
Exploring Semi-Quantitative Metagenomic Studies Using Oxford Nanopore Sequencing: A Computational and Experimental Protocol

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. ONT Microbiome-Like Simulated Data
2.3. Bioinformatics Workflow for Taxonomic Binning of ONT Sequencing
- Centrifuge-based taxonomic binning: Centrifuge [28] was used for the taxonomic binning of individual ONT reads using their comprehensive reference database of more than 8000 reference genomes from prokaryotes and viruses (including human reference genome). This step allows for excluding human sequence reads. To remove spurious taxonomic assignments, we additionally mapped a read bin product of the initial Centrifuge classification against the corresponding reference genome from the centrifuge database using minimap2 with the map-ont option optimized for ONT reads [29]. Based on simulation experiment results, only sequences with a minimum mapQ score of 5 were retained for subsequent analyses (see results). A species relative abundance table was generated by summing the counts of each taxonomic bin (NCBI taxonomy identifiers) from the filtered Centrifuge results. This relative abundance table was combined with the experiment metadata information and a reference taxonomic table reconstructed from Centrifuge NCBI taxonomy identifiers using the R package taxize v0.9.95 [30] using phyloseq v1.30.0 [31], generating a phyloseq-class R object. This object can be used for microbial ecology analyses (rarefaction, alpha-diversity, beta-diversity, and differential abundance analysis).
