
A Review of Probabilistic Genotyping Systems: EuroForMix, DNAStatistX and STRmix™



Abstract
:1. Introduction
2. Probabilistic Genotyping in Generality
2.1. Probabilistic Genotyping Software
2.2. Investigative vs. Evaluative Forensic Genetics
2.3. Probabilistic Genotyping to Detect Contamination Events
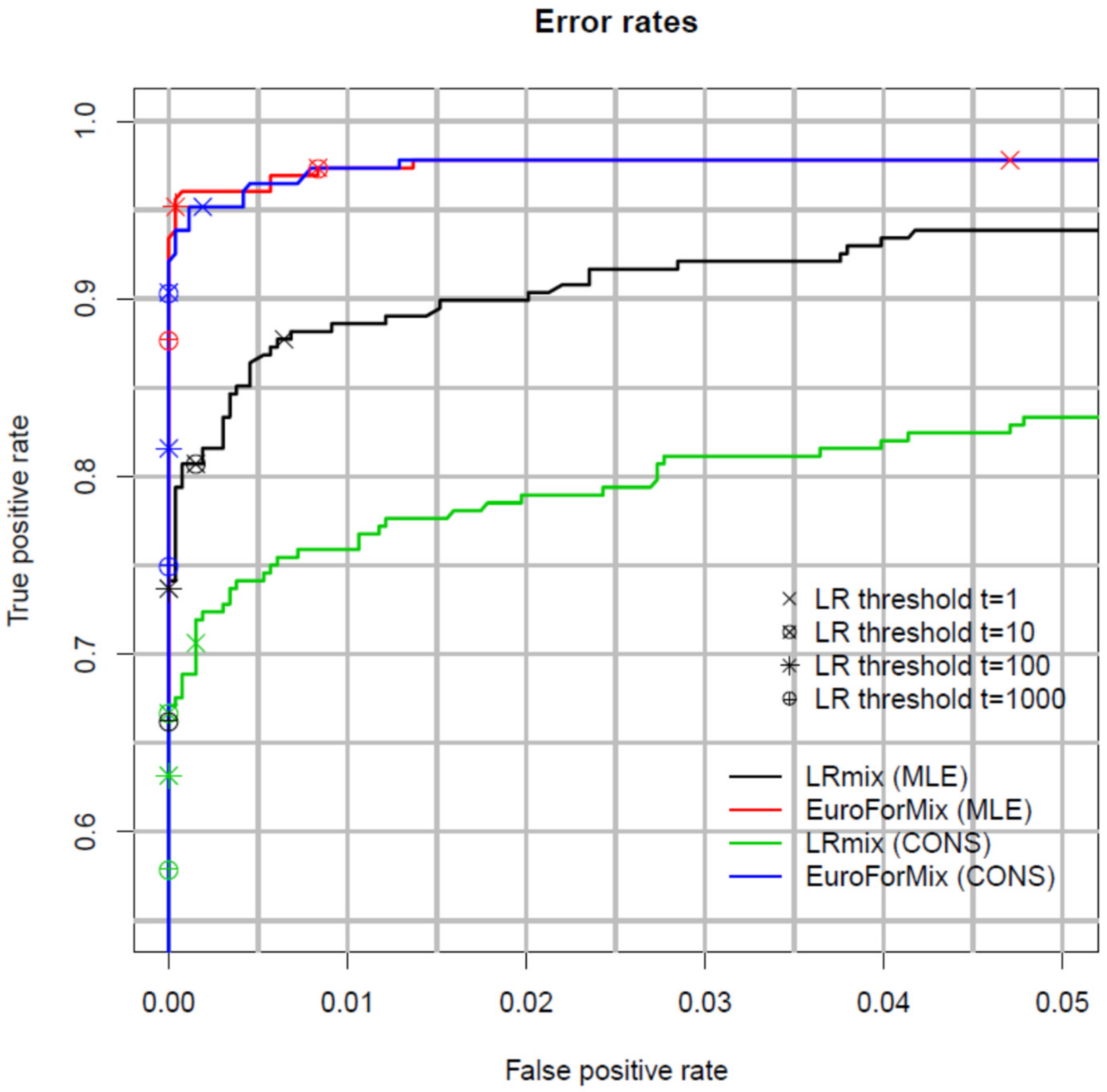
2.4. Inter and Intra-Laboratory Studies
Non-Contributor Tests and Calibration of the LR
2.5. Number of Contributors (NOC)
2.6. Proposition Setting/Hierarchy of Propositions
2.7. Validation of PG Systems
- (a)
- Sensitivity (demonstrate the range of LRs that can be expected for true contributors)
- (b)
- Specificity (demonstrate the range of LRs that can be expected for non-contributors)
- (c)
- Precision (variation in LRs from repeated software analyses of the same input data)
- (a)
- H1 = true: where we know the POI is a contributor.
