
The Importance of Extended Analysis Using Current Molecular Genetic Methods Based on the Example of a Cohort of 228 Patients with Hereditary Breast and Ovarian Cancer Syndrome

 , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Collection and Risk Assessment
2.2. Sequencing Methods and Bioinformatics Programs
3. Results
3.1. Statistical Evaluation
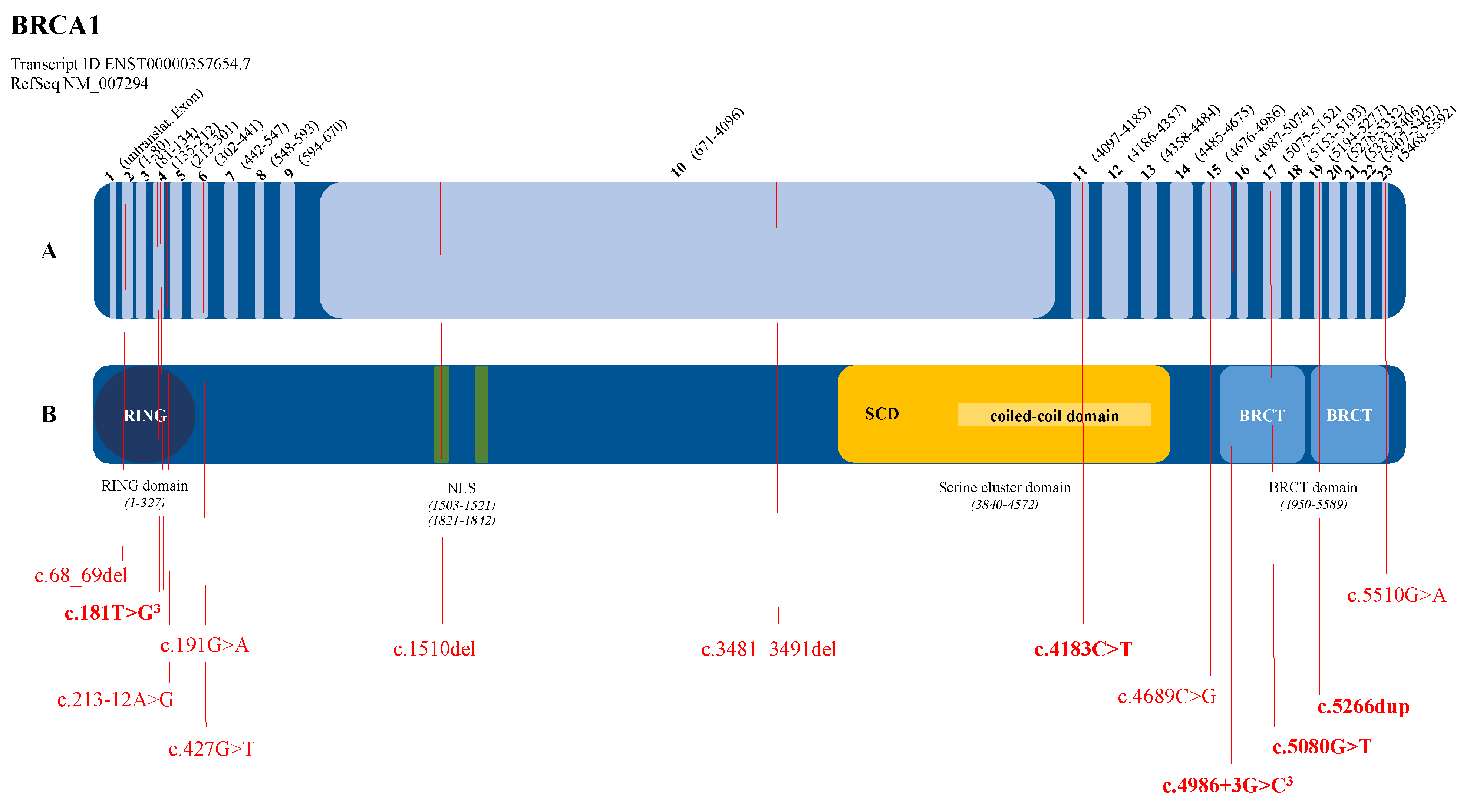
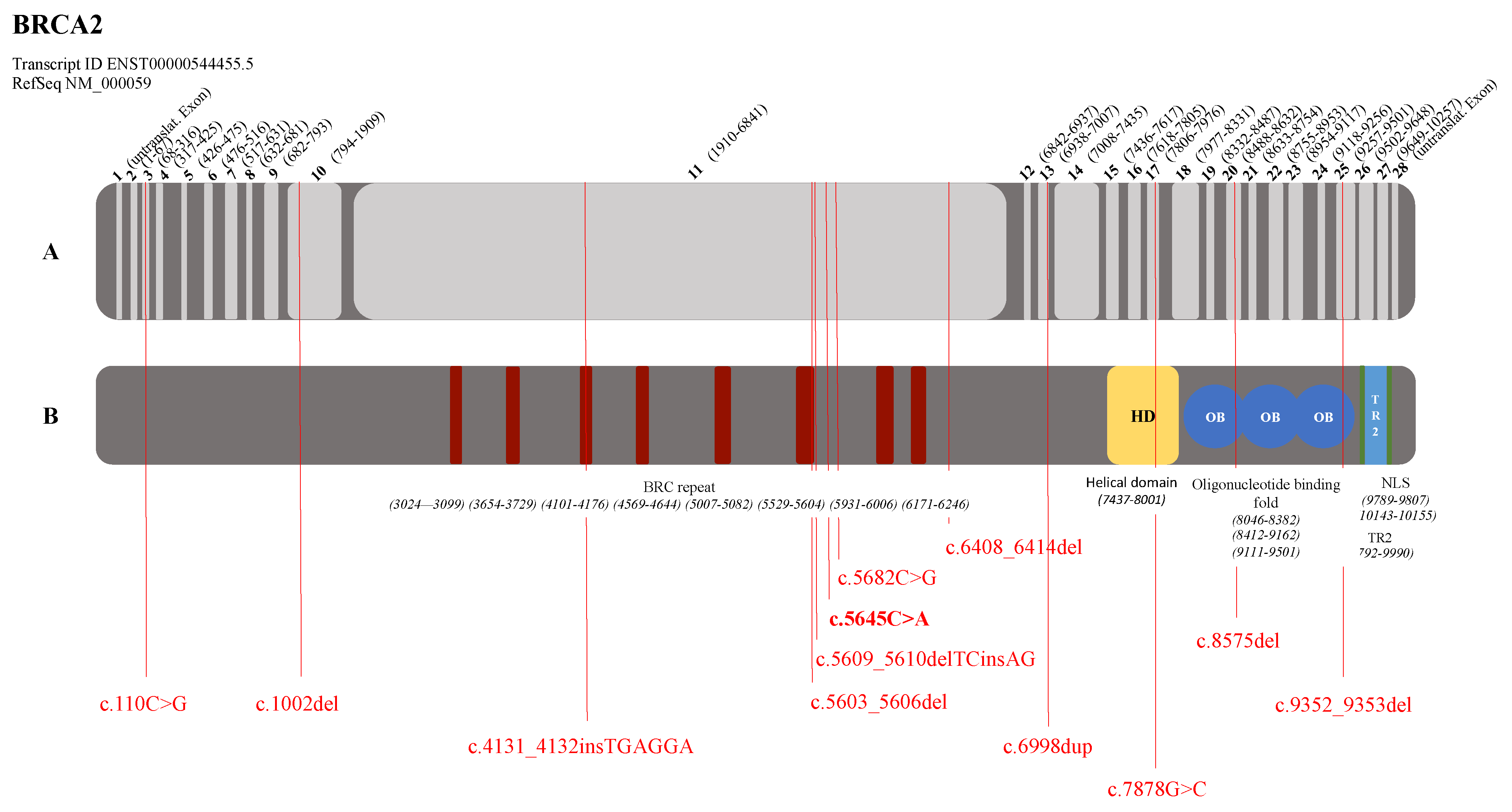
3.2. Graphical Representation of the Pathogenic Variants in the BRCA1 and BRCA2 Genes
3.3. Evaluation of the Molecular Genetic Analyses of the High-Risk Patient Group
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Leitlinienprogramm Onkologie (Deutsche Krebsgesellschaft, Deutsche Krebshilfe, AWMF) S3-Leitlinie Früherkennung, Diagnose, Therapie Und Nachsorge Des Mammakarzinoms, Version 4.1, AWMF Registernummer: 032-045OL. Available online: http://www.leitlinienprogramm-onkologie.de/leitlinien/mammakarzinom/ (accessed on 12 February 2019).
- Deutsches Konsortium Familiärer Brust-und Eierstockkrebs Standard Operating Procedures Im Deutschen Konsortium Familiärer Brust-Und Eierstockkrebs 2019. As of 28.05.2019; Annex to the consortium agreement. not public.
- Die TruRisk® Genpanel-Analyse Bei Familiärem Brust-Und Eierstockkrebs. Available online: https://www.konsortium-familiaerer-brustkrebs.de/betreuungskonzept/molekulare-diagnostik/truriskr-genpanel-analyse/ (accessed on 19 July 2021).
