
Non-Recombinogenic Functions of Rad51, BRCA2, and Rad52 in DNA Damage Tolerance

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. DSB Repair by HR
3. ssDNA Gap Filling by HR
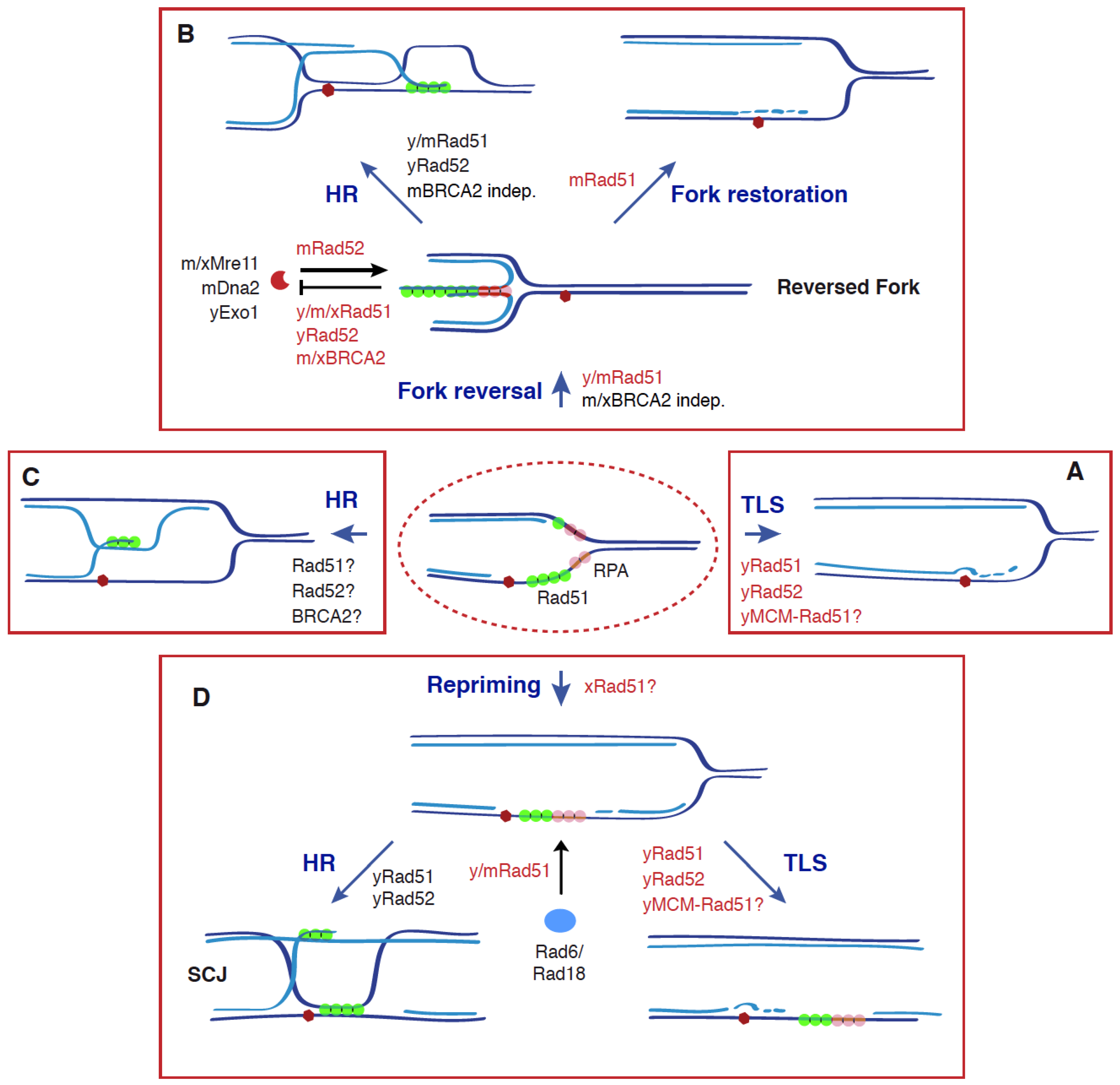
4. Non-Recombinogenic Roles of Rad51, BRCA2 and Rad52 in the Dynamics of Reversed Forks
5. Non-Recombinogenic Roles of Rad51 and Rad52 in TLS
6. Concluding Remarks
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Heyer, W.-D.; Ehmsen, K.T.; Liu, J. Regulation of Homologous Recombination in Eukaryotes. Annu. Rev. Genet. 2010, 44, 113–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhat, K.P.; Cortez, D. RPA and RAD51: Fork Reversal, Fork Protection, and Genome Stability. Nat. Struct. Mol. Biol. 2018, 25, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Qiu, S.; Jiang, G.; Cao, L.; Huang, J. Replication Fork Reversal and Protection. Front. Cell. Dev. Biol. 2021, 9, 670392. [Google Scholar] [CrossRef]
- Chen, X.; Bosques, L.; Sung, P.; Kupfer, G.M. A Novel Role for Non-Ubiquitinated FANCD2 in Response to Hydroxyurea-Induced DNA Damage. Oncogene 2016, 35, 22–34. [Google Scholar] [CrossRef]
- Cano-Linares, M.I.; Yáñez-Vilches, A.; García-Rodríguez, N.; Barrientos-Moreno, M.; González-Prieto, R.; San-Segundo, P.; Ulrich, H.D.; Prado, F. Non-recombinogenic Roles for Rad52 in Translesion Synthesis during DNA Damage Tolerance. EMBO Rep. 2021, 22, e50410. [Google Scholar] [CrossRef]
- Cabello-Lobato, M.J.; González-Garrido, C.; Cano-Linares, M.I.; Wong, R.P.; Yáñez-Vílchez, A.; Morillo-Huesca, M.; Roldán-Romero, J.M.; Vicioso, M.; González-Prieto, R.; Ulrich, H.D.; et al. Physical Interactions between MCM and Rad51 Facilitate Replication Fork Lesion Bypass and ssDNA Gap Filling by Non-Recombinogenic Functions. Cell Rep. 2021, 36, 109440. [Google Scholar] [CrossRef]
- Zhao, L.; Washington, M. Translesion Synthesis: Insights into the Selection and Switching of DNA Polymerases. Genes 2017, 8, 24. [Google Scholar] [CrossRef]
- Prakash, R.; Zhang, Y.; Feng, W.; Jasin, M. Homologous Recombination and Human Health: The Roles of BRCA1, BRCA2, and Associated Proteins. Cold Spring Harb. Perspect. Biol. 2015, 7, a016600. [Google Scholar] [CrossRef] [Green Version]
- Symington, L.S. Mechanism and Regulation of DNA End Resection in Eukaryotes. Crit. Rev. Biochem. Mol. 2016, 51, 195–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Zhao, L.; Xu, Y.; Zhao, W.; Sung, P.; Wang, H.-W. Cryo-EM Structures of Human RAD51 Recombinase Filaments during Catalysis of DNA-Strand Exchange. Nat. Struct. Mol. Biol. 2017, 24, 40–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benson, F.E.; Stasiak, A.; West, S.C. Purification and Characterization of the Human Rad51 Protein, an Analogue of E. Coli RecA. EMBO J. 1994, 13, 5764–5771. [Google Scholar] [CrossRef]
- Sung, P.; Robberson, D.L. DNA Strand Exchange Mediated by a RAD51-SsDNA Nucleoprotein Filament with Polarity Opposite to That of RecA. Cell 1995, 82, 453–461. [Google Scholar] [CrossRef] [Green Version]
- Qi, Z.; Redding, S.; Lee, J.Y.; Gibb, B.; Kwon, Y.; Niu, H.; Gaines, W.A.; Sung, P.; Greene, E.C. DNA Sequence Alignment by Microhomology Sampling during Homologous Recombination. Cell 2015, 856–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.Y.; Terakawa, T.; Qi, Z.; Steinfeld, J.B.; Redding, S.; Kwon, Y.; Gaines, W.A.; Zhao, W.; Sung, P.; Greene, E.C. Base Triplet Stepping by the Rad51/RecA Family of Recombinases. Science 2015, 349, 977–981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cloud, V.; Chan, Y.