- IGC-based taxonomic binning: A complementary approach consisting of quantifying the abundance of microbial genes. Here, ONT reads were aligned against the Integrated Gene Catalog of reference genes of the human gut microbiome (IGC) catalog [24] using minimap2 with the map-ont option [29]. The alignment of long ONT reads over short or fragmented IGC genes provided two different types of multiple mappings (an ONT read mapped over several genes). First, a long ONT read could cover a genomic region harboring more than one gene, so different genes can be mapped over nonoverlapping regions of an ONT read, providing a structural annotation of the corresponding DNA region. Second, multiple genes can also be mapped in overlapping regions of a read. These second multiple mappings were filtered out using the GenomicRanges and plyrRangesR packages [32,33], allowing for retaining the genes with the highest mapQ score and sequence identity across each alignment region. The raw gene abundance table was reconstructed by counting the number of times each gene was mapped by ONT reads. From this gene count table, the abundance of metagenomic species (MGS; coabundant gene groups clustered from 1267 human gut metagenomes used to construct the IGC [25]) was estimated as the mean value of the 50 most connected genes in each MGS as proposed in the original study [25].
2.4. Bioinformatics Workflow for Functional Profiling of ONT Sequencing
2.5. Study Participants for Wet-Lab Experiments
2.6. Sample Collection and Bacterial DNA Extraction for Preanalytic Protocol Experiments: Fresh Stools Were Collected with Two Different Methods
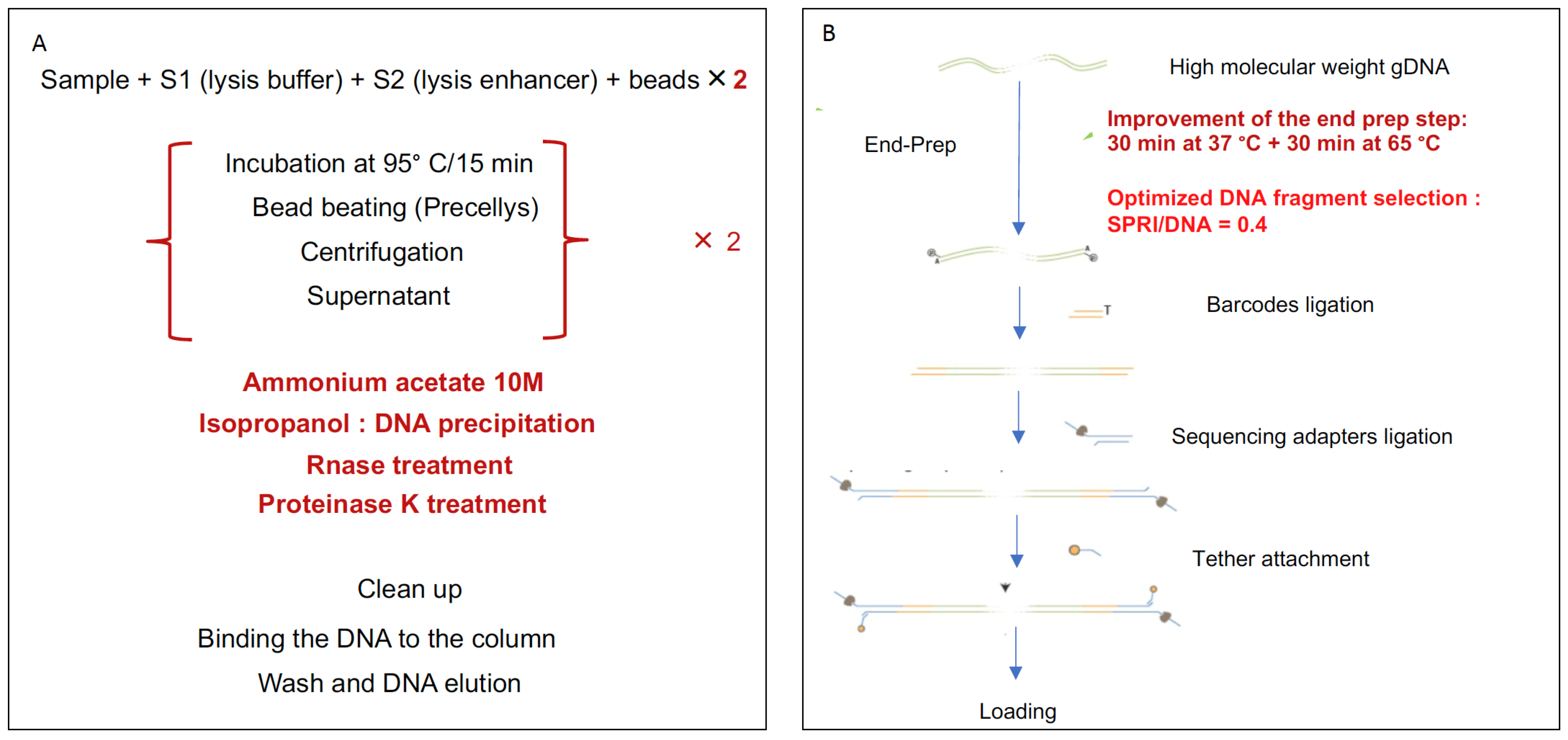
2.7. Optimization of DNA Extraction, DNA Fragmentation, and End Repair
2.8. Library Preparation and Sequencing
2.9. Statistical–Ecological Analyses
3. Results
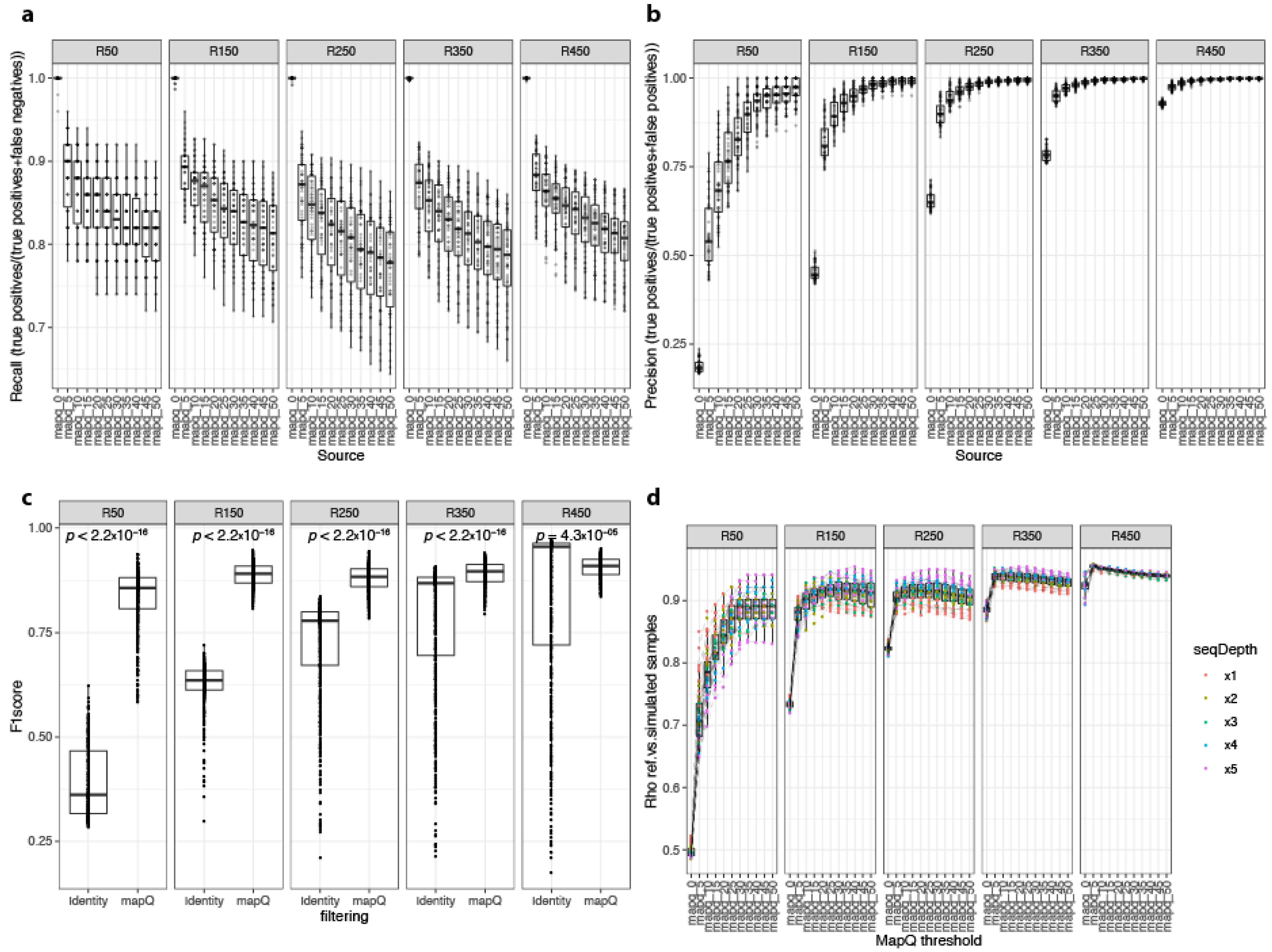