- (b)
- H2 = true: where we know that the POI is not a contributor
3. Evolution of EuroForMix and DNAStatistX
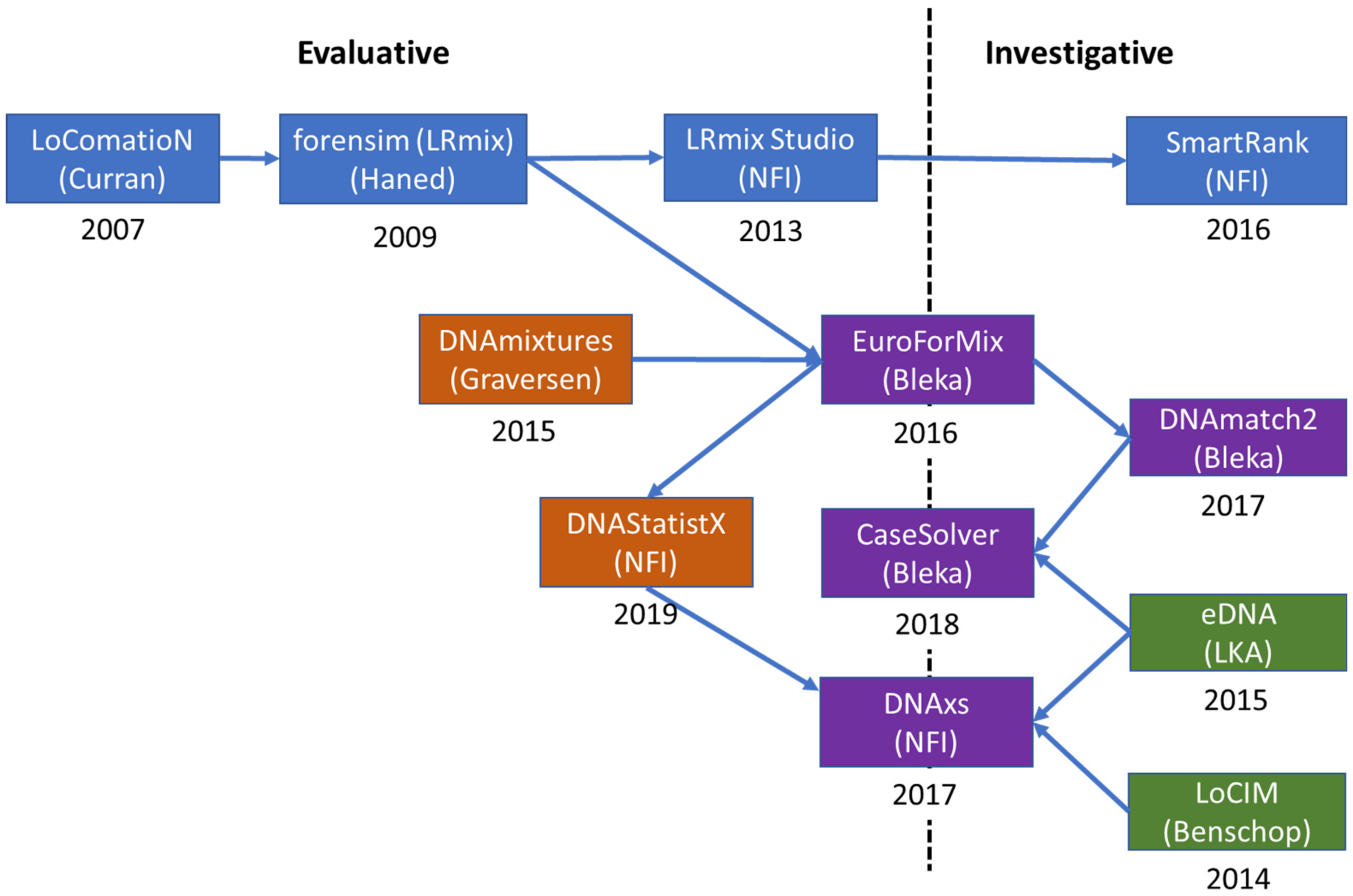
3.1. Evolution
3.1.1. Qualitative Software
3.1.2. Quantitative Software
3.1.3. DNAxs and Related Modules
- (1)
- By aggregating replicate profiles into one composite view (bar graphs)
- (2)
- By viewing the trace profile as bar graphs underneath which alleles of reference profiles are comparedThrough the match matrix option
- (3)
- By sending a DNA profile for a SmartRank search against the DNA database
- (4)
- By calculating LRs using DNAStatistX for a comparison of a person of interest to a trace DNA profile [128]
3.2. The γ Model
3.3. An Outline of the γ Model Incorporated into Euroformix and DNAStatistX
3.3.1. Model Features
- The software accommodates degradation, allele drop-out, allele drop-in, ‘n − 1’ and ‘n + 1’ stutters and sub-population structure (Fst correction). Note that stutters are not accommodated in the current version of DNAStatistX, but is under development for a future version.