-L.; Grubb, J.; Budke, B.; Bishop, D.K. Rad51 Is an Accessory Factor for Dmc1-Mediated Joint Molecule Formation during Meiosis. Science 2012, 337, 1222–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mason, J.M.; Chan, Y.-L.; Weichselbaum, R.W.; Bishop, D.K. Non-Enzymatic Roles of Human RAD51 at Stalled Replication Forks. Nat. Commun. 2019, 10, 4410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solinger, J.A.; Kiianitsa, K.; Heyer, W.-D. Rad54, a Swi2/Snf2-like Recombinational Repair Protein, Disassembles Rad51:DsDNA Filaments. Mol. Cell 2002, 10, 1175–1188. [Google Scholar] [CrossRef]
- Prado, F.; Cortés-Ledesma, F.; Huertas, P.; Aguilera, A. Mitotic Recombination in Saccharomyces Cerevisiae. Curr. Genet. 2003, 42, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Malkova, A.; Ivanov, E.L.; Haber, J.E. Double-Strand Break Repair in the Absence of RAD51 in Yeast: A Possible Role for Break-Induced DNA Replication. Proc. Natl. Acad. Sci. USA 1996, 93, 7131–7136. [Google Scholar] [CrossRef] [Green Version]
- Prado, F.; Aguilera, A. Role of Reciprocal Exchange, One-Ended Invasion Crossover and Single-Strand Annealing on Inverted and Direct Repeat Recombination in Yeast: Different Requirements for the RAD1, RAD10, and RAD52 Genes. Genetics 1995, 139, 109–123. [Google Scholar] [CrossRef]
- Mortensen, U.H.; Bendixen, C.; Sunjevaric, I.; Rothstein, R. DNA Strand Annealing Is Promoted by the Yeast Rad52 Protein. Proc. Natl. Acad. Sci. USA 1996, 93, 10729–10734. [Google Scholar] [CrossRef] [Green Version]
- Petukhova, G.; Stratton, S.A.; Sung, P. Single Strand DNA Binding and Annealing Activities in the Yeast Recombination Factor Rad59*. J. Biol. Chem. 1999, 274, 33839–33842. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-C.; Feng, W.; Lim, P.X.; Kass, E.M.; Jasin, M. Homology-Directed Repair and the Role of BRCA1, BRCA2, and Related Proteins in Genome Integrity and Cancer. Annu. Rev. Cancer Biol. 2017, 2, 313–336. [Google Scholar] [CrossRef]
- Feng, Z.; Scott, S.P.; Bussen, W.; Sharma, G.G.; Guo, G.; Pandita, T.K.; Powell, S.N. Rad52 Inactivation Is Synthetically Lethal with BRCA2 Deficiency. Proc. Natl. Acad. Sci. USA 2011, 108, 686–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Heyer, W.D. Who’s Who in Human Recombination: BRCA2 and RAD52. Proc. Natl. Acad. Sci. USA 2011, 108, 441–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mortensen, U.H.; Lisby, M.; Rothstein, R. Rad52. Curr. Biol. 2009, 19, R676–R677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carreira, A.; Hilario, J.; Amitani, I.; Baskin, R.J.; Shivji, M.K.K.; Venkitaraman, A.R.; Kowalczykowski, S.C. The BRC Repeats of BRCA2 Modulate the DNA-Binding Selectivity of RAD51. Cell 2009, 136, 1032–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellegrini, L.; Yu, D.S.; Lo, T.; Anand, S.; Lee, M.; Blundell, T.L.; Venkitaraman, A.R. Insights into DNA Recombination from the Structure of a RAD51–BRCA2 Complex. Nature 2002, 420, 287–293. [Google Scholar] [CrossRef]
- Yang, H.; Li, Q.; Fan, J.; Holloman, W.K.; Pavletich, N.P. The BRCA2 Homologue Brh2 Nucleates RAD51 Filament Formation at a DsDNA–SsDNA Junction. Nature 2005, 433, 653–657. [Google Scholar] [CrossRef]
- Esashi, F.; Galkin, V.E.; Yu, X.; Egelman, E.H.; West, S.C. Stabilization of RAD51 Nucleoprotein Filaments by the C-Terminal Region of BRCA2. Nat. Struct. Mol. Biol. 2007, 14, 468–474. [Google Scholar] [CrossRef]