3.1. Metagenome Simulations Identified Key Pipeline Parameters for ONT Microbiome Quantification
3.2. Alignment Identity and Alignment Quality Affect Workflow Precision
3.3. Validation of the Bioinformatics Pipeline with ZymoBIOMICS Mock Community
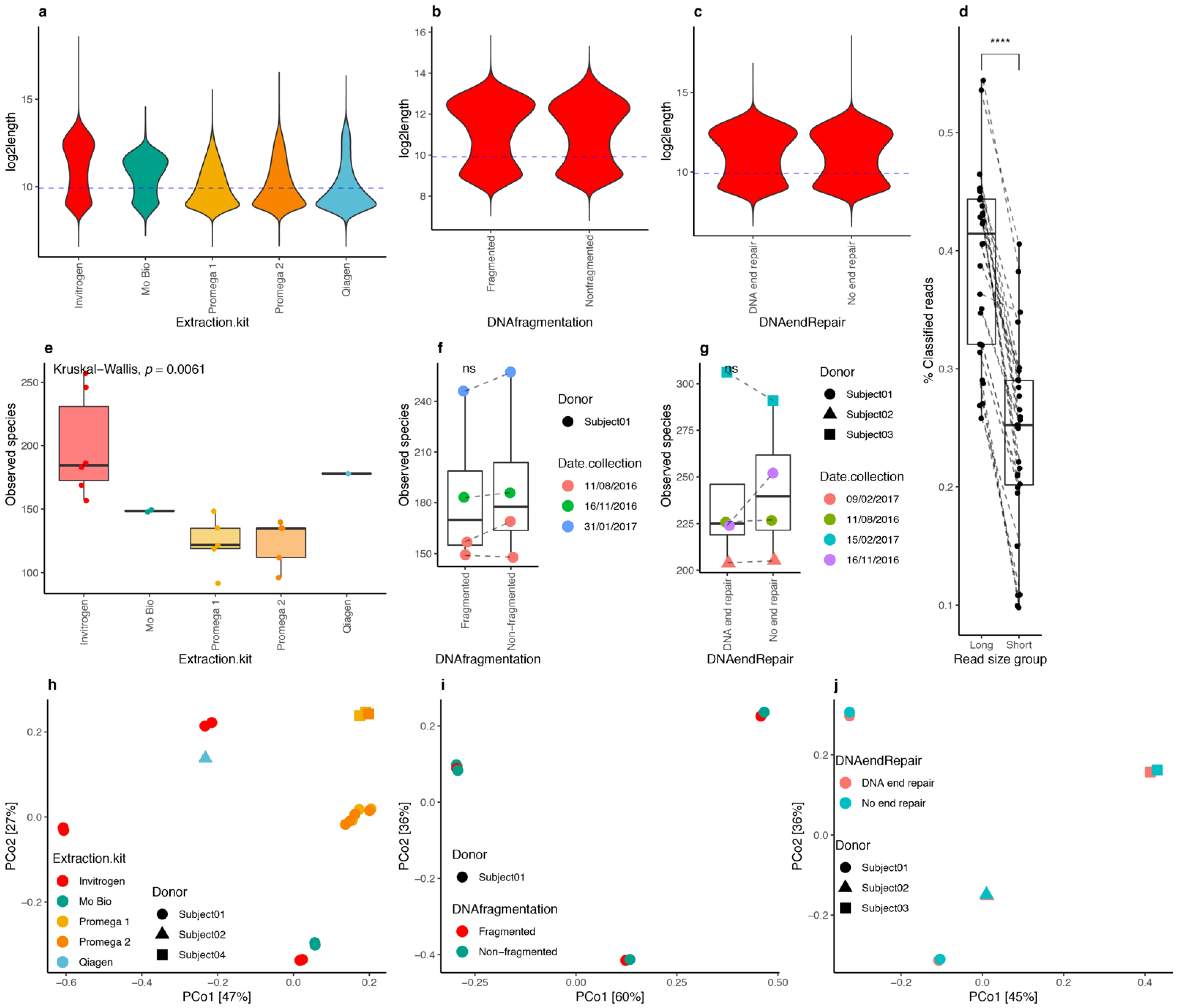
3.4. DNA Extraction Kits Influenced Read Length Distribution
3.5. DNA Fragmentation and End Repair
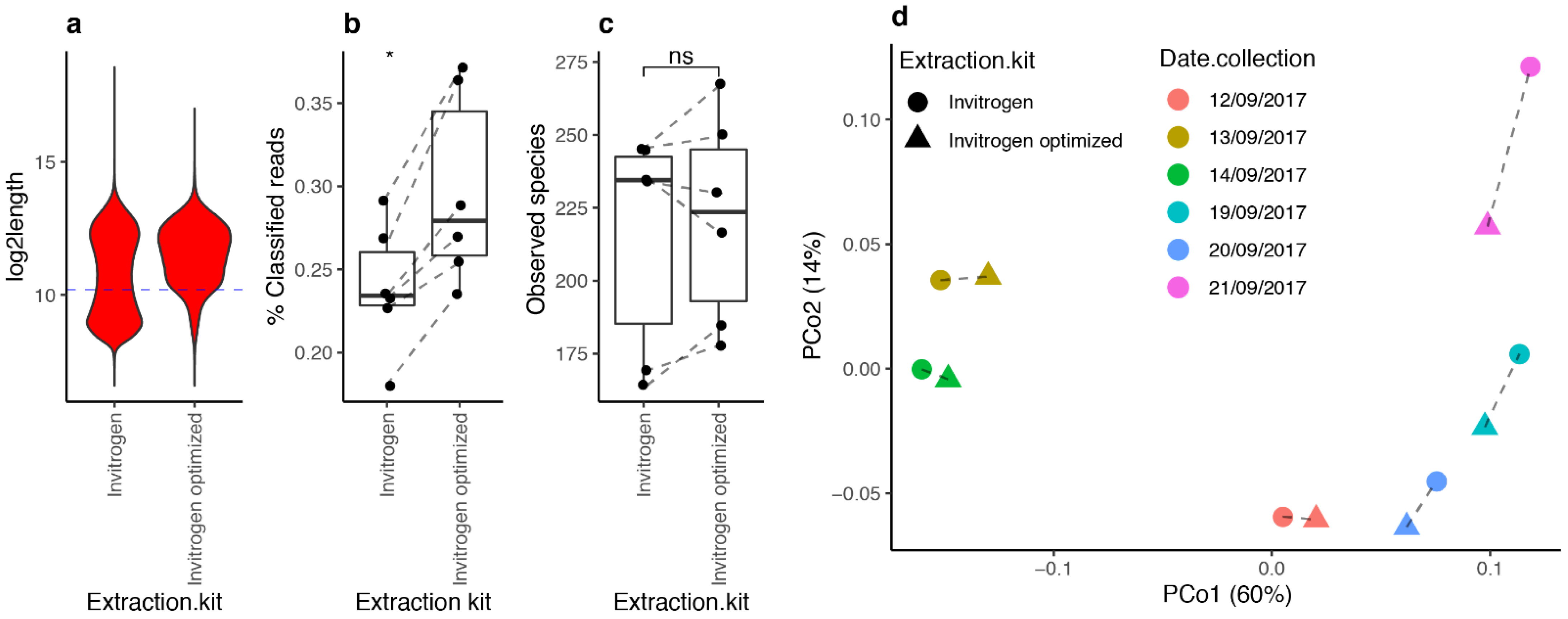
3.6. Optimized DNA Extraction Protocol Improved ONT Read Length and Microbial Diversity Estimation
3.7. Impact of Stool Sampling and Storage on Sequence Length and Diversity
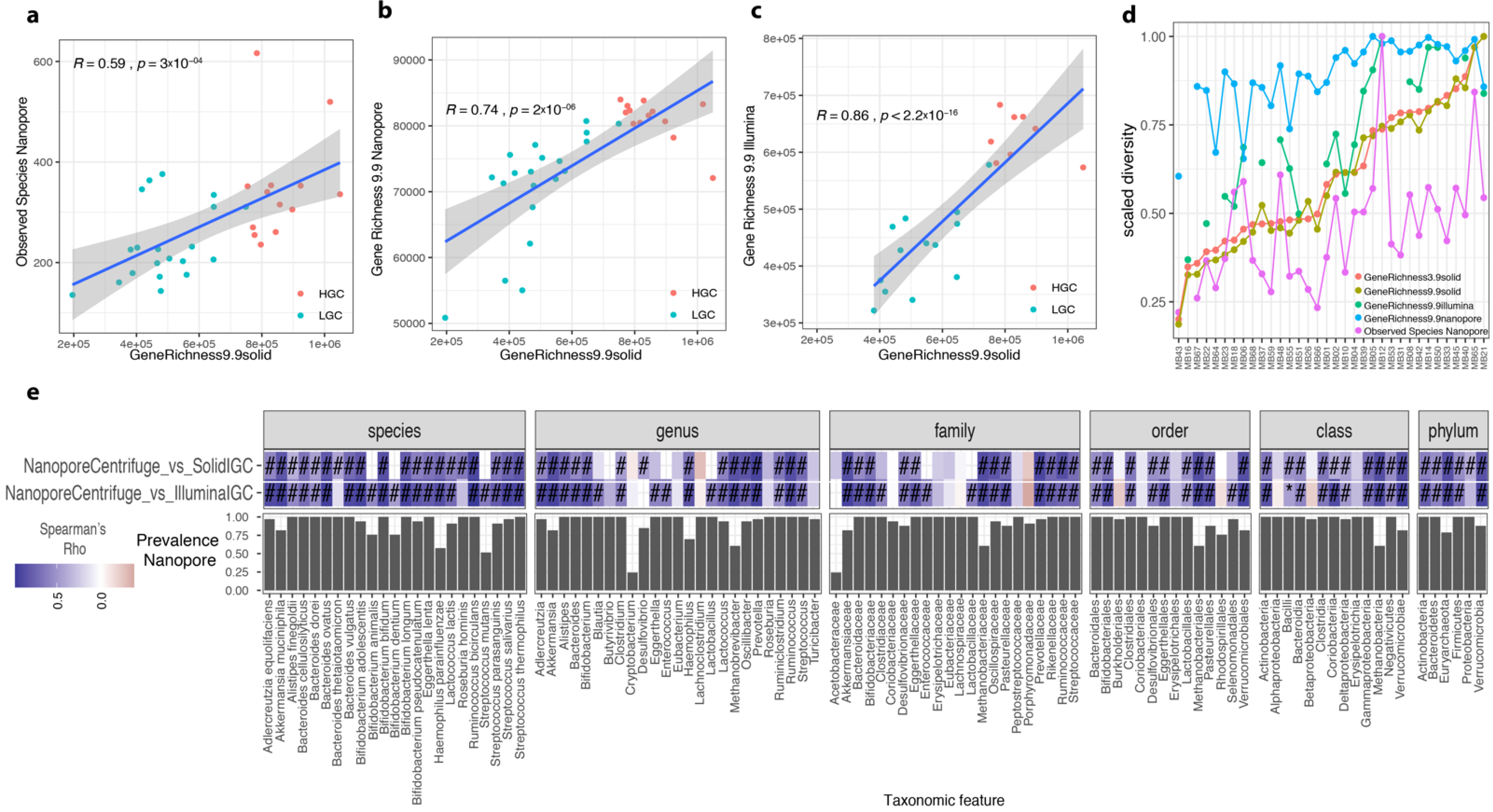
3.8. Optimized ONT Protocol Compared with Illumina SOLiD Sequencing
3.9. ONT Pipeline Detects Target Species and Functional Profiles
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Prifti, E.; Chevaleyre, Y.; Hanczar, B.; Belda, E.; Danchin, A.; Clément, K.; Zucker, J.D. Interpretable and accurate prediction models for metagenomics data. Gigascience 2020, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vieira-Silva, S.; Falony, G.; Belda, E.; Nielsen, T.; Aron-Wisnewsky, J.; Chakaroun, R.; Forslund, S.K.; Assmann, K.; Valles-Colomer, M.; Nguyen, T.T.D.; et al. Statin Therapy Is Associated with Lower Prevalence of Gut Microbiota Dysbiosis. Nature 2020, 581, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Aron-Wisnewsky, J.; Gaborit, B.; Dutour, A.; Clement, K. Gut microbiota and non-alcoholic fatty liver disease: New insights. Clin. Microbiol. Infect. 2013, 19, 338–348. [Google Scholar] [CrossRef] [Green Version]