- Engel, C.; Zachariae, S.; Fischer, C. Familiärer Brustkrebs—Empirische Erkrankungsrisiken und Risikoberechnungsmodelle. Med. Genet. 2015, 27, 217–222. [Google Scholar] [CrossRef]
- Kuchenbaecker, K.B.; Hopper, J.L.; Barnes, D.R.; Phillips, K.-A.; Mooij, T.M.; Roos-Blom, M.-J.; Jervis, S.; Van Leeuwen, F.E.; Milne, R.L.; Andrieu, N.; et al. Risks of breast, ovarian, and contralateral breast cancer for BRCA1 and BRCA2 mutation carriers. JAMA 2017, 317, 2402. [Google Scholar] [CrossRef] [Green Version]
- Deutsches Konsortium Familiärer Brust- und Eierstockkrebs Checkliste Zur Risikoerfassung. Available online: https://www.konsortium-familiaerer-brustkrebs.de/informationen/checkliste-zur-risikoerfassung/ (accessed on 23 April 2019).
- Resch, L.D. Molekulargenetische Diagnostik Bei Erblichem Brust-und Eierstockkrebs; Albert-Ludwigs-Universität: Freiburg im Breisgau, Germany, 2020. [Google Scholar]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation Prediction for the Deep-Sequencing Age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shihab, H.A.; Gough, J.; Mort, M.; Cooper, D.N.; Day, I.N.; Gaunt, T.R. Ranking non-synonymous single nucleotide polymorphisms based on disease concepts. Hum. Genom. 2014, 8, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, P.C.; Henikoff, S. Predicting deleterious amino acid substitutions. Genome Res. 2001, 11, 863–874. [Google Scholar] [CrossRef] [Green Version]
- Hebsgaard, S.M.; Korning, P.G.; Tolstrup, N.; Engelbrecht, J.; Rouzé, P.; Brunak, S. Splice site prediction in Arabidopsis thaliana pre-mRNA by combining local and global sequence information. Nucleic Acids Res. 1996, 24, 3439–3452. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Antoniou, A.C.; Pharoah, P.D.P.; McMullan, G.; Day, N.E.; Stratton, M.R.; Peto, J.; Ponder, B.J.; Easton, D.F. A comprehensive model for familial breast cancer incorporating BRCA1, BRCA2 and other genes. Br. J. Cancer 2002, 86, 76–83. [Google Scholar] [CrossRef]
- Prat, J.; Ribé, A.; Gallardo, A. Hereditary ovarian cancer. Hum. Pathol. 2005, 36, 861–870. [Google Scholar] [CrossRef]
- Struewing, J.P.; Brody, L.C.; Erdos, M.R.; Kase, R.G.; Giambarresi, T.R.; Smith, S.A.; Collins, F.S.; Tucker, M.A. Detection of eight BRCA1 mutations in 10 breast/ovarian cancer families, including 1 family with male breast cancer. Am. J. Hum. Genet. 1995, 57, 1–7. [Google Scholar] [PubMed]
- Friedman, L.S.; Ostermeyer, E.A.; Szabo, C.I.; Dowd, P.; Lynch, E.D.; Rowell, S.E.; King, M.C. Confirmation of BRCA1 by analysis of germline mutations linked to breast and ovarian cancer in ten families. Nat. Genet. 1994, 8, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Couch, F.J.; Weber, B.L. Mutations and polymorphisms in the familial early-onset breast cancer (BRCA1) gene. Breast cancer information core. Hum. Mutat. 1996, 8, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, J.D.; Hallam, S.E.; Venne, V.L.; Lyon, E.; Ward, K. Implications of a novel cryptic splice site in the BRCA1 gene. Am. J. Med. Genet. 1998, 80, 140–144. [Google Scholar] [CrossRef]
- Shattuck-Eidens, D.; Oliphant, A.; McClure, M.; McBride, C.; Gupte, J.; Rubano, T.; Pruss, D.; Tavtigian, S.V.; Teng, D.H.; Adey, N.; et al. BRCA1 sequence analysis in women at high risk for susceptibility mutations: Risk factor analysis and implications for genetic testing. JAMA 1997, 278, 1242–1250. [Google Scholar] [CrossRef] [PubMed]