- Prado, F.; Maya, D. Regulation of Replication Fork Advance and Stability by Nucleosome Assembly. Genes 2017, 8, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saini, N.; Ramakrishnan, S.; Elango, R.; Ayyar, S.; Zhang, Y.; Deem, A.; Ira, G.; Haber, J.E.; Lobachev, K.S.; Malkova, A. Migrating Bubble during Break-Induced Replication Drives Conservative DNA Synthesis. Nature 2013, 502, 389–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, M.A.; Kwon, Y.; Xu, Y.; Chung, W.-H.; Chi, P.; Niu, H.; Mayle, R.; Chen, X.; Malkova, A.; Sung, P.; et al. Pif1 Helicase and Polδ Promote Recombination-Coupled DNA Synthesis via Bubble Migration. Nature 2013, 502, 393–396. [Google Scholar] [CrossRef] [PubMed]
- Zou, H.; Rothstein, R. Holliday Junctions Accumulate in Replication Mutants via a RecA Homolog-Independent Mechanism. Cell 1997, 90, 87–96. [Google Scholar] [CrossRef] [Green Version]
- Clemente-Ruiz, M.; Prado, F. Chromatin Assembly Controls Replication Fork Stability. EMBO Rep. 2009, 10, 790–796. [Google Scholar] [CrossRef] [Green Version]
- Berti, M.; Cortez, D.; Lopes, M. The Plasticity of DNA Replication Forks in Response to Clinically Relevant Genotoxic Stress. Nat. Rev. Mol. Cell. Biol. 2020, 21, 633–651. [Google Scholar] [CrossRef] [PubMed]
- Jossen, R.; Bermejo, R. The DNA Damage Checkpoint Response to Replication Stress: A Game of Forks. Front. Genet. 2013, 4, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prado, F. Homologous Recombination: To Fork and Beyond. Genes 2018, 9, 603. [Google Scholar] [CrossRef] [Green Version]
- Pagès, V.; Fuchs, R.P. Uncoupling of Leading- and Lagging-Strand DNA Replication during Lesion Bypass in Vivo. Science 2003, 300, 1300–1303. [Google Scholar] [CrossRef]
- Lopes, M.; Foiani, M.; Sogo, J.M. Multiple Mechanisms Control Chromosome Integrity after Replication Fork Uncoupling and Restart at Irreparable UV Lesions. Mol. Cell 2006, 21, 15–27. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Chaudhuri, A.R.; Lopes, M.; Costanzo, V. Rad51 Protects Nascent DNA from Mre11-Dependent Degradation and Promotes Continuous DNA Synthesis. Nat. Struct. Mol. Biol. 2010, 17, 1305–1311. [Google Scholar] [CrossRef] [Green Version]
- Hoege, C.; Pfander, B.; Moldovan, G.-L.; Pyrowolakis, G.; Jentsch, S. RAD6-Dependent DNA Repair Is Linked to Modification of PCNA by Ubiquitin and SUMO. Nature 2002, 419, 135–141. [Google Scholar] [CrossRef]
- Niimi, A.; Brown, S.; Sabbioneda, S.; Kannouche, P.L.; Scott, A.; Yasui, A.; Green, C.M.; Lehmann, A.R. Regulation of Proliferating Cell Nuclear Antigen Ubiquitination in Mammalian Cells. Proc. Natl. Acad. Sci. USA 2008, 105, 16125–16130. [Google Scholar] [CrossRef] [Green Version]
- Davies, A.A.; Huttner, D.; Daigaku, Y.; Chen, S.; Ulrich, H.D. Activation of Ubiquitin-Dependent DNA Damage Bypass Is Mediated by Replication Protein A. Mol. Cell 2008, 29, 625–636. [Google Scholar] [CrossRef]
- Prado, F. Homologous Recombination Maintenance of Genome Integrity during DNA Damage Tolerance. Mol. Cell. Oncol. 2014, 1, e957039. [Google Scholar] [CrossRef] [Green Version]
- Neelsen, K.J.; Lopes, M. Replication Fork Reversal in Eukaryotes: From Dead End to Dynamic Response. Nat. Rev. Mol. Cell. Biol. 2015, 16, 207–220. [Google Scholar] [CrossRef]
- Miyabe, I.; Mizuno, K.; Keszthelyi, A.; Daigaku, Y.; Skouteri, M.; Mohebi, S.; Kunkel, T.A.; Murray, J.M.; Carr, A.M. Polymerase δ Replicates Both Strands after Homologous Recombination–Dependent Fork Restart. Nat. Struct. Mol. Biol. 2015, 22, 932–938. [Google Scholar] [CrossRef]