- Aron-Wisnewsky, J.; Vigliotti, C.; Witjes, J.; Le, P.; Holleboom, A.G.; Verheij, J.; Nieuwdorp, M.; Clément, K. Gut microbiota and human NAFLD: Disentangling microbial signatures from metabolic disorders. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 279–297. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Qin, N.; Yang, F.; Li, A.; Prifti, E.; Chen, Y.; Shao, L.; Guo, J.; Chatelier, E.; Yao, J.; Wu, L.; et al. Alterations of the human gut microbiome in liver cirrhosis. Nature 2014, 513, 59–64. [Google Scholar] [CrossRef]
- Cotillard, A.; Kennedy, S.P.; Kong, L.C.; Prifti, E.; Pons, N.; Le Chatelier, E.; Almeida, M.; Quinquis, B.; Levenez, F.; Galleron, N.; et al. Dietary intervention impact on gut microbial gene richness. Nature 2013, 500, 585–588. [Google Scholar] [CrossRef]
- Aron-Wisnewsky, J.; Prifti, E.; Belda, E.; Ichou, F.; Kayser, B.D.; Dao, M.C.; Verger, E.; Hedjazil, L.; Bouillot, J.-L.; Chevallier, J.-M.; et al. Major microbiota dysbiosis in severe obesity: Fate after bariatric surgery. Gut 2019, 68, 70–82. [Google Scholar] [CrossRef]
- Moss, E.L.; Maghini, D.G.; Bhatt, A.S. Complete, closed bacterial genomes from microbiomes using nanopore sequencing. Nat. Biotechnol. 2020, 38, 701–707. [Google Scholar] [CrossRef] [Green Version]
- Maghini, D.G.; Moss, E.L.; Vance, S.E.; Bhatt, A.S. Improved high-molecular-weight DNA extraction, nanopore sequencing and metagenomic assembly from the human gut microbiome. Nat. Protoc. 2021, 16, 458–471. [Google Scholar] [CrossRef]
- Deng, X.; Achari, A.; Federman, S.; Yu, G.; Somasekar, S.; Bártolo, I.; Yagi, S.; Mbala-Kingebeni, P.; Kapetshi, J.; Ahuka-Mundeke, S.; et al. Metagenomic sequencing with spiked primer enrichment for viral diagnostics and genomic surveillance. Nat. Microbiol. 2020, 5, 443–454. [Google Scholar] [CrossRef] [Green Version]