- Machackova, E.; Foretova, L.; Lukesova, M.; Vasickova, P.; Navratilova, M.; Coene, I.; Pavlu, H.; Kosinova, V.; Kuklova, J.; Claes, K. Spectrum and characterisation of BRCA1 and BRCA2 deleterious mutations in high-risk czech patients with breast and/or ovarian cancer. BMC Cancer 2008, 8, 140. [Google Scholar] [CrossRef] [Green Version]
- Langston, A.A.; Malone, K.E.; Thompson, J.D.; Daling, J.R.; Ostrander, E.A. BRCA1 mutations in a population-based sample of young women with breast cancer. N. Engl. J. Med. 1996, 334, 137–142. [Google Scholar] [CrossRef]
- Serova, O.; Montagna, M.; Torchard, D.; Narod, S.A.; Tonin, P.; Sylla, B.; Lynch, H.T.; Feunteun, J.; Lenoir, G.M. A high incidence of BRCA1 mutations in 20 breast-ovarian cancer families. Am. J. Hum. Genet. 1996, 58, 42–51. [Google Scholar]
- Adem, C.; Reynolds, C.; Soderberg, C.L.; Slezak, J.M.; McDonnell, S.K.; Sebo, T.J.; Schaid, D.J.; Myers, J.L.; Sellers, T.A.; Hartmann, L.C.; et al. Pathologic characteristics of breast parenchyma in patients with hereditary breast carcinoma, including BRCA1 and BRCA2 mutation carriers. Cancer 2003, 97, 1–11. [Google Scholar] [CrossRef]
- Simard, J.; Tonin, P.; Durocher, F.; Morgan, K.; Rommens, J.; Gingras, S.; Samson, C.; Leblanc, J.F.; Bélanger, C.; Dion, F. Common origins of BRCA1 mutations in canadian breast and ovarian cancer families. Nat. Genet. 1994, 8, 392–398. [Google Scholar] [CrossRef]
- Tung, N.; Battelli, C.; Allen, B.; Kaldate, R.; Bhatnagar, S.; Bowles, K.; Timms, K.; Garber, J.E.; Herold, C.; Ellisen, L.; et al. Frequency of mutations in individuals with breast cancer referred for BRCA1 and BRCA2 testing using next-generation sequencing with a 25-gene panel. Cancer 2015, 121, 25–33. [Google Scholar] [CrossRef]
- Borg, A.; Haile, R.W.; Malone, K.E.; Capanu, M.; Diep, A.; Törngren, T.; Teraoka, S.; Begg, C.B.; Thomas, D.C.; Concannon, P.; et al. Characterization of BRCA1 and BRCA2 deleterious mutations and variants of unknown clinical significance in unilateral and bilateral breast cancer: The WECARE study. Hum. Mutat. 2010, 31, E1200–E1240. [Google Scholar] [CrossRef] [Green Version]
- Delgado, L.; Fernández, G.; González, A.; Bressac-de Paillerets, B.; Gualco, G.; Bombled, J.; Cataldi, S.; Sabini, G.; Roca, R.; Musé, I.M. Hereditary breast cancer associated with a germline BRCA2 mutation in identical female twins with similar disease expression. Cancer Genet. Cytogenet. 2002, 133, 24–28. [Google Scholar] [CrossRef]
- Li, J.-Y.; Jing, R.; Wei, H.; Wang, M.; Xiaowei, Q.; Liu, H.; Jian, L.; Ou, J.-H.; Jiang, W.-H.; Tian, F.-G.; et al. Germline mutations in 40 cancer susceptibility genes among Chinese patients with high hereditary risk breast cancer. Int. J. Cancer 2019, 144, 281–289. [Google Scholar] [CrossRef]
- Howlett, N.G.; Taniguchi, T.; Olson, S.; Cox, B.; Waisfisz, Q.; De Die-Smulders, C.; Persky, N.; Grompe, M.; Joenje, H.; Pals, G.; et al. Biallelic inactivation of BRCA2 in fanconi anemia. Science 2002, 297, 606–609. [Google Scholar] [CrossRef] [PubMed]
- Tea, M.-K.M.; Kroiss, R.; Muhr, D.; Fuerhauser-Rappaport, C.; Oefner, P.; Wagner, T.M.; Singer, C.F. Central European BRCA2 mutation carriers: Birth cohort status correlates with onset of breast cancer. Maturitas 2014, 77, 68–72. [Google Scholar] [CrossRef] [PubMed]
- De Benedetti, V.M.; Radice, P.; Pasini, B.; Stagi, L.; Pensotti, V.; Mondini, P.; Manoukian, S.; Conti, A.; Spatti, G.; Rilke, F.; et al. Characterization of ten novel and 13 recurring BRCA1 and BRCA2 germline mutations in italian breast and/or ovarian carcinoma patients. Mutations in brief No. 178. Online. Hum. Mutat. 1998, 12, 215. [Google Scholar]