- García-Gómez, S.; Reyes, A.; Martínez-Jiménez, M.I.; Chocrón, E.S.; Mourón, S.; Terrados, G.; Powell, C.; Salido, E.; Méndez, J.; Holt, I.J.; et al. PrimPol, an Archaic Primase/Polymerase Operating in Human Cells. Mol. Cell 2013, 52, 541–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mourón, S.; Rodriguez-Acebes, S.; Martínez-Jiménez, M.I.; García-Gómez, S.; Chocrón, S.; Blanco, L.; Méndez, J. Repriming of DNA Synthesis at Stalled Replication Forks by Human PrimPol. Nat. Struct. Mol. Biol. 2013, 20, 1383–1389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fumasoni, M.; Zwicky, K.; Vanoli, F.; Lopes, M.; Branzei, D. Error-Free DNA Damage Tolerance and Sister Chromatid Proximity during DNA Replication Rely on the Polα/Primase/Ctf4 Complex. Mol. Cell 2015, 57, 812–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez, N.G.; Wong, R.P.; Ulrich, H.D. The Helicase Pif1 Functions in the Template Switching Pathway of DNA Damage Bypass. Nucleic Acids Res. 2018, 46, 8347–8356. [Google Scholar] [CrossRef]
- Rodríguez, N.G.; Morawska, M.; Wong, R.P.; Daigaku, Y.; Ulrich, H.D. Spatial Separation between Replisome- and Template-Induced Replication Stress Signaling. EMBO J. 2018, 37, 779. [Google Scholar] [CrossRef]
- Vanoli, F.; Fumasoni, M.; Szakal, B.; Maloisel, L.; Branzei, D. Replication and Recombination Factors Contributing to Recombination-Dependent Bypass of DNA Lesions by Template Switch. PLoS Genet. 2010, 6, e1001205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mankouri, H.W.; Ngo, H.-P.; Hickson, I.D. Shu Proteins Promote the Formation of Homologous Recombination Intermediates That Are Processed by Sgs1-Rmi1-Top3. Mol. Biol. Cell 2007, 18, 4062–4073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Branzei, D.; Vanoli, F.; Foiani, M. SUMOylation Regulates Rad18-Mediated Template Switch. Nature 2008, 456, 915–920. [Google Scholar] [CrossRef] [PubMed]
- Liberi, G.; Maffioletti, G.; Lucca, C.; Chiolo, I.; Baryshnikova, A.; Cotta-Ramusino, C.; Lopes, M.; Pellicioli, A.; Haber, J.E.; Foiani, M. Rad51-Dependent DNA Structures Accumulate at Damaged Replication Forks in Sgs1 Mutants Defective in the Yeast Ortholog of BLM RecQ Helicase. Genes Dev. 2005, 19, 339–350. [Google Scholar] [CrossRef] [Green Version]
- Shor, E.; Weinstein, J.; Rothstein, R. A Genetic Screen for Top3 Suppressors in Saccharomyces Cerevisiae Identifies SHU1, SHU2, PSY3 and CSM2: Four Genes Involved in Error-Free DNA Repair. Genetics 2005, 169, 1275–1289. [Google Scholar] [CrossRef] [Green Version]
- Giannattasio, M.; Zwicky, K.; Lopes, M.; Branzei, D. Visualization of Recombination-Mediated Damage Bypass by Template Switching. Nat. Struct. Mol. Biol. 2014, 21, 884–892. [Google Scholar] [CrossRef] [PubMed]
- Zellweger, R.; Dalcher, D.; Mutreja, K.; Berti, M.; Schmid, J.A.; Herrador, R.; Vindigni, A.; Lopes, M. Rad51-Mediated Replication Fork Reversal Is a Global Response to Genotoxic Treatments in Human Cells. J. Cell Biol. 2015, 208, 563–579. [Google Scholar] [CrossRef]
- Sogo, J.M.; Lopes, M.; Foiani, M. Fork Reversal and SsDNA Accumulation at Stalled Replication Forks Owing to Checkpoint Defects. Science 2002, 297, 599–602. [Google Scholar] [CrossRef]
- Vujanovic, M.; Krietsch, J.; Raso, M.C.; Terraneo, N.; Zellweger, R.; Schmid, J.A.; Taglialatela, A.; Huang, J.-W.; Holland, C.L.; Zwicky, K.; et al. Replication Fork Slowing and Reversal upon DNA Damage Require PCNA Polyubiquitination and ZRANB3 DNA Translocase Activity. Mol. Cell 2017, 67, 882–890.e5. [Google Scholar] [CrossRef] [Green Version]
- Krishnamoorthy, A.; Jackson, J.; Mohamed, T.; Adolph, M.; Vindigni, A.; Cortez, D. RADX Prevents Genome Instability by Confining Replication Fork Reversal to Stalled Forks. Mol. Cell 2021. [Google Scholar] [CrossRef]