- Replicated samples can be analysed. Consensus or composite profiles, a feature of pre-PG software, are not used.
- The model assumes same contributors and the same peak height properties for each replicate.
- Optional Locus specific settings (DNAStatistX from v1, EuroForMix v3 onwards) are as follows:
- (a)
- Analytical threshold
- (b)
- Drop-in model
- (c)
- Fst correction
3.3.2. Exploratory Data Analysis
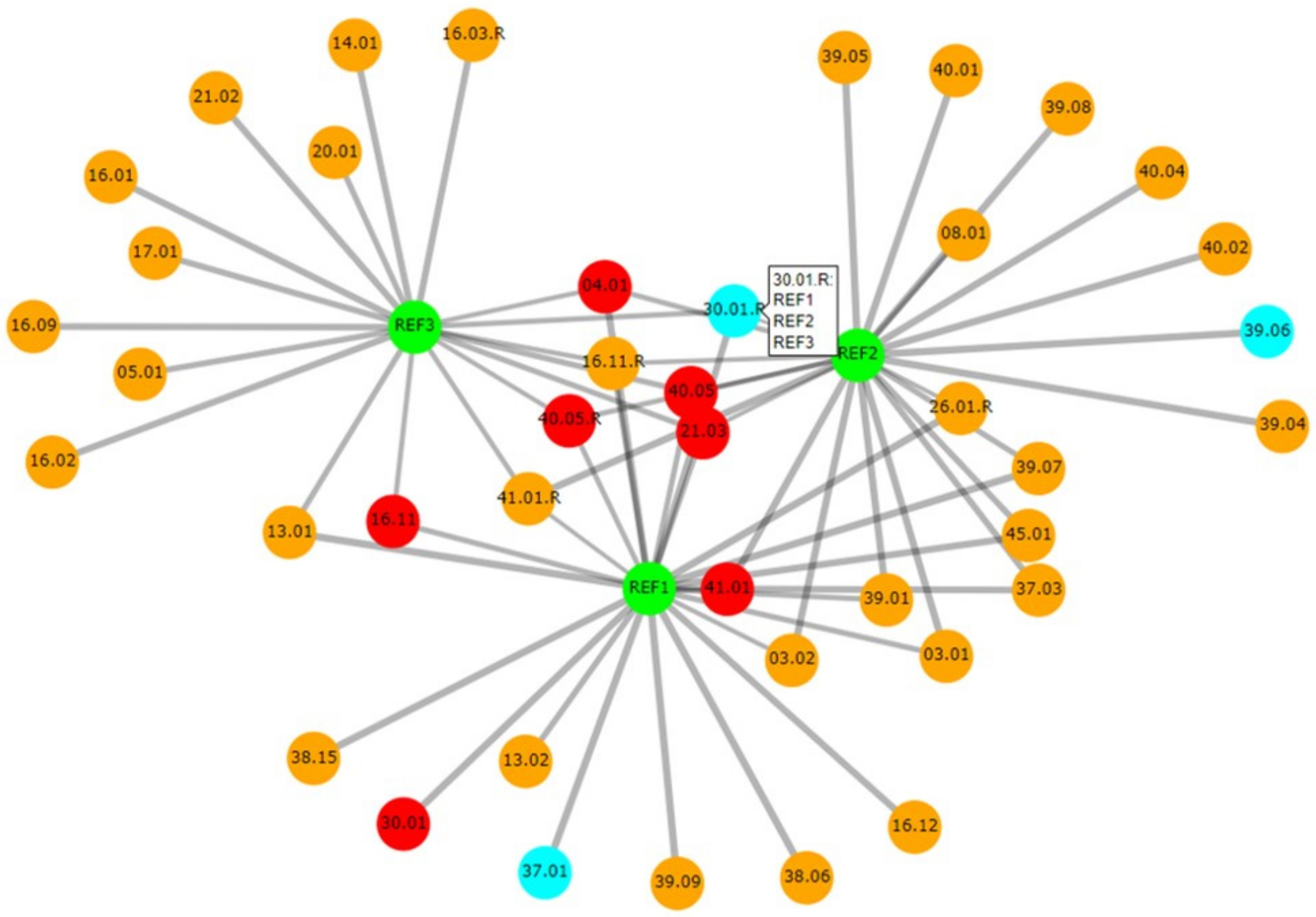
3.3.3. Relatedness
3.3.4. Deconvolution
3.4. Investigative Forensic Genetics
Probabilistic Genotyping to Carry out Searches of National DNA Databases
3.5. Massively Parallel Sequencing (MPS)