- Risch, H.A.; McLaughlin, J.R.; Cole, D.E.; Rosen, B.; Bradley, L.; Kwan, E.; Jack, E.; Vesprini, D.J.; Kuperstein, G.; Abrahamson, J.L.; et al. Prevalence and penetrance of germline BRCA1 and BRCA2 mutations in a population series of 649 women with ovarian cancer. Am. J. Hum. Genet. 2001, 68, 700–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamboom, K.; Kaasik, K.; Aršavskaja, J.; Tekkel, M.; Lilleorg, A.; Padrik, P.; Metspalu, A.; Veidebaum, T. BRCA1 mutations in women with familial or early-onset breast cancer and BRCA2 mutations in familial cancer in Estonia. Hered. Cancer Clin. Pract. 2010, 8, 4. [Google Scholar] [CrossRef] [Green Version]
- Tedaldi, G.; Tebaldi, M.; Zampiga, V.; Danesi, R.; Arcangeli, V.; Ravegnani, M.; Cangini, I.; Pirini, F.; Petracci, E.; Rocca, A.; et al. Multiple-gene panel analysis in a case series of 255 women with hereditary breast and ovarian cancer. Oncotarget 2017, 8, 47064–47075. [Google Scholar] [CrossRef]
- Barber, L.M.; Barlow, R.A.; Meyer, S.; White, D.J.; Will, A.M.; Eden, T.O.B.; Taylor, G.M. Inherited FANCD1/BRCA2 exon 7 splice mutations associated with acute myeloid leukaemia in Fanconi anaemia D1 are not found in sporadic childhood leukaemia. Br. J. Haematol. 2005, 130, 796–797. [Google Scholar] [CrossRef] [PubMed]
- Lindor, N.M.; Guidugli, L.; Wang, X.; Vallée, M.P.; Monteiro, A.N.A.; Tavtigian, S.; Goldgar, D.E.; Couch, F.J. A review of a multifactorial probability-based model for classification of BRCA1 and BRCA2 variants of uncertain significance (VUS). Hum. Mutat. 2012, 33, 8–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, A.M.; Blackwood, M.A.; Antin-Ozerkis, D.; Shih, H.A.; Calzone, K.; Colligon, T.A.; Seal, S.; Collins, N.; Stratton, M.R.; Weber, B.L.; et al. Germline mutations in BRCA1 and BRCA2 in breast-ovarian families from a breast cancer risk evaluation clinic. J. Clin. Oncol. 2001, 19, 2247–2253. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Meng, H.; Yao, L.; Lv, M.; Bai, J.; Zhang, J.; Wang, L.; Ouyang, T.; Li, J.; Wang, T.; et al. Germline mutations in cancer susceptibility genes in a large series of unselected breast cancer patients. Clin. Cancer Res. 2017, 23, 6113–6119. [Google Scholar] [CrossRef] [Green Version]
- Bell, D.W.; Varley, J.M.; Szydlo, T.E.; Kang, D.H.; Wahrer, D.C.; Shannon, K.E.; Lubratovich, M.; Verselis, S.J.; Isselbacher, K.J.; Fraumeni, J.F.; et al. Heterozygous germ line HCHK2 mutations in Li-Fraumeni syndrome. Science 1999, 286, 2528–2531. [Google Scholar] [CrossRef] [PubMed]
- Schubert, S.; Van Luttikhuizen, J.L.; Auber, B.; Schmidt, G.; Hofmann, W.; Penkert, J.; Davenport, C.F.; Hille-Betz, U.; Wendeburg, L.; Bublitz, J.; et al. The identification of pathogenic variants in BRCA1/2 negative, high risk, hereditary breast and/or ovarian cancer patients: High frequency of FANCM pathogenic variants. Int. J. Cancer 2019, 144, 2683–2694. [Google Scholar] [CrossRef]
- Marabelli, M.; Cheng, S.-C.; Parmigiani, G. Penetrance of ATM gene mutations in breast cancer: A meta-analysis of different measures of risk. Genet. Epidemiol. 2016, 40, 425–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schumacher, F.R.; Al Olama, A.A.; Berndt, S.I.; Benlloch, S.; Ahmed, M.; Saunders, E.J.; Dadaev, T.; Leongamornlert, D.; Anokian, E.; Cieza-Borrella, C.; et al. Association analyses of more than 140,000 men identify 63 new prostate cancer susceptibility loci. Nat. Genet. 2018, 50, 928–936. [Google Scholar] [CrossRef] [Green Version]
