chemical cleavage method: A new splice mutation. Am. J. Hum. Genet. 1991, 48, 631–635. [Google Scholar]
- Lichter-Konecki, U.; Konecki, D.S.; DiLella, A.G.; Brayton, K.; Marvit, J.; Hahn, T.M.; Trefz, F.K.; Woo, S.L.C. Phenylalanine Hydroxylase Deficiency Caused by a Single Base Substitution in an Exon of the Human Phenylalanine Hydroxylase Gene. Biochemistry 1988, 27, 2881–2885. [Google Scholar] [CrossRef]
- Guldberg, P.; Rey, F.; Zschocke, J.; Romano, V.; François, B.; Michiels, L.; Ullrich, K.; Hoffmann, G.F.; Burgard, P.; Schmidt, H.; et al. A European Multicenter Study of Phenylalanine Hydroxylase Deficiency: Classification of 105 Mutations and a General System for Genotype-Based Prediction of Metabolic Phenotype. Am. J. Hum. Genet. 1998, 63, 71–79. [Google Scholar] [CrossRef] [Green Version]
- Rozen, R.; Mascisch, A.; Lambert, M.; Laframboise, R.; Scriver, C.R. Mutation profiles of phenylketonuria in Quebec populations: Evidence of stratification and novel mutations. Am. J. Hum. Genet. 1994, 55, 321–326. [Google Scholar]
- Dworniczak, B.; Aulehla-Scholz, C.; Kalaydjieva, L.; Bartholomé, K.; Grudda, K.; Horst, J. Aberrant splicing of phenylalanine hydroxylase mRNA: The major cause for phenylketonuria in parts of southern Europe. Genomics 1991, 11, 242–246. [Google Scholar] [CrossRef]
- Guldberg, P.; Henriksen, K.F.; Güttler, F. Molecular Analysis of Phenylketonuria in Denmark: 99% of the Mutations Detected by Denaturing Gradient Gel Electrophoresis. Genomics 1993, 17, 141–146. [Google Scholar] [CrossRef]
- Guldberg, P.; Henriksen, K.F.; Thöny, B.; Blau, N.; Güttler, F. Molecular Heterogeneity of Nonphenylketonuria Hyperphenylalaninemia in 25 Danish Patients. Genomics 1994, 21, 453–455. [Google Scholar] [CrossRef]
- DiLella, A.G.; Marvit, J.; Brayton, K.; Woo, S.L. An amino-acid substitution involved in phenylketonuria is in linkage disequilibrium with DNA haplotype 2. Nature 1987, 327, 333–336. [Google Scholar] [CrossRef]
- Okano, Y.; Eisensmith, R.C.; Güttler, F.; Lichter-Konecki, U.; Konecki, D.S.; Trefz, F.K.; Dasovich, M.; Wang, T.; Henriksen, K.; Lou, H.; et al. Molecular Basis of Phenotypic Heterogeneity in Phenylketonuria. N. Engl. J. Med. 1991, 324, 1232–1238. [Google Scholar] [CrossRef] [PubMed]
- DiLella, A.G.; Kwok, S.C.M.; Ledley, F.D.; Marvit, J.; Woo, S.L.C. Molecular Structure and Polymorphic Map of the Human Phenylalanine Hydroxylase Gene. Biochemistry 1986, 25, 743–749. [Google Scholar] [CrossRef] [PubMed]
- Romani, C.; Palermo, L.; MacDonald, A.; Limback, E.; Hall, S.K.; Geberhiwot, T. The impact of phenylalanine levels on cognitive outcomes in adults with phenylketonuria: Effects across tasks and developmental stages. Neuropsychology 2017, 31, 242–254. [Google Scholar] [CrossRef]
- Hillert, A.; Anikster, Y.; Belanger-Quintana, A.; Burlina, A.; Burton, B.K.; Carducci, C.; Chiesa, A.E.; Christodoulou, J.; Đorđević, M.; Desviat, L.R.; et al. The Genetic Landscape and Epidemiology of Phenylketonuria. Am. J. Hum. Genet. 2020, 107, 234–250. [Google Scholar] [CrossRef] [PubMed]
- Vieira Neto, E.; Maia Filho, H.S.; Monteiro, C.B.; Carvalho, L.M.; Tonon, T.; Vanz, A.P.; Schwartz, I.V.D.; Ribeiro, M.G. Quality of life and adherence to treatment in early-treated Brazilian phenylketonuria pediatric patients. Braz. J. Med. Biol. Res. 2018, 51, 1–10. [Google Scholar] [CrossRef]
- Trevisan, L.M.; Nalin, T.; Tonon, T.; Veiga, L.M.; Vargas, P.; Krug, B.C.; Leivas, P.G.C.; Schwartz, I.V.D. Access to treatment for phenylketonuria by judicial means in Rio Grande do Sul, Brazil. Cien. Saude Colet. 2015, 20, 1607–1616. [Google Scholar] [CrossRef] [Green Version]
- Ministério da Saúde do Brasil. Portaria No 822, de 06 de Junho de 2001; Ministério da Saúde: Brasília, Brazil, 2001.