- Chaudhuri, A.R.; Hashimoto, Y.; Herrador, R.; Neelsen, K.J.; Fachinetti, D.; Bermejo, R.; Cocito, A.; Costanzo, V.; Lopes, M. Topoisomerase I Poisoning Results in PARP-Mediated Replication Fork Reversal. Nat. Struct. Mol. Biol. 2012, 19, 417–423. [Google Scholar] [CrossRef]
- Wong, R.P.; Rodríguez, N.G.; Zilio, N.; Hanulová, M.; Ulrich, H.D. Processing of DNA Polymerase-Blocking Lesions during Genome Replication Is Spatially and Temporally Segregated from Replication Forks. Mol. Cell 2020, 77, 3–16.e4. [Google Scholar] [CrossRef]
- Alabert, C.; Bianco, J.N.; Pasero, P. Differential Regulation of Homologous Recombination at DNA Breaks and Replication Forks by the Mrc1 Branch of the S-Phase Checkpoint. EMBO J. 2009, 28, 1131–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vázquez, M.V.; Rojas, V.; Tercero, J.A. Multiple Pathways Cooperate to Facilitate DNA Replication Fork Progression through Alkylated DNA. DNA Repair 2008, 7, 1693–1704. [Google Scholar] [CrossRef] [PubMed]
- González-Prieto, R.; Muñoz-Cabello, A.M.; Cabello-Lobato, M.J.; Prado, F. Rad51 Replication Fork Recruitment Is Required for DNA Damage Tolerance. EMBO J. 2013, 32, 1307–1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlacher, K.; Christ, N.; Siaud, N.; Egashira, A.; Wu, H.; Jasin, M. Double-Strand Break Repair-Independent Role for BRCA2 in Blocking Stalled Replication Fork Degradation by MRE11. Cell 2011, 145, 529–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlacher, K.; Wu, H.; Jasin, M. A Distinct Replication Fork Protection Pathway Connects Fanconi Anemia Tumor Suppressors to RAD51-BRCA1/2. Cancer Cell 2012, 22, 106–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Somyajit, K.; Saxena, S.; Babu, S.; Mishra, A.; Nagaraju, G. Mammalian RAD51 Paralogs Protect Nascent DNA at Stalled Forks and Mediate Replication Restart. Nucleic Acids Res. 2015, 43, gkv880-21. [Google Scholar] [CrossRef]
- Wang, A.T.; Kim, T.; Wagner, J.E.; Conti, B.A.; Lach, F.P.; Huang, A.L.; Molina, H.; Sanborn, E.M.; Zierhut, H.; Cornes, B.K.; et al. A Dominant Mutation in Human RAD51 Reveals Its Function in DNA Interstrand Crosslink Repair Independent of Homologous Recombination. Mol. Cell 2015, 59, 478–490. [Google Scholar] [CrossRef] [Green Version]
- Ait Saada, A.A.; Teixeira-Silva, A.; Iraqui, I.; Costes, A.; Hardy, J.; Paoletti, G.; Fréon, K.; Lambert, S.A.E. Unprotected Replication Forks Are Converted into Mitotic Sister Chromatid Bridges. Mol. Cell 2017, 66, 398–410.e4. [Google Scholar] [CrossRef] [Green Version]
- Mijic, S.; Zellweger, R.; Chappidi, N.; Berti, M.; Jacobs, K.; Mutreja, K.; Ursich, S.; Chaudhuri, A.R.; Nussenzweig, A.; Janscak, P.; et al. Replication Fork Reversal Triggers Fork Degradation in BRCA2-Defective Cells. Nat. Commun. 2017, 8, 859. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, A.R.; Callen, E.; Ding, X.; Gogola, E.; Duarte, A.A.; Lee, J.-E.; Wong, N.; Lafarga, V.; Calvo, J.A.; Panzarino, N.J.; et al. Replication Fork Stability Confers Chemoresistance in BRCA-Deficient Cells. Nature 2016, 535, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Jasin, M. BRCA2 Suppresses Replication Stress-Induced Mitotic and G1 Abnormalities through Homologous Recombination. Nat. Commun. 2017, 8, 525. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Petermann, E.; Schultz, N.; Jemth, A.; Loseva, O.; Issaeva, N.; Johansson, F.; Fernandez, S.; McGlynn, P.; Helleday, T. PARP Is Activated at Stalled Forks to Mediate Mre11-dependent Replication Restart and Recombination. EMBO J. 2009, 28, 2601–2615. [Google Scholar] [CrossRef] [Green Version]