- Roberts, N.J.; Jiao, Y.; Yu, J.; Kopelovich, L.; Petersen, G.M.; Bondy, M.L.; Gallinger, S.; Schwartz, A.G.; Syngal, S.; Cote, M.L.; et al. ATM mutations in patients with hereditary pancreatic cancer. Cancer Discov. 2012, 2, 41–46. [Google Scholar] [CrossRef] [Green Version]
- Brohl, A.S.; Patidar, R.; Turner, C.E.; Wen, X.; Song, Y.K.; Wei, J.S.; Calzone, K.A.; Khan, J. Frequent inactivating germline mutations in DNA repair genes in patients with Ewing sarcoma. Genet. Med. 2017, 19, 955–958. [Google Scholar] [CrossRef] [Green Version]
- Ballinger, M.L.; Goode, D.L.; Ray-Coquard, I.; James, P.A.; Mitchell, G.; Niedermayr, E.; Puri, A.; Schiffman, J.D.; Dite, G.S.; Cipponi, A.; et al. Monogenic and polygenic determinants of sarcoma risk: An international genetic study. Lancet Oncol. 2016, 17, 1261–1271. [Google Scholar] [CrossRef]
- Pugh, T.J.; Morozova, O.; Attiyeh, E.F.; Asgharzadeh, S.; Wei, J.S.; Auclair, D.; Carter, S.L.; Cibulskis, K.; Hanna, M.; Kiezun, A.; et al. The Genetic landscape of high-risk neuroblastoma. Nat. Genet. 2013, 45, 279–284. [Google Scholar] [CrossRef] [Green Version]
- Win, A.K.; Reece, J.C.; Dowty, J.G.; Buchanan, D.D.; Clendenning, M.; Rosty, C.; Southey, M.C.; Young, J.P.; Cleary, S.P.; Kim, H.; et al. Risk of extracolonic cancers for people with biallelic and monoallelic mutations in MUTYH. Int. J. Cancer 2016, 139, 1557–1563. [Google Scholar] [CrossRef] [Green Version]
- Tischkowitz, M.; Huang, S.; Banerjee, S.; Hague, J.; Hendricks, W.P.D.; Huntsman, D.G.; Lang, J.D.; Orlando, K.A.; Oza, A.M.; Pautier, P.; et al. Small-Cell Carcinoma of the Ovary, Hypercalcemic Type-Genetics, New Treatment Targets, and Current Management Guidelines. Clin. Cancer Res. 2020, 26, 3908–3917. [Google Scholar] [CrossRef] [Green Version]
- Schoenfeld, A.J.; Bandlamudi, C.; Lavery, J.A.; Montecalvo, J.; Namakydoust, A.; Rizvi, H.; Egger, J.; Concepcion, C.P.; Paul, S.; Arcila, M.E.; et al. The genomic landscape of SMARCA4 alterations and associations with outcomes in patients with lung Cancer. Clin. Cancer Res. 2020, 26, 5701–5708. [Google Scholar] [CrossRef]
- Kolin, D.L.; Quick, C.M.; Dong, F.; Fletcher, C.D.M.; Stewart, C.J.R.; Soma, A.; Hornick, J.L.; Nucci, M.R.; Howitt, B.E. SMARCA4-deficient uterine sarcoma and undifferentiated endometrial carcinoma are distinct clinicopathologic entities. Am. J. Surg. Pathol. 2020, 44, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Lhota, F.; Zemankova, P.; Kleiblova, P.; Soukupova, J.; Vocka, M.; Stranecky, V.; Janatova, M.; Hartmannova, H.; Hodanova, K.; Kmoch, S.; et al. Hereditary truncating mutations of DNA repair and other genes in BRCA1/BRCA2/PALB2-negatively tested breast cancer patients. Clin. Genet. 2016, 90, 324–333. [Google Scholar] [CrossRef]
- Carter, N.J.; Marshall, M.L.; Susswein, L.R.; Zorn, K.K.; Hiraki, S.; Arvai, K.J.; Torene, R.I.; McGill, A.K.; Yackowski, L.; Murphy, P.D.; et al. Germline pathogenic variants identified in women with ovarian tumors. Gynecol. Oncol. 2018, 151, 481–488. [Google Scholar] [CrossRef]
- Lee, C.; Banerjee, T.; Gillespie, J.; Ceravolo, A.; Parvinsmith, M.R.; Starita, L.; Fields, S.; Toland, A.E.; Parvin, J.D. Functional analysis of BARD1 missense variants in homology directed repair of DNA double strand breaks. Hum. Mutat. 2015, 36, 1205–1214. [Google Scholar] [CrossRef] [PubMed]