- Li, N.; Jia, H.; Liu, Z.; Tao, J.; Chen, S.; Li, X.; Deng, Y.; Jin, X.; Song, J.; Zhang, L.; et al. Molecular characterisation of phenylketonuria in a Chinese mainland population using next-generation sequencing. Sci. Rep. 2015, 5, 15769. [Google Scholar] [CrossRef] [Green Version]
- Karačić, I.; Meili, D.; Sarnavka, V.; Heintz, C.; Thöny, B.; Ramadža, D.P.; Fumić, K.; Mardešić, D.; Barić, I.; Blau, N. Genotype-predicted tetrahydrobiopterin (BH4)-responsiveness and molecular genetics in Croatian patients with phenylalanine hydroxylase (PAH) deficiency. Mol. Genet. Metab. 2009, 97, 165–171. [Google Scholar] [CrossRef]
- Zschocke, J. Phenylketonuria mutations in Europe. Hum. Mutat. 2003, 21, 345–356. [Google Scholar] [CrossRef]
- Desviat, L.R.; Pérez, B.; De Lucca, M.; Cornejo, V.; Schmidt, B.; Ugarte, M. Evidence in Latin America of recurrence of V388M, a phenylketonuria mutation with high in vitro residual activity. Am. J. Hum. Genet. 1995, 57, 337–342. [Google Scholar] [PubMed]
- Aulehla-Scholz, C.; Heilbronner, H. Mutational spectrum in German patients with phenylalanine hydroxylase deficiency. Hum. Mutat. 2003, 21, 399–400. [Google Scholar] [CrossRef] [PubMed]
- Pena, S.D.J.; di Pietro, G.; Fuchshuber-Moraes, M.; Genro, J.P.; Hutz, M.H.; de Kehdy, F.S.G.; Kohlrausch, F.; Magno, L.A.V.; Montenegro, R.C.; Moraes, M.O.; et al. The genomic ancestry of individuals from different geographical regions of Brazil is more uniform than expected. PLoS ONE 2011, 6, e17063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Souza, A.M.; Resende, S.S.; de Sousa, T.N.; de Brito, C.F.A. A systematic scoping review of the genetic ancestry of the brazilian population. Genet. Mol. Biol. 2019, 42, 495–508. [Google Scholar] [CrossRef] [PubMed]
- Acosta, A.X.; Silva, W.A.; Carvalho, T.M.; Gomes, M.; Zago, M.A. Mutations of the phenylalanine hydroxylase (PAH) gene in Brazilian patients with phenylketonuria. Hum. Mutat. 2001, 17, 122–130. [Google Scholar] [CrossRef]
- Costa, R.D.; Galera, B.B.; Rezende, B.C.; Venâncio, A.C.; Galera, M.F. Identification of mutations in the PAH gene in PKU patients in the state of Mato Grosso. Rev. Paul. Pediatr. 2020, 38. [Google Scholar] [CrossRef]
- dos Santos, L.L.; Magalhães, M.D.C.; Reis, A.D.O.; Starling, A.L.P.; Januário, J.N.; da Fonseca, C.G.; de Aguiar, M.J.B.; Carvalho, M.R.S. Frequencies of phenylalanine hydroxylase mutations I65T, R252W, R261Q, R261X, IVS10nt11, V388M, R408W, Y414C, and IVS12nt1 in Minas Gerais, Brazil. Genet. Mol. Res. 2006, 5, 16–23. [Google Scholar]