- Wilhelm, T.; Magdalou, I.; Barascu, A.; Técher, H.; Debatisse, M.; Lopez, B.S. Spontaneous Slow Replication Fork Progression Elicits Mitosis Alterations in Homologous Recombination-Deficient Mammalian Cells. Proc. Natl. Acad. Sci. USA 2014, 111, 763–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, X.; Broderick, R.; Bergoglio, V.; Zimmer, J.; Badie, S.; Niedzwiedz, W.; Hoffmann, J.-S.; Tarsounas, M. MUS81 Nuclease Activity Is Essential for Replication Stress Tolerance and Chromosome Segregation in BRCA2-Deficient Cells. Nat. Commun. 2017, 8, 15983. [Google Scholar] [CrossRef] [Green Version]
- Berti, M.; Teloni, F.; Mijic, S.; Ursich, S.; Fuchs, J.; Palumbieri, M.D.; Krietsch, J.; Schmid, J.A.; Garcin, E.B.; Gon, S.; et al. Sequential Role of RAD51 Paralog Complexes in Replication Fork Remodeling and Restart. Nat. Commun. 2020, 11, 3531. [Google Scholar] [CrossRef] [PubMed]
- Bugreev, D.V.; Rossi, M.J.; Mazin, A.V. Cooperation of RAD51 and RAD54 in Regression of a Model Replication Fork. Nucleic Acids Res. 2011, 39, 2153–2164. [Google Scholar] [CrossRef] [Green Version]
- Ciccia, A.; Nimonkar, A.V.; Hu, Y.; Hajdu, I.; Achar, Y.J.; Izhar, L.; Petit, S.A.; Adamson, B.; Yoon, J.C.; Kowalczykowski, S.C.; et al. Polyubiquitinated PCNA Recruits the ZRANB3 Translocase to Maintain Genomic Integrity after Replication Stress. Mol. Cell 2012, 47, 396–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, G.; Kermi, C.; Stoy, H.; Schiltz, C.J.; Bacal, J.; Zaino, A.M.; Hadden, M.K.; Eichman, B.F.; Lopes, M.; Cimprich, K.A. HLTF Promotes Fork Reversal, Limiting Replication Stress Resistance and Preventing Multiple Mechanisms of Unrestrained DNA Synthesis. Mol. Cell 2020, 78, 1237–1251.e7. [Google Scholar] [CrossRef] [PubMed]
- Blastyak, A.; Hajdu, I.; Unk, I.; Haracska, L. Role of Double-Stranded DNA Translocase Activity of Human HLTF in Replication of Damaged DNA. Mol. Cell. Biol. 2010, 30, 684–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bétous, R.; Couch, F.B.; Mason, A.C.; Eichman, B.F.; Manosas, M.; Cortez, D. Substrate-Selective Repair and Restart of Replication Forks by DNA Translocases. Cell Rep. 2013, 3, 1958–1969. [Google Scholar] [CrossRef] [Green Version]
- Schubert, L.; Ho, T.; Hoffmann, S.; Haahr, P.; Guérillon, C.; Mailand, N. RADX Interacts with Single-stranded DNA to Promote Replication Fork Stability. EMBO Rep. 2017, 18, 1991–2003. [Google Scholar] [CrossRef] [PubMed]
- Dungrawala, H.; Bhat, K.P.; Meur, R.L.; Chazin, W.J.; Ding, X.; Sharan, S.K.; Wessel, S.R.; Sathe, A.A.; Zhao, R.; Cortez, D. RADX Promotes Genome Stability and Modulates Chemosensitivity by Regulating RAD51 at Replication Forks. Mol. Cell 2017, 67, 374–386.e5. [Google Scholar] [CrossRef] [PubMed]
- Bhat, K.P.; Krishnamoorthy, A.; Dungrawala, H.; Garcin, E.B.; Modesti, M.; Cortez, D. RADX Modulates RAD51 Activity to Control Replication Fork Protection. Cell Rep. 2018, 24, 538–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adolph, M.B.; Mohamed, T.M.; Balakrishnan, S.; Xue, C.; Morati, F.; Modesti, M.; Greene, E.C.; Chazin, W.J.; Cortez, D. RADX Controls RAD51 Filament Dynamics to Regulate Replication Fork Stability. Mol. Cell 2021. [Google Scholar] [CrossRef]
- Bhowmick, R.; Minocherhomji, S.; Hickson, I.D. RAD52 Facilitates Mitotic DNA Synthesis Following Replication Stress. Mol. Cell 2016, 64, 1117–1126. [Google Scholar] [CrossRef] [Green Version]