- Sanderson, N.D.; Street, T.L.; Foster, D.; Swann, J.; Atkins, B.L.; Brent, A.J.; McNally, M.A.; Oakley, S.; Taylor, A.; Peto, T.E.A.; et al. Real-time analysis of nanopore-based metagenomic sequencing from infected orthopaedic devices. BMC Genom. 2018, 19, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urban, L.; Holzer, A.; Baronas, J.J.; Hall, M.B.; Braeuninger-Weimer, P.; Scherm, M.J.; Kunz, D.J.; Perera, S.N.; Martin-Herranz, D.E.; Tipper, E.T.; et al. Freshwater monitoring by nanopore sequencing. eLife 2021, 10, e61504. [Google Scholar] [CrossRef] [PubMed]
- Raes, J.; Bork, P. MAronolecular eco-systems biology: Towards an understanding of community function. Nat. Rev. Genet. 2008, 6, 693–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, M.B.; Highlander, S.; Anderson, E.L.; Li, W.; Dayrit, M.; Klitgord, N.; Fabani, M.M.; Seguritan, V.; Green, J.; Pride, D.T.; et al. Library preparation methodology can influence genomic and functional predictions in human microbiome research. Proc. Natl. Acad. Sci. USA 2015, 112, 14024–14029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, N.; Walker, A.; Berry, S.H.; Duncan, S.; Farquarson, F.M.; Louis, P.; Thomson, J.M.; Satsangi, J.; Flint, H.J.; Parkhill, J.; et al. The Impact of Different DNA Extraction Kits and Laboratories upon the Assessment of Human Gut Microbiota Composition by 16S rRNA Gene Sequencing. PLoS ONE 2014, 9, e88982. [Google Scholar] [CrossRef]
- Voigt, A.Y.; Costea, P.I.; Kultima, J.R.; Li, S.S.; Zeller, G.; Sunagawa, S.; Bork, P. Temporal and technical variability of human gut metagenomes. Genome Biol. 2015, 16, 73. [Google Scholar] [CrossRef] [Green Version]
- Turnbaugh, P.J.; Ley, R.E.; Hamady, M.; Fraser-Liggett, C.M.; Knight, R.; Gordon, J.I. The Human Microbiome Project. Nature 2007, 449, 804–810. [Google Scholar] [CrossRef]