3.6. Validation, Guidelines for Best Practice and Quality
4. STRmix™
4.1. History of STRmix™ Creation
4.2. Probabilistic Genotyping and STRmix™
4.3. Capabilities of STRmix™
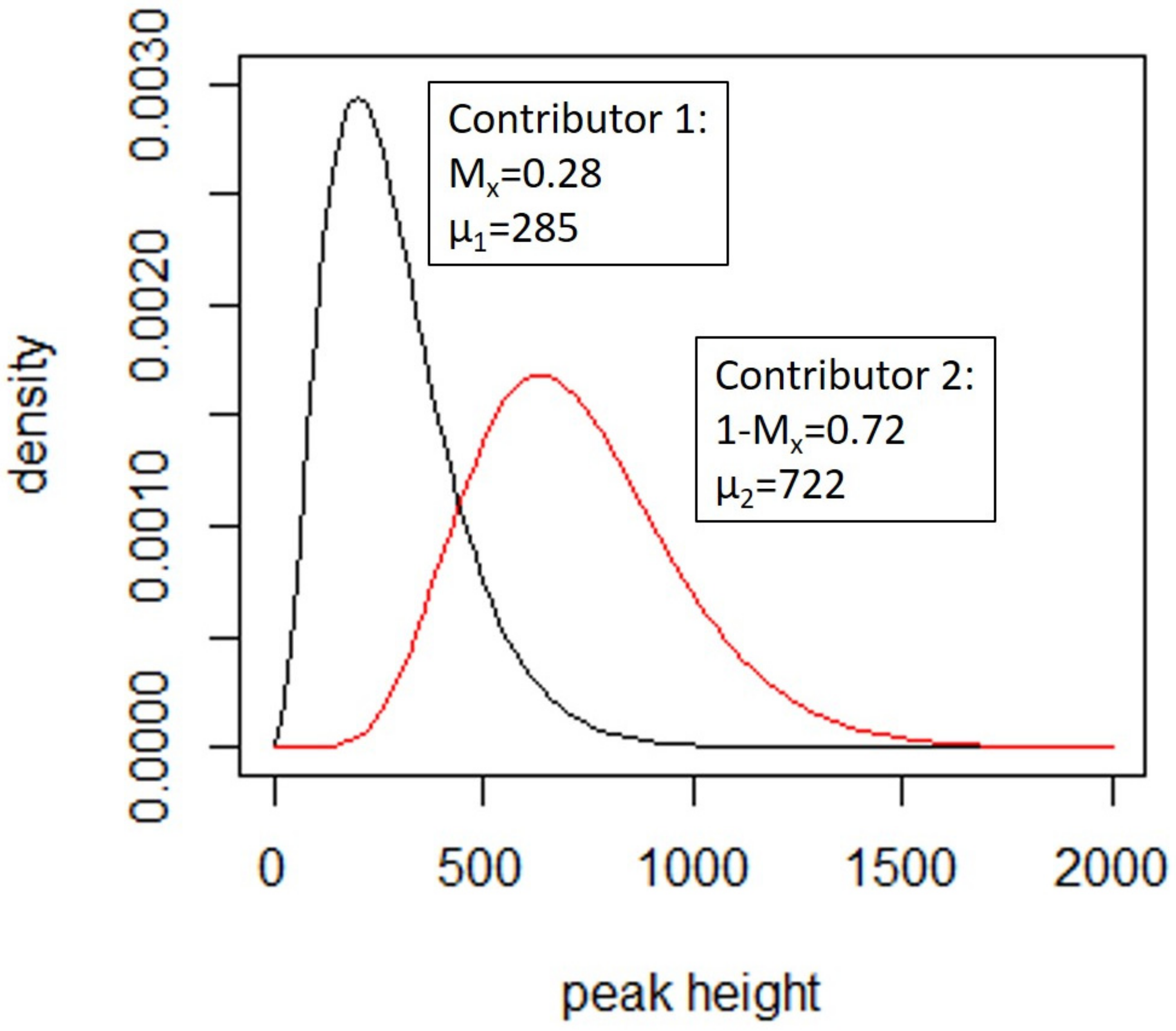
4.3.1. Deconvolution
- A template amount for each of the n contributors,
- A degradation (described in [154]) which models the decay with respect to molecular weight (m) in the template for each of the contributors,
- Amplification efficiency at each locus to allow for the observed amplification levels of each locus,
- Replicate multipliers, which scales all peaks up or down between PCR replicates.