- Santos, L.L.; Castro-Magalhães, M.; Fonseca, C.G.; Starling, A.L.P.; Januário, J.N.; Aguiar, M.J.B.; Carvalho, M.R.S. PKU in Minas Gerais State, Brazil: Mutation Analysis. Ann. Hum. Genet. 2008, 72, 774–779. [Google Scholar] [CrossRef]
- Enacán, R.E.; Miñana, M.N.; Fernandez, L.; Valle, M.G.; Salerno, M.; Fraga, C.I.; Santos-Simarro, F.; Prieto, L.; Lapunzina, P.; Specola, N.; et al. Phenylalanine Hydroxylase (PAH) Genotyping in PKU Argentine Patients. J. Inborn Errors Metab. Screen. 2019, 7. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, V.; Santa María, L.; Fuenzalida, K.; Morales, P.; Desviat, L.R.; Ugarte, M.; Pérez, B.; Cabello, J.F.; Cornejo, V. Characterization of Phenyalanine Hydroxylase Gene Mutations in Chilean PKU Patients. In JIMD Reports; Springer: Heidelberg, Germany, 2018; Volume 4, pp. 71–77. ISBN 978-3-642-32441-3. [Google Scholar]
- Betts, M.J.; Russell, R.B. Amino-Acid Properties and Consequences of Substitutions. In Bioinformatics for Geneticists; John Wiley & Sons, Ltd.: Chichester, UK, 2006; Volume 9, pp. 311–342. ISBN 9780470026199. [Google Scholar]
- Dobrowolski, S.F.; Heintz, C.; Miller, T.; Ellingson, C.; Ellingson, C.; Özer, I.; Gökçay, G.; Baykal, T.; Thöny, B.; Demirkol, M.; et al. Molecular genetics and impact of residual in vitro phenylalanine hydroxylase activity on tetrahydrobiopterin responsiveness in Turkish PKU population. Mol. Genet. Metab. 2011, 102, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Sarkissian, C.N.; Gamez, A.; Scott, P.; Dauvillier, J.; Dorenbaum, A.; Scriver, C.R.; Stevens, R.C. Chaperone-Like Therapy with Tetrahydrobiopterin in Clinical Trials for Phenylketonuria: Is Genotype a Predictor of Response. In JIMD Reports; Springer: Heidelberg, Germany, 2011; Volume 4, pp. 59–70. ISBN 978-3-642-32441-3. [Google Scholar]



{kind=link}
{kind=link}
| Patient | State | Gender | First Phe Level (µMol/L) | Age at Diagnosis (Days) | Age at Treatment Starting (Days) | Type of PKU | NGS Genotype | Previous Genotype | Type of PKU According to BioPKU 1 | BH4 Responsiveness According to the Test | BH4 Responsiveness According to BioPKU 2 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 * | RS | F | 1566 | 26 | 26 | C | c.1222C>T(;)1222C>T p.Arg408Trp(;)Arg408Trp | c.1222C>T(;)1222C>T p.Arg408Trp(;)Arg408Trp | Classic (1832/1845) | NP | No (95/98) Yes (2/98) Slow (1/98) |
| 2 ** | RS | M | 847 | 28 | 58 | M | c.524C>G(;)754C>T p.(Pro175Arg)(;)Arg252Trp | c.754C>T(;)? p.Arg252Trp(;)? | NA | No ***** | NA |
| 3 | RS | F | 1784 | 36 | 36 | C | c.1042C>G(;)1315+1G>A p.Leu348Val(;)? | c.1042C>G(;)1315+1G>A p.Leu348Val(;)? | Classic (11/16) | NP | No (5/6)Yes (1/6) |