- Costea, P.I.; Zeller, G.; Sunagawa, S.; Pelletier, E.; Alberti, A.; Levenez, F.; Tramontano, M.; Driessen, M.; Hercog, R.; Jung, F.-E.; et al. Towards standards for human fecal sample processing in metagenomic studies. Nat. Biotechnol. 2017, 35, 1069–1076. [Google Scholar] [CrossRef]
- Harstad, H.; Ahmad, R.; Bredberg, A. Nanopore-based DNA sequencing in clinical microbiology: Preliminary assessment of basic requirements. bioRxiv 2018. [Google Scholar] [CrossRef]
- Nayfach, S.; Pollard, K.S. Toward Accurate and Quantitative Comparative Metagenomics. Cell 2016, 166, 1103–1116. [Google Scholar] [CrossRef] [Green Version]
- Cranmer, K.; Brehmer, J.; Louppe, G. The frontier of simulation-based inference. Proc. Natl. Acad. Sci. USA 2020, 117, 30055–30062. [Google Scholar] [CrossRef]
- Li, J.; Jia, H.; Cai, X.; Zhong, H.; Feng, Q.; Sunagawa, S.; Arumugam, M.; Kultima, J.R.; Prifti, E.; Nielsen, T.; et al. Supporting data for the paper: “An integrated catalog of reference genes in the human gut microbiome”. Nat. Biotechnol. 2014, 32, 834–841. [Google Scholar] [CrossRef]
- Nielsen, H.B.; Almeida, M.; Juncker, A.S.; Rasmussen, S.; Li, J.; Sunagawa, S.; Plichta, D.R.; Gautier, L.; Pedersen, A.G.; Le Chatelier, E.; et al. Identification and assembly of genomes and genetic elements in complex metagenomic samples without using reference genomes. Nat. Biotechnol. 2014, 32, 822–828. [Google Scholar] [CrossRef]
- Fritz, A.; Hofmann, P.; Majda, S.; Dahms, E.; Dröge, J.; Fiedler, J.; Lesker, T.R.; Belmann, P.; DeMaere, M.Z.; Darling, A.E.; et al. CAMISIM: Simulating metagenomes and microbial communities. Microbiome 2019, 7, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Available online: https://nanoporetech.com/community (accessed on 18 November 2020).