- (1)
- Choose a locus at random and propose a genotype set at that locus.
- (2)
- Choose new values for all mass parameters by stepping a small distance from the values in the current set (known as a random walk, and with step size dictated by a Gaussian distribution). Propose these values.
- (3)
- Calculate the expected peak heights using the proposed sample values.
- (4)
- Calculate the likelihood value of the proposed sample values.
- (5)
- Use a Metropolis-Hastings algorithm to accept or reject the proposed sample. If the proposed sample is accepted, then the proposed set of parameter values becomes the current set. If the proposed sample is rejected, then the proposal is discarded.
- (6)
- Repeat steps 1 to 6 until a defined number of proposal accepts have been attained.
4.3.2. LR Calculation
- (1)
- sampling variation in allele frequency database
- (2)
- sampling variation in the iterations of the MCMC leading to the assignment of weights
- (3)
- uncertainty in the value of θ
- sub-sub-source proposition pair,
- the sub-source proposition pair considering the alternate DNA donor as
- ○
- unrelated,
- ○
- sibling,
- ○
- half-sibling,
- ○
- parent/child,
- ○
- aunt/uncle/niece/nephew,
- ○
- grandparent,
- ○
- cousin, or
- ○
- unified across all relationship types,
- all of the above for:
- ○
- each ethnic population in the local geographical region, and
- ○
- stratified across all populations,
- all of the above for:
- ○
- each NOC in a chosen range, and
- ○
- stratified across the range (or with bespoke NOC choice for proposition),
The probability of observing a likelihood ratio of LRPOI or larger from an unrelated non-donor is less than or equal to 1 in LRPOI.
4.4. Implementation of STRmix™
4.5. DBLR—A Companion Product to STRmix™
- How many common donors are in a mixture?
- Are any donors of the multiple mixtures related (see [203] for an investigation into the effect of not recognising relatedness in mixtures)?
- If I assume a relative of a POI to one mixture does that assist in resolving the other components?
- If I use multiple mixed samples from a disaster victim identification (DVI) together in a single analysis will that help to better resolve the genotypes of the donors?
4.6. Validation of STRmix™
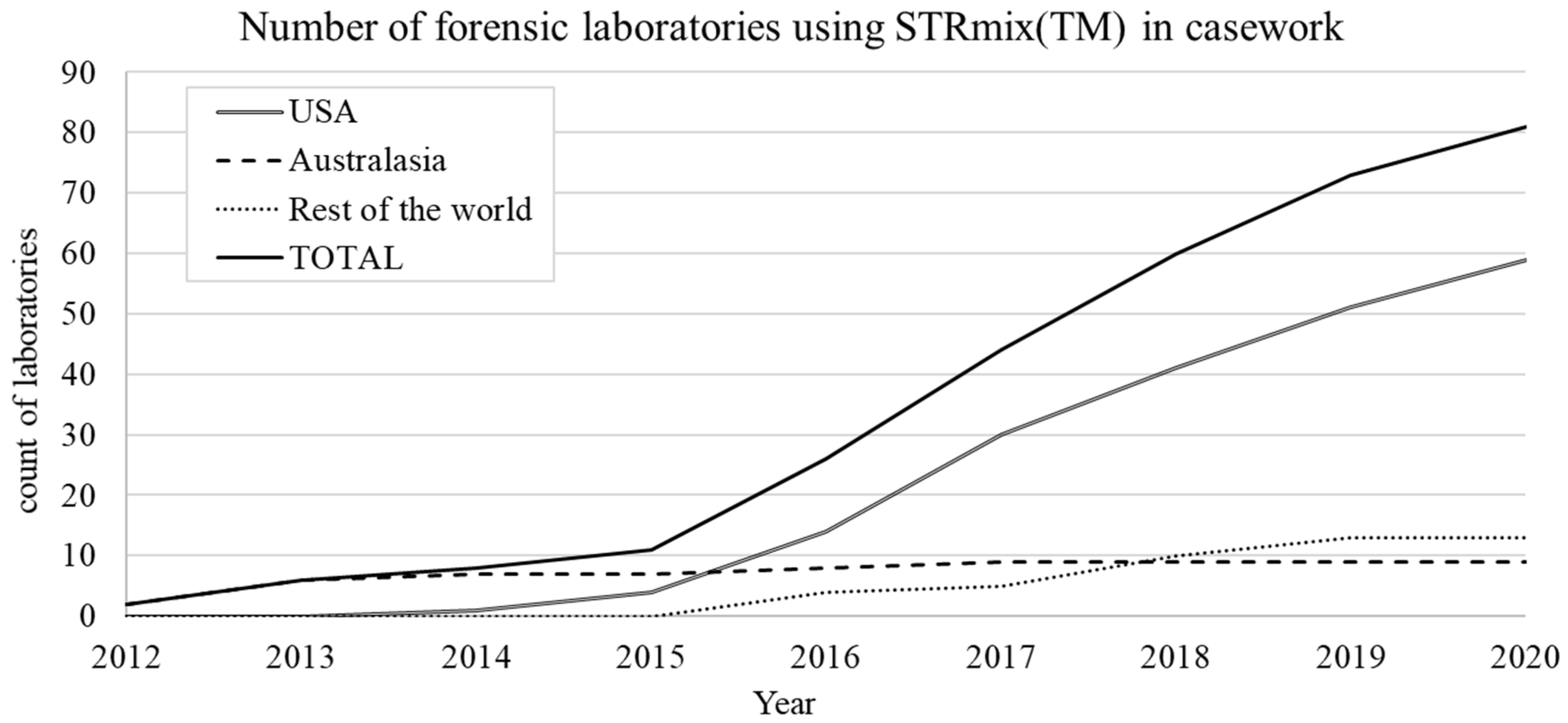
4.7. Growth of STRmix™
4.8. Admissibility Experiences with STRmix™
4.8.1. Independence of Validation
4.8.2. Run to Run Variability
4.8.3. Code Access
4.8.4. Code Quality
4.8.5. Validation
Supplementary Materials
Conflicts of Interest
References