| 4 | RS | F | 1478 | 43 | 43 | C | c.932T>C(;)1315+1G>A p.Leu311Pro(;)? | c.1315+1G>A(;)? p.?(;)? | Classic (2/2) | NP | NA |
| 5 | RS | M | 417 | 28 | 50 | M | c.1162G>A(;)1169A>G p.Val388Met(;)Glu390Gly | c.1162G>T(;)? p.Val388Met(;)? | Mild (8/12) | NP | Yes (11/11) |
| 6 | RS | F | 375 | 32 | 66 | M | c.1066-11G>A(;)1169A>G p.Gln355_Tyr356insGlyLeuGln(;)Glu390Gly | NP | Mild (8/14) | NP | Yes (8/8) |
| 7 ** | RS | M | NA | 60 | 60 | U | c.842+1G>A(;)1162G>A p.(?)(;)Val388Met | c.842+1G>A(;)1162G>Ap.?(;)Val388Met | NA | No ***** | NA |
| 8 | RS | F | 562 | 74 | 82 | M | c.1169A>G(;)1222C>T p.Glu390Gly(;)Arg408Trp | NP | Mild (54/84) | Yes | Yes (23/23) |
| 9 | RS | M | 1845 | 102 | 102 | M | c.722G>A(;)1222C>T p.Arg241His(;)Arg408Trp | c.1222C>T(;)1222C>T p.Arg408Trp(;)Arg408Trp | Mild (25/28) | No ****** | Yes (3/6) Slow (2/6) No (1/6) |
| 10 * | RS | M | 877 | 128 | 156 | M | c.473G>A(;)1162G>A p.Arg158Gln(;)Val388Met | c.1162G>A(;)? p.Val388Met(;)? | Classic (5/7) | No | Yes (2/3) Slow (1/3) |
| 11 *** | RS | M | 1022 | 195 | 292 | M | c.[1241A>G];[1042C>G] p.[Leu348Val];[Tyr414Cys] | NP | Mild (4/5) | Yes ***** | Yes (3/3) |
| 12 | RS | F | 3245 | 5 | 44 | C | c.745C>T(;)838G>A p.Leu249Phe(;)Glu280Lys | NP | Classic (1/1) | NP | NA |
| 13 | RS | F | 1361 | 15 | 19 | C | c.754C>T(;)1222C>T p.Arg252Trp(;)Arg408Trp | c.1222C>T(;)? p.Arg408Trp(;)? | Classic (103/103) | NP | No (4/4) |
| 14 *** | RS | F | 1736 | 16 | 16 | C | c.[473G>A];[1055delG] p.[Arg158Gln];[Gly352ValfsTer48] | NP | Classic (1/1) | NP | NA |
| 15 | RS | F | 2329 | 24 | 24 | C | c.712A>C(;)814G>T p.Thr238Pro(;)Gly272Ter | NP | Classic (1/1) | NP | NA |
| 16 | RS | M | 2716 | 27 | 27 | C | c.194T>C(;)472C>T p.Ile65Thr(;)Arg158Trp | c.194T>C(;)? p.Ile65Thr(;)? | Classic (2/2) | NP | No (1/1) |
| 17 | RS | M | 1697 | 30 | 30 | C | c.754C>T(;)1024delG p.Arg252Trp(;)Ala342HisfsTer58 | NP | NA | NP | NA |
| 18 ** | RS | F | 2178 | 27 | 48 | C | c.754C>T(;)1315+1G>A p.Arg242Trp(;)? | c.1315+1G>A(;)? p.?(;)? | Classic (9/9) | NP | No (1/1) |
| 19 | RS | M | 2904 | 73 | 101 | M | c.473G>A(;)1162G>A p.Arg158Gln(;)Val388Met | NP | Classic (5/7) | NP | No (2/3) Slow (1/3) |
| 20 | RS | M | 2323 | 4 | 17 | C | c.1222C>T(;)1315+1G>A p.Arg408Trp(;)? | c.1222C>T(;)1315+1G>A p.Arg408Trp(;)? | Classic (265/265) | NP | No (40/40) |
| 21 | RS | M | 2081 | 227 | 233 | C | c.473G>A(;)1315+1G>A p.Arg158Gln | NP | Classic (29/29) | No ***** | No (6/6) |
| 22.1 **** | RS | M | 1455 | 670 | 670 | C | c.782G>A(;)1315+1G>A p.Arg261Gln(;)? | c.782G>A(;)1315+1G>A p.Arg261Gln(;)? | Classic (47/66) | No ***** | No (24/25) Slow (1/25) |