- Kim, D.; Song, L.; Breitwieser, F.P.; Salzberg, S.L. Centrifuge: Rapid and sensitive classification of metagenomic sequences. Genome Res. 2016, 26, 1721–1729. [Google Scholar] [CrossRef] [Green Version]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, S.; Szöcs, E. Taxize: Taxonomic search and retrieval in R. F1000Research 2013, 2, 191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrence, M.; Huber, W.; Pagès, H.; Aboyoun, P.; Carlson, M.; Gentleman, R.; Morgan, M.; Carey, V.J. Software for Computing and Annotating Genomic Ranges. PLoS Comput. Biol. 2013, 9, e1003118. [Google Scholar] [CrossRef]
- Lee, S.; Cook, D.; Lawrence, M. Plyranges: A grammar of genomic data transformation. Genome Biol. 2019, 20, 1–10. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Sato, Y.; Furumichi, M.; Tanabe, M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012, 40, D109–D114. [Google Scholar] [CrossRef] [Green Version]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; Stevens, M.H.H.; et al. Vegan: Community Ecology Package. 2019. Available online: https://cran.r-project.org/web/packages/vegan/index.html (accessed on 18 November 2020).
- Marcelino, V.R.; Clausen, P.T.L.C.; Buchmann, J.P.; Wille, M.; Iredell, J.R.; Meyer, W.; Lund, O.; Sorrell, T.C.; Holmes, E.C. CCMetagen: Comprehensive and accurate identification of eukaryotes and prokaryotes in metagenomic data. Genome Biol. 2020, 21, 1–15. [Google Scholar] [CrossRef] [PubMed]
- James@cancer. CoreGenomics: How Do SPRI Beads Work? CoreGenomics. 2012. Available online: http://core-genomics.blogspot.com/2012/04/how-do-spri-beads-work.html (accessed on 18 November 2020).