- Weber-Lassalle, N.; Borde, J.; Weber-Lassalle, K.; Horváth, J.; Niederacher, D.; Arnold, N.; Kaulfuß, S.; Ernst, C.; Paul, V.G.; Honisch, E.; et al. Germline loss-of-function variants in the BARD1 gene are associated with early-onset familial breast cancer but not ovarian cancer. Breast Cancer Res. 2019, 21, 55. [Google Scholar] [CrossRef] [Green Version]
- Thompson, D.; Easton, D.; Breast Cancer Linkage Consortium. Variation in BRCA1 Cancer Risks by Mutation Position. Cancer Epidemiol. Biomark. Prev. 2002, 11, 329–336. [Google Scholar] [PubMed]
- Rebbeck, T.R.; Mitra, N.; Wan, F.; Sinilnikova, O.M.; Healey, S.; McGuffog, L.; Mazoyer, S.; Chenevix-Trench, G.; Easton, D.F.; Antoniou, A.C.; et al. Association of type and location of BRCA1 and BRCA2 mutations with risk of breast and ovarian cancer. JAMA 2015, 313, 1347–1361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antoniou, A.C.; Goldgar, D.E.; Andrieu, N.; Chang-Claude, J.; Brohet, R.; Rookus, M.A.; Easton, D.F. A weighted cohort approach for analysing factors modifying disease risks in carriers of high-risk susceptibility genes. Genet. Epidemiol. 2005, 29, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Terry, M.B.; Liao, Y.; Whittemore, A.S.; Leoce, N.; Buchsbaum, R.; Zeinomar, N.; Dite, G.S.; Chung, W.K.; Knight, J.A.; Southey, M.C.; et al. 10-year performance of four models of breast cancer risk: A validation study. Lancet Oncol. 2019, 20, 504–517. [Google Scholar] [CrossRef]
- Kramer, I.; Hooning, M.J.; Mavaddat, N.; Hauptmann, M.; Keeman, R.; Steyerberg, E.W.; Giardiello, D.; Antoniou, A.C.; Pharoah, P.D.P.; Canisius, S.; et al. Breast cancer polygenic risk score and contralateral breast cancer risk. Am. J. Hum. Genet. 2020, 107, 837–848. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Risk Score | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 20 |
| Number of patients | 1 | 2 | 15 | 47 | 46 | 36 | 25 | 12 | 9 | 8 | 2 | 5 | 1 | 1 | 1 | 1 | 1 |
| ∑ | 18 | 154 | 41 | ||||||||||||||
| Percentage [%] | 0.5 | 0.9 | 7.0 | 22.0 | 21.6 | 17.0 | 11.7 | 5.6 | 4.2 | 3.8 | 0.9 | 2.3 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 |
| ∑ | 8.4% | 72.3% | 19.3% | ||||||||||||||
| Classification | Low Risk | Intermediate Risk | High Risk | ||||||||||||||
| Gene | DNA Level | Protein Level | Cancer Type | Reference |
|---|---|---|---|---|
| BRCA1 (NM_007294.4) | c.68_69del | p.(Glu23Valfs*17) | BC, 1 patient | Struewing et al., 1995 [16] |
| c.181T>G | p.(Cys61Gly) | BC, 3 patients | Friedman et al., 1994 [17] | |
| c.191G>A | p.(Cys64Tyr) | BC + OC, 1 patient | Couch et al., 1996 [18] | |
| c.213-12A>G | OC, 1 patient | Hoffman et al., 1998 [19] | ||
| c.427G>T | p.(Glu143*) | BC, 1 patient | Shattuck-Eidens et al., 1997 [20] | |
| c.1510del | p.(Arg504Valfs*28) | BC + OC, 1 patient | Machackova et al., 2008 [21] | |
| c.3481_3491del | p.(Glu1161Phefs*3) | OC, 1 patient | Struewing et al., 1995 [16] | |
| c.4183C>T | p.(Gln1395*) | OC, 2 patients | Langston et al., 1996 [22] | |
| c.4689C>G | p.(Tyr1563*) | BC, 1 patient | Serova et al., 1996 [23] | |
| c.4986+3G>C | p.(Met1663Valfs*14) | BC, 2 patients | Adem et al., 2003 [24] | |
| c.5080G>T | p.(Glu1694*) | BC, 1 patient unknown, 1 patient | Shattuck-Eidens et al., 1997 [20] | |
| c.5266dup | p.(Gln1756Profs*74) | BC, 2 patients | Simard et al., 1994 [25] | |
| c.5510G>A | p.(Trp1837*) | OC, 1 patient | Couch et al., 1996 [18] | |