| 22.2 **** | RS | M | 2196 | 2555 | 2677 | C | c.782G>A(;)1315+1G>A p.Arg261Gln(;)? | c.782G>A(;)1315+1G>A p.Arg261Gln(;)? | Classic (47/66) | No ***** | No (24/25) Slow (1/25) |
| 23 | DF | M | 1978 | 39 | 39 | C | c.754C>T(;)1066-11G>A p.Arg252Trp(;) Gln355_Tyr356insGlyLeuGln | NP | Classic (19/19) | No | No (6/6) |
| 24 | DF | M | 1857 | 18 | 22 | C | c.168+5G>A(;)782G>A p.?(;)Arg261Gln | NP | Mild (4/5) | Slow | Yes (2/2) |
| 25 | DF | M | 768 | 47 | 47 | M | c.184delC(;)1169A>G p.Leu62Ter(;)Glu390Gly | NP | NA | Yes | NA |
| 26 | DF | F | 344 | 41 | 41 | M | c.184delC(;)1169A>G p.Leu62Ter(;)Glu390Gly | NP | NA | Yes | NA |
| 27 | DF | F | 3133 | 38 | 38 | C | c.1066-11G>A(;)1066-11G>A p.Gln355_Tyr356insGlyLeuGln(;)Gln355_Tyr356insGlyLeuGln | NP | Classic (420/427) | Yes | No (107/114) Slow (7/114) |
| 28 | DF | F | 1724 | 41 | 41 | C | c.728G>A(;)728G>A p.Arg243Gln(;)Arg243Gln | NP | Classic (140/141) | No | No (13/14) Slow (1/14) |
| 29 | DF | F | 1936 | 44 | 44 | C | c.441+5G>T(;)473G>A p.Arg158Gln(;)? | NP | Classic (20/21) | No | No (9/12) Slow (3/12) |
| 30 | DF | F | 2299 | 22 | 22 | C | c.184delC(;)184delC p.Leu62Ter(;)Leu62Ter | NP | NA | No | NA |
| 31 | DF | F | 1754 | 57 | 57 | C | c.473G>A(;)782G>A p.Arg158Gln(;)Arg261Gln | NP | Mild (21/36) | Yes | Yes (8/13) No (4/13) Slow (1/13) |
| 32 | DF | F | NA | 60 | NA | U | c.1162G>A(;)1162G>A p.Val388Met(;)Val388Met | NP | Classic (23/41) | Yes | Yes (9/15) No (4/15) Slow (2/15 |
| 33 | DF | F | 1361 | 30 | 30 | C | c.782G>A(;)1315+1G>A p.Arg261Gln(;)? | NP | Classic (47/66) | Yes | No (24/25) Slow (1/25) |
| Allele | Protein | Location | Reference | ACMG | Effect |
|---|---|---|---|---|---|
| c.168+5G>A | p.? | I 2 | [24] | PP5 | VUS |
| c.184delC | p.Leu62Ter | E 3 | [25] | PVS1, PM2, PP3, PM4 | Pathogenic |
| c.194T>C | p.Ile65Thr | E 3 | [26] | PS3, PP2, PP5 | Likely pathogenic |
| c.441+5G>T | p.? | I 4 | [24] | PP5 | VUS |
| c.472C>T | p.Arg158Trp | E 5 | [27] | PS1, PP2, PP3, PP5 | Likely pathogenic |
| c.473G>A | p.Arg158Gln | E 5 | [28] | PS1, PP2, PP3, PP5 | Likely pathogenic |
| c.524C>G | p.(Pro175Arg) | E 6 | This article | PM2, PM5, PP2, PP3 | Likely pathogenic |
| c.712A>C | p.Thr238Pro | E 7 | [29] | PM2, PP2, PP3, PP5 | VUS |
| c.722G>A | p.Arg241His | E 7 | [30] | PS1, PS3, PP2, PP3, PP5 | Pathogenic |
| c.728G>A | p.Arg243Gln | E 7 | [31] | PS1, PS3, PP2, PP3, PP5 | Pathogenic |
| c.745C>T | p.Leu249Phe | E 7 | [32] | PS1, PP2, PP3, PP5 | Likely pathogenic |
| c.754C>T | p.Arg252Trp | E 7 | [33] | PS1, PS3, PP2, PP3, PP5 | Pathogenic |
| c.782G>A | p.Arg261Gln | E 7 | [33] | PS1, PS3, PP2, PP3, PP5 | Pathogenic |