- Gill, P.; Brenner, C.; Buckleton, J.; Carracedo, A.; Krawczak, M.; Mayr, W.; Morling, N.; Prinz, M.; Schneider, P.M.; Weir, B. DNA commission of the International Society of Forensic Genetics: Recommendations on the interpretation of mixtures. Forensic Sci. Int. 2006, 160, 90–101. [Google Scholar] [CrossRef]
- Gill, P.; Gusmão, L.; Haned, H.; Mayr, W.; Morling, N.; Parson, W.; Prieto, L.; Prinz, M.; Schneider, H.; Schneider, P.; et al. DNA commission of the International Society of Forensic Genetics: Recommendations on the evaluation of STR typing results that may include drop-out and/or drop-in using probabilistic methods. Forensic Sci. Int. Genet. 2012, 6, 679–688. [Google Scholar] [CrossRef] [Green Version]
- Gill, P.; Hicks, T.; Butler, J.M.; Connolly, E.; Gusmão, L.; Kokshoorn, B.; Morling, N.; van Oorschot, R.A.; Parson, W.; Prinz, M.; et al. DNA commission of the International society for forensic genetics: Assessing the value of forensic biological evidence—Guidelines highlighting the importance of propositions. Forensic Sci. Int. Genet. 2018, 36, 189–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clayton, T.; Whitaker, J.; Sparkes, R.; Gill, P. Analysis and interpretation of mixed forensic stains using DNA STR profiling. Forensic Sci. Int. 1998, 91, 55–70. [Google Scholar] [CrossRef]
- Gill, P.; Whitaker, J.; Flaxman, C.; Brown, N.; Buckleton, J. An investigation of the rigor of interpretation rules for STRs derived from less than 100 pg of DNA. Forensic Sci. Int. 2000, 112, 17–40. [Google Scholar] [CrossRef]
- Balding, D. Evaluation of mixed-source, low-template DNA profiles in forensic science. Proc. Natl. Acad. Sci. USA 2013, 110, 12241–12246. [Google Scholar] [CrossRef] [Green Version]
- Balding, J.D.; Buckleton, J. Interpreting low template DNA profiles. Forensic Sci. Int. Genet. 2009, 4, 1–10. [Google Scholar] [CrossRef]
- Puch-Solis, R.; Clayton, T. Evidential evaluation of DNA profiles using a discrete statistical model implemented in the DNA LiRa software. Forensic Sci. Int. Genet. 2014, 11, 220–228. [Google Scholar] [CrossRef]
- Gill, P.; Haned, H. A new methodological framework to interpret complex DNA profiles using likelihood ratios. Forensic Sci. Int. Genet. 2013, 7, 251–263. [Google Scholar] [CrossRef]
- Curran, J.; Gill, P.; Bill, M. Interpretation of repeat measurement DNA evidence allowing for multiple contributors and population substructure. Forensic Sci. Int. 2005, 148, 47–53. [Google Scholar] [CrossRef]
- Slooten, K. Accurate assessment of the weight of evidence for DNA mixtures by integrating the likelihood ratio. Forensic Sci. Int. Genet. 2017, 27, 1–16. [Google Scholar] [CrossRef]