- Minocherhomji, S.; Ying, S.; Bjerregaard, V.A.; Bursomanno, S.; Aleliunaite, A.; Wu, W.; Mankouri, H.W.; Shen, H.; Liu, Y.; Hickson, I.D. Replication Stress Activates DNA Repair Synthesis in Mitosis. Nature 2015, 528, 286–290. [Google Scholar] [CrossRef]
- Wassing, I.E.; Graham, E.; Saayman, X.; Rampazzo, L.; Ralf, C.; Bassett, A.; Esashi, F. The RAD51 Recombinase Protects Mitotic Chromatin in Human Cells. Nat. Commun. 2021, 12, 5380. [Google Scholar] [CrossRef] [PubMed]
- Waters, L.S.; Walker, G.C. The Critical Mutagenic Translesion DNA Polymerase Rev1 Is Highly Expressed during G(2)/M Phase Rather than S Phase. Proc. Natl. Acad. Sci. USA 2006, 103, 8971–8976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motegi, A.; Kuntz, K.; Majeed, A.; Smith, S.; Myung, K. Regulation of Gross Chromosomal Rearrangements by Ubiquitin and SUMO Ligases in Saccharomyces Cerevisiae. Mol. Cell. Biol. 2006, 26, 1424–1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zgaga, Z. Transformation of Saccharomyces Cerevisiae with UV-Irradiated Single-Stranded Plasmid. Mutat. Res. 1991, 263, 211–215. [Google Scholar] [CrossRef]
- Ball, L.G.; Zhang, K.; Cobb, J.A.; Boone, C.; Xiao, W. The Yeast Shu Complex Couples Error-Free Post-Replication Repair to Homologous Recombination. Mol. Microbiol. 2009, 73, 89–102. [Google Scholar] [CrossRef]
- Paulovich, A.G.; Armour, C.D.; Hartwell, L.H. The Saccharomyces Cerevisiae RAD9, RAD17, RAD24 and MEC3 Genes Are Required for Tolerating Irreparable, Ultraviolet-Induced DNA Damage. Genetics 1998, 150, 75–93. [Google Scholar] [CrossRef]
- Schiestl, R.H.; Prakash, S.; Prakash, L. The SRS2 Suppressor of Rad6 Mutations of Saccharomyces Cerevisiae Acts by Channeling DNA Lesions into the RAD52 DNA Repair Pathway. Genetics 1990, 124, 817–831. [Google Scholar] [CrossRef]
- Pfander, B.; Moldovan, G.-L.; Sacher, M.; Hoege, C.; Jentsch, S. SUMO-Modified PCNA Recruits Srs2 to Prevent Recombination during S Phase. Nature 2005, 436, 428–433. [Google Scholar] [CrossRef]
- Parker, J.L.; Ulrich, H.D. A SUMO-Interacting Motif Activates Budding Yeast Ubiquitin Ligase Rad18 towards SUMO-Modified PCNA. Nucleic Acids Res. 2012, 40, 11380–11388. [Google Scholar] [CrossRef]
- Tsuji, Y.; Watanabe, K.; Araki, K.; Shinohara, M.; Yamagata, Y.; Tsurimoto, T.; Hanaoka, F.; Yamamura, K.; Yamaizumi, M.; Tateishi, S. Recognition of Forked and Single-stranded DNA Structures by Human RAD18 Complexed with RAD6B Protein Triggers Its Recruitment to Stalled Replication Forks. Genes Cells 2008, 13, 343–354. [Google Scholar] [CrossRef]
- Bailly, V.; Lamb, J.; Sung, P.; Prakash, S.; Prakash, L. Specific Complex Formation between Yeast RAD6 and RAD18 Proteins: A Potential Mechanism for Targeting RAD6 Ubiquitin-Conjugating Activity to DNA Damage Sites. Genes Dev. 1994, 8, 811–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailly, V.; Lauder, S.; Prakash, S.; Prakash, L. Yeast DNA Repair Proteins Rad6 and Rad18 Form a Heterodimer That Has Ubiquitin Conjugating, DNA Binding, and ATP Hydrolytic Activities. J. Biol. Chem. 1997, 272, 23360–23365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bienko, M.; Green, C.M.; Crosetto, N.; Rudolf, F.; Zapart, G.; Coull, B.; Kannouche, P.; Wider, G.; Peter, M.; Lehmann, A.R.; et al. Ubiquitin-Binding Domains in Y-Family Polymerases Regulate Translesion Synthesis. Science 2005, 310, 1821–1824. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Bazán, M.Á.; Gallo-Fernández, M.; Saugar, I.; Jiménez-Martín, A.; Vázquez, M.V.; Tercero, J.A. Rad5 Plays a Major Role in the Cellular Response to DNA Damage during Chromosome Replication. Cell Rep. 2014, 9, 460–468. [Google Scholar] [CrossRef] [Green Version]