| c.814G>T | p.Gly272Ter | E 7 | [34] | PVS1, PM4, PP3, PP5 | Pathogenic |
| c.838G>A | p.Glu280Lys | E 7 | [35] | PS1, PS3, PP2, PP3, PP5 | Pathogenic |
| c.842+1G>A | p.? | I 7 | [36] | PVS1, PP5 | VUS |
| c.932T>C | p.Leu311Pro | E 9 | [37] | PS1, PS3, PP2, PP3, PP5 | Pathogenic |
| c.1024delG | p.Ala342HisfsTer58 | E 10 | [38] | PVS1, PM2, PM4, PP3, PP5 | Pathogenic |
| c.1042C>G | p.Leu348Val | E 10 | [26] | PS3, PP2, PP3, PP5 | Likely pathogenic |
| c.1055delG | p.Gly352ValfsTer48 | E 10 | [39] | PVS1, PM4, PP3, PP5 | Pathogenic |
| c.1066-11G>A | p.Gln355_Tyr356insGlyLeuGln | I 10 | [40] | PS3, PP5 | VUS |
| c.1162G>A | p.Val388Met | E11 | [41] | PS1, PS3, PP2, PP3, PP5 | Pathogenic |
| c.1169A>G | p.Glu390Gly | E 11 | [42] | PS3, PS1, PP2, PP3, PP5 | Pathogenic |
| c.1222C>T | p.Arg408Trp | E 12 | [43] | PS3, PP2, PP3, PP5 | Likely pathogenic |
| c.1241A>G | p.Tyr414Cys | E 12 | [44] | PS1, PS3, PP2, PP3, PP5 | Pathogenic |
| c.1315+1G>A | p.? | I 12 | [45] | PVS1, PP5 | VUS |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tresbach, R.H.; Sperb-Ludwig, F.; Ligabue-Braun, R.; Tonon, T.; de Oliveira Cardoso, M.T.; Heredia, R.S.; da Silva Rosa, M.T.A.; Martins, B.C.; Poubel, M.O.; da Silva, L.C.S.; et al. Phenylketonuria Diagnosis by Massive Parallel Sequencing and Genotype-Phenotype Association in Brazilian Patients. Genes 2021, 12, 20. https://doi.org/10.3390/genes12010020
Tresbach RH, Sperb-Ludwig F, Ligabue-Braun R, Tonon T, de Oliveira Cardoso MT, Heredia RS, da Silva Rosa MTA, Martins BC, Poubel MO, da Silva LCS, et al. Phenylketonuria Diagnosis by Massive Parallel Sequencing and Genotype-Phenotype Association in Brazilian Patients. Genes. 2021; 12(1):20. https://doi.org/10.3390/genes12010020
Chicago/Turabian StyleTresbach, Rafael Hencke, Fernanda Sperb-Ludwig, Rodrigo Ligabue-Braun, Tássia Tonon, Maria Teresinha de Oliveira Cardoso, Romina Soledad Heredia, Maria Teresa Alves da Silva Rosa, Bárbara Cátia Martins, Monique Oliveira Poubel, Luiz Carlos Santana da Silva, and et al. 2021. "Phenylketonuria Diagnosis by Massive Parallel Sequencing and Genotype-Phenotype Association in Brazilian Patients" Genes 12, no. 1: 20. https://doi.org/10.3390/genes12010020
APA StyleTresbach, R. H., Sperb-Ludwig, F., Ligabue-Braun, R., Tonon, T., de Oliveira Cardoso, M. T., Heredia, R. S., da Silva Rosa, M. T. A., Martins, B. C., Poubel, M. O., da Silva, L. C. S., Maillot, F., & Schwartz, I. V. D. (2021). Phenylketonuria Diagnosis by Massive Parallel Sequencing and Genotype-Phenotype Association in Brazilian Patients. Genes, 12(1), 20. https://doi.org/10.3390/genes12010020