- Puch-Solis, R.; Rodgers, L.; Mazumder, A.; Pope, S.; Evett, I.; Curran, J.; Balding, D. Evaluating forensic DNA profiles using peak heights, allowing for multiple donors, allelic dropout and stutters. Forensic Sci. Int. Genet. 2013, 7, 555–563. [Google Scholar] [CrossRef]
- Puch-Solis, R. A dropin peak height model. Forensic Sci. Int. Genet. 2014, 11, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Perlin, M.W.; Legler, M.W.; Spencer, C.E.; Smith, J.L.; Allan, W.P.; Belrose, J.L.; Duceman, B.W. Validating TrueAllele® DNA mixture interpretation. J. Forensic Sci. 2011, 56, 1430–1447. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.; Bright, J.-A.; Buckleton, J. The interpretation of single source and mixed DNA profiles. Forensic Sci. Int. Genet. 2013, 7, 516–528. [Google Scholar] [CrossRef] [PubMed]
- Robert, G.C. Validation of an STR peak area model. Forensic Sci. Int. Genet. 2009, 3, 193–199. [Google Scholar]
- Bleka, Ø.; Storvik, G.O.; Gill, P. EuroForMix: An open source software based on a continuous model to evaluate STR DNA profiles from a mixture of contributors with artefacts. Forensic Sci. Int. Genet. 2016, 21, 35–44. [Google Scholar] [CrossRef] [Green Version]
- Cowell, R.; Lauritzen, S.; Mortera, J. Probabilistic expert systems for handling artifacts in complex DNA mixtures. Forensic Sci. Int. Genet. 2011, 5, 202–209. [Google Scholar] [CrossRef] [Green Version]
- ENFSI Guideline for Evaluative Reporting in Forensic Science. European Network of Forensic Science Institutes. 2015. Available online: https://enfsi.eu/wp-content/uploads/2016/09/m1_guideline.pdf (accessed on 28 September 2021).
- Slooten, K. Identifying common donors in DNA mixtures, with applications to database searches. Forensic Sci. Int. Genet. 2017, 26, 40–47. [Google Scholar] [CrossRef]
- Bleka, Ø.; Prieto, L.; Gill, P. CaseSolver: An investigative open source expert system based on EuroForMix. Forensic Sci. Int. Genet. 2019, 41, 83–92. [Google Scholar] [CrossRef]
- Benschop, C.C.; van de Merwe, L.; de Jong, J.; Vanvooren, V.; Kempenaers, M.; van der Beek, C.; Barni, F.; Reyes, E.L.; Moulin, L.; Pene, L.; et al. Validation of SmartRank: A likelihood ratio software for searching national DNA databases with complex DNA profiles. Forensic Sci. Int. Genet. 2017, 29, 145–153. [Google Scholar] [CrossRef]
- Benschop, C.; Jong, J.; Merwe, L.; Haned, H. Adapting a likelihood ratio model to enable searching DNA databases with complex STR DNA profiles. In Proceedings of the 2016 27th International Symposium on Human Identification, Nagoya, Japan, 28–30 November 2016; Available online: https://promega.media/-/media/files/products-and-services/genetic-identity/ishi-27-oral-abstracts/4-benschop.pdf (accessed on 28 September 2021).