- Frei, C.; Gasser, S.M. The Yeast Sgs1p Helicase Acts Upstream of Rad53p in the DNA Replication Checkpoint and Colocalizes with Rad53p in S-Phase-Specific Foci. Genes Dev. 2000, 14, 81–96. [Google Scholar] [CrossRef]
- Pasero, P.; Duncker, B.P.; Schwob, E.; Gasser, S.M. A Role for the Cdc7 Kinase Regulatory Subunit Dbf4p in the Formation of Initiation-Competent Origins of Replication. Genes Dev. 1999, 13, 2159–2176. [Google Scholar] [CrossRef]
- Shah, P.P.; Zheng, X.; Epshtein, A.; Carey, J.N.; Bishop, D.K.; Klein, H.L. Swi2/Snf2-Related Translocases Prevent Accumulation of Toxic Rad51 Complexes during Mitotic Growth. Mol. Cell 2010, 39, 862–872. [Google Scholar] [CrossRef] [Green Version]
- Muraszko, J.; Kramarz, K.; Argunhan, B.; Ito, K.; Baranowska, G.; Kurokawa, Y.; Murayama, Y.; Tsubouchi, H.; Lambert, S.; Iwasaki, H.; et al. Rrp1 Translocase and Ubiquitin Ligase Activities Restrict the Genome Destabilising Effects of Rad51 in Fission Yeast. Nucleic Acids Res. 2021, 49, 6832–6848. [Google Scholar] [CrossRef]
- Bailis, J.M.; Luche, D.D.; Hunter, T.; Forsburg, S.L. Minichromosome Maintenance Proteins Interact with Checkpoint and Recombination Proteins To Promote S-Phase Genome Stability. Mol. Cell. Biol. 2008, 28, 1724–1738. [Google Scholar] [CrossRef] [Green Version]
- Shukla, A.; Navadgi, V.M.; Mallikarjuna, K.; Rao, B.J. Interaction of HRad51 and HRad52 with MCM Complex: A Cross-Talk between Recombination and Replication Proteins. Biochem. Biophys. Res. Commu. 2005, 329, 1240–1245. [Google Scholar] [CrossRef]
- Hesketh, E.L.; Knight, J.R.P.; Wilson, R.H.C.; Chong, J.P.J.; Coverley, D. Transient Association of MCM Complex Proteins with the Nuclear Matrix during Initiation of Mammalian DNA Replication. Cell Cycle 2015, 14, 333–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, R.H.C.; Coverley, D. Relationship between DNA Replication and the Nuclear Matrix. Genes Cells 2012, 18, 17–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Indiani, C.; Patel, M.; Goodman, M.F.; O’Donnell, M.E. RecA Acts as a Switch to Regulate Polymerase Occupancy in a Moving Replication Fork. Proc. Natl. Acad. Sci. USA 2013, 110, 5410–5415. [Google Scholar] [CrossRef] [Green Version]
- Kolinjivadi, A.M.; Sannino, V.; Antoni, A.D.; Zadorozhny, K.; Kilkenny, M.; Técher, H.; Baldi, G.; Shen, R.; Ciccia, A.; Pellegrini, L.; et al. Smarcal1-Mediated Fork Reversal Triggers Mre11- Dependent Degradation of Nascent DNA in the Absence of Brca2 and Stable Rad51 Nucleofilaments. Mol. Cell 2017, 67, 867–881.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinet, A.; Tirman, S.; Cybulla, E.; Meroni, A.; Vindigni, A. To Skip or Not to Skip: Choosing Repriming to Tolerate DNA Damage. Mol. Cell 2021, 81, 649–658. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prado, F. Non-Recombinogenic Functions of Rad51, BRCA2, and Rad52 in DNA Damage Tolerance. Genes 2021, 12, 1550. https://doi.org/10.3390/genes12101550
Prado F. Non-Recombinogenic Functions of Rad51, BRCA2, and Rad52 in DNA Damage Tolerance. Genes. 2021; 12(10):1550. https://doi.org/10.3390/genes12101550
Chicago/Turabian StylePrado, Félix. 2021. "Non-Recombinogenic Functions of Rad51, BRCA2, and Rad52 in DNA Damage Tolerance" Genes 12, no. 10: 1550. https://doi.org/10.3390/genes12101550
APA StylePrado, F. (2021). Non-Recombinogenic Functions of Rad51, BRCA2, and Rad52 in DNA Damage Tolerance. Genes, 12(10), 1550. https://doi.org/10.3390/genes12101550