- Browne, H.P.; Forster, S.C.; Anonye, B.O.; Kumar, N.; Neville, B.A.; Stares, M.D.; Goulding, D.; Lawley, T.D. Culturing of ‘unculturable’ human microbiota reveals novel taxa and extensive sporulation. Nature 2016, 533, 543–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krajmalnik-Brown, R.; Ilhan, Z.-E.; Kang, D.-W.; DiBaise, J.K. Effects of Gut Microbes on Nutrient Absorption and Energy Regulation. Nutr. Clin. Pract. 2012, 27, 201–214. [Google Scholar] [CrossRef] [Green Version]
- Ammer-Herrmenau, C.; Pfisterer, N.; Berg, T.V.D.; Gavrilova, I.; Amanzada, A.; Singh, S.K.; Khalil, A.; Alili, R.; Belda, E.; Clement, K.; et al. Comprehensive Wet-Bench and Bioinformatics Workflow for Complex Microbiota Using Oxford Nanopore Technologies. mSystems 2021, 6, e00750-21. [Google Scholar] [CrossRef] [PubMed]
- Bäckhed, F.; Roswall, J.; Peng, Y.; Feng, Q.; Jia, H.; Kovatcheva-Datchary, P.; Li, Y.; Xia, Y.; Xie, H.; Zhong, H.; et al. Dynamics and Stabilization of the Human Gut Microbiome during the First Year of Life. Cell Host Microbe 2015, 17, 690–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yachida, S.; Mizutani, S.; Shiroma, H.; Shiba, S.; Nakajima, T.; Sakamoto, T.; Watanabe, H.; Masuda, K.; Nishimoto, Y.; Kubo, M.; et al. Metagenomic and metabolomic analyzes reveal distinct stage-specific phenotypes of the gut microbiota in colorectal cancer. Nat. Med. 2019, 25, 968–976. [Google Scholar] [CrossRef]
- Wesolowska-Andersen, A.; Bahl, M.I.; Carvalho, V.; Kristiansen, K.; Sicheritz-Pontén, T.; Gupta, R.; Licht, T.R. Choice of bacterial DNA extraction method from fecal material influences community structure as evaluated by metagenomic analysis. Microbiome 2014, 2, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McOrist, A.L.; Jackson, M.; Bird, A.R. A comparison of five methods for extraction of bacterial DNA from human faecal samples. J. Microbiol. Methods 2002, 50, 131–139. [Google Scholar] [CrossRef]
- Ariefdjohan, M.W.; Savaiano, D.A.; Nakatsu, C.H. Comparison of DNA extraction kits for PCR-DGGE analysis of human intestinal microbial communities from fecal specimens. Nutr. J. 2010, 9, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Available online: http://www.microbiome-standards.org/ (accessed on 18 November 2020).
- Ott, S.J.; Musfeldt, M.; Timmis, K.N.; Hampe, J.; Wenderoth, D.F.; Schreiber, S. In vitro alterations of intestinal bacterial microbiota in fecal samples during storage. Diagn. Microbiol. Infect. Dis. 2004, 50, 237–245. [Google Scholar] [CrossRef]
- Cardona, S.; Eck, A.; Cassellas, M.; Gallart, M.; Alastrue, C.; Dore, J.; Azpiroz, F.; Roca, J.; Guarner, F.; Manichanh, C. Storage conditions of intestinal microbiota matter in metagenomic analysis. BMC Microbiol. 2012, 12, 158. [Google Scholar] [CrossRef] [Green Version]
- Hillmann, B.; Al-Ghalith, G.A.; Shields-Cutler, R.R.; Zhu, Q.; Gohl, D.; Beckman, K.B.; Knight, R.; Knights, D. Evaluating the Information Content of Shallow Shotgun Metagenomics. mSystems 2018, 3, e00069-18. [Google Scholar] [CrossRef] [Green Version]
- Tedersoo, L.; Drenkhan, R.; Anslan, S.; Morales-Rodriguez, C.; Cleary, M. High-throughput identification and diagnostics of pathogens and pests: Overview and practical recommendations. Mol. Ecol. Resour. 2018, 19, 47–76. [Google Scholar] [CrossRef] [Green Version]
- Fox, E.J.; Reid-Bayliss, K.S.; Emond, M.J.; Loeb, L.A. Accuracy of Next Generation Sequencing Platforms. J. Next Gener. Seq. Appl. 2014, 1, 1000106. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alili, R.; Belda, E.; Le, P.; Wirth, T.; Zucker, J.-D.; Prifti, E.; Clément, K. Exploring Semi-Quantitative Metagenomic Studies Using Oxford Nanopore Sequencing: A Computational and Experimental Protocol. Genes 2021, 12, 1496. https://doi.org/10.3390/genes12101496
Alili R, Belda E, Le P, Wirth T, Zucker J-D, Prifti E, Clément K. Exploring Semi-Quantitative Metagenomic Studies Using Oxford Nanopore Sequencing: A Computational and Experimental Protocol. Genes. 2021; 12(10):1496. https://doi.org/10.3390/genes12101496
Chicago/Turabian StyleAlili, Rohia, Eugeni Belda, Phuong Le, Thierry Wirth, Jean-Daniel Zucker, Edi Prifti, and Karine Clément. 2021. "Exploring Semi-Quantitative Metagenomic Studies Using Oxford Nanopore Sequencing: A Computational and Experimental Protocol" Genes 12, no. 10: 1496. https://doi.org/10.3390/genes12101496
APA StyleAlili, R., Belda, E., Le, P., Wirth, T., Zucker, J. -D., Prifti, E., & Clément, K. (2021). Exploring Semi-Quantitative Metagenomic Studies Using Oxford Nanopore Sequencing: A Computational and Experimental Protocol. Genes, 12(10), 1496. https://doi.org/10.3390/genes12101496