| BRCA2 (NM_000059.3) | c.110C>G | p.(Ser37*) | BC, 1 patient | Tung et al., 2015 [26] |
| c.1002del | p.(His334Glnfs*15) | BC + OC, 1 patient | Borg et al., 2010 [27] | |
| c.4131_4132insTGAGGA | p.(Thr1378*) | BC, 1 patient | Delgado et al., 2002 [28] | |
| c.5603_5606del | p.(Asp1868Valfs*5) | BC, 1 patient | Li et al., 2019 [29] | |
| c.5609_5610delTCinsA | p.(Phe1870*) | BC, 1 patient | Howlett et al., 2002 (Fanconi Anemia) [30] Tea et al., 2014 (Breast Cancer) [31] | |
| c.5645C>A | p.(Ser1882*) | BC, 1 patient unknown, 1 patient | De Benedetti et al., 1998 [32] | |
| c.5682C>G | p.(Tyr1894*) | PC, 1 patient | Risch et al., 2001 [33] | |
| c.6408_6414del | p.(Asn2137Lysfs*29) | BC, 1 patient | Tamboom et al., 2010 [34] | |
| c.6998dup | p.(Pro2334Thrfs*6) | BC, 1 patient | Tedaldi et al., 2017 [35] | |
| c.7878G>C | p.(Trp2626Cys) | BC, 2 patients | Barber et al., 2005 (Fanconi Anemia) [36] Lindor et al., 2012 (Breast Cancer) [37] | |
| c.8575del | p.(Gln2859Lysfs*4) | BC, 1 patient | Martin et al., 2001 [38] | |
| c.9352_9353del | p.(Met3118Valfs*31) | BC, 1 patient | Sun et al., 2017 [39] | |
| CHEK2 (NM_007194.4) | c.1100del | p.(Thr367Metfs*15) | BC, 2 patients | Bell et al., 1999 [40] |
| RAD51C | Deletion Exon 5-9 | BC, 1 patient | Schubert et al., 2019 [41] | |
| Gene | Transcript ID/RefSeq | DNA Level | Protein Level | dbSNP | MAF | Classification ACMG |
|---|---|---|---|---|---|---|
| ATM | ENST00000675843.1/NM_000051.4 | c.7327C>T | p.(Arg2443*) | rs121434220 | 3.983 × 10−6 | Class 5 |
| BARD1 | ENST00000260947.9/NM_000465.4 | c.212G>T | p.(Cys71Phe) | rs1064793959 | 3.185 × 10−5 | Class 3 |
| MUTYH | ENST00000456914.7/NM_001048174.2 | c.919C>T | p.(Arg307Trp) | rs759822330 | 2.388 × 10−5 | Class 3 |
| SMARCA4 | ENST00000344626.10/NM_003072.5 | c.2275-3C>A | rs117611401 | 2.521 × 10−3 | Class 3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Resch, L.D.; Hotz, A.; Zimmer, A.D.; Komlosi, K.; Singh, N.; Tzschach, A.; Windfuhr-Blum, M.; Juhasz-Boess, I.; Erbes, T.; Fischer, J.; et al. The Importance of Extended Analysis Using Current Molecular Genetic Methods Based on the Example of a Cohort of 228 Patients with Hereditary Breast and Ovarian Cancer Syndrome. Genes 2021, 12, 1483. https://doi.org/10.3390/genes12101483
Resch LD, Hotz A, Zimmer AD, Komlosi K, Singh N, Tzschach A, Windfuhr-Blum M, Juhasz-Boess I, Erbes T, Fischer J, et al. The Importance of Extended Analysis Using Current Molecular Genetic Methods Based on the Example of a Cohort of 228 Patients with Hereditary Breast and Ovarian Cancer Syndrome. Genes. 2021; 12(10):1483. https://doi.org/10.3390/genes12101483
Chicago/Turabian StyleResch, Luise D., Alrun Hotz, Andreas D. Zimmer, Katalin Komlosi, Nina Singh, Andreas Tzschach, Marisa Windfuhr-Blum, Ingolf Juhasz-Boess, Thalia Erbes, Judith Fischer, and et al. 2021. "The Importance of Extended Analysis Using Current Molecular Genetic Methods Based on the Example of a Cohort of 228 Patients with Hereditary Breast and Ovarian Cancer Syndrome" Genes 12, no. 10: 1483. https://doi.org/10.3390/genes12101483
APA StyleResch, L. D., Hotz, A., Zimmer, A. D., Komlosi, K., Singh, N., Tzschach, A., Windfuhr-Blum, M., Juhasz-Boess, I., Erbes, T., Fischer, J., & Alter, S. (2021). The Importance of Extended Analysis Using Current Molecular Genetic Methods Based on the Example of a Cohort of 228 Patients with Hereditary Breast and Ovarian Cancer Syndrome. Genes, 12(10), 1483. https://doi.org/10.3390/genes12101483