- Bleka, Ø.; Bouzga, M.; Fonneløp, A.; Gill, P. dnamatch2: An open source software to carry out large scale database searches of mixtures using qualitative and quantitative models. Forensic Sci. Int. Genet. Suppl. Ser. 2017, 6, e404–e406. [Google Scholar] [CrossRef] [Green Version]
- Taylor, D.; Rowe, E.; Kruijver, M.; Abarno, D.; Bright, J.-A.; Buckleton, J. Inter-sample contamination detection using mixture deconvolution comparison. Forensic Sci. Int. Genet. 2019, 40, 160–167. [Google Scholar] [CrossRef] [Green Version]
- Kloosterman, A.; Sjerps, M.; Quak, A. Error rates in forensic DNA analysis: Definition, numbers, impact and communication. Forensic Sci. Int. Genet. 2014, 12, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Gill, P. Misleading DNA Evidence: Reasons for Miscarriages of Justice. Int. Comment. Évid. 2012, 10, 55–71. [Google Scholar] [CrossRef]
- Duewer, D.L.; Kline, M.C.; Redman, J.W.; Butler, J.M. NIST Mixed Stain Study 3: Signal Intensity Balance in Commercial Short Tandem Repeat Multiplexes. Anal. Chem. 2004, 76, 6928–6934. [Google Scholar] [CrossRef] [PubMed]
- Benschop, C.C.; Sijen, T. LoCIM-tool: An expert’s assistant for inferring the major contributor’s alleles in mixed consensus DNA profiles. Forensic Sci. Int. Genet. 2014, 11, 154–165. [Google Scholar] [CrossRef] [PubMed]
- Butler, J.M. Scientific Working Group on DNA Analysis Methods (SWGDAM) Mixture Interpretation Issues & Insights. 2007. Available online: https://strbase.nist.gov/pub_pres/SWGDAM_Jan2007_MixtureInterpretation.pdf (accessed on 28 September 2021).
- Coble, M.D. MIX13: An Interlaboratory Study on the Present State of DNA Mixture Interpretation in the U.S. In Proceedings of the 5th Annual Prescription for Criminal Justice Forensics, New York, NY, USA, 6 June 2014; Available online: http://www.cstl.nist.gov/strbase/pub_pres/Coble-ABA2014-MIX13.pdf (accessed on 28 September 2021).
- Crespillo, M.; Barrio, P.A.; Luque, J.A.; Alves, C.; Aler, M.; Alessandrini, F.; Andrade, L.; Barretto, R.; Bofarull, A.; Costa, S.; et al. GHEP-ISFG collaborative exercise on mixture profiles of autosomal STRs (GHEP-MIX01, GHEP-MIX02 and GHEP-MIX03): Results and evaluation. Forensic Sci. Int. Genet. 2014, 10, 64–72. [Google Scholar] [CrossRef]
- Cooper, S.; McGovern, C.; Bright, J.-A.; Taylor, D.; Buckleton, J. Investigating a common approach to DNA profile interpretation using probabilistic software. Forensic Sci. Int. Genet. 2015, 16, 121–131. [Google Scholar] [CrossRef]
- Torres, Y.; Flores, I.; Prieto, V.; López-Soto, M.; Farfán, M.J.; Carracedo, A.; Sanz, P. DNA mixtures in forensic casework: A 4-year retrospective study. Forensic Sci. Int. 2003, 134, 180–186. [Google Scholar] [CrossRef]
- Benschop, C.C.; Haned, H.; de Blaeij, T.J.; Meulenbroek, A.J.; Sijen, T. Assessment of mock cases involving complex low template DNA mixtures: A descriptive study. Forensic Sci. Int. Genet. 2012, 6, 697–707. [Google Scholar] [CrossRef] [PubMed]
- Benschop, C.C.G.; Connolly, E.; Ansell, R.; Kokshoorn, B. Results of an inter and intra laboratory exercise on the assessment of complex autosomal DNA pro-files. Sci. Justice 2017, 57, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Benschop, C.C.; Hoogenboom, J.; Bargeman, F.; Hovers, P.; Slagter, M.; van der Linden, J.; Parag, R.; Kruise, D.; Drobnic, K.; Klucevsek, G.; et al. Multi-laboratory validation of DNAxs including the statistical library DNAStatistX. Forensic Sci. Int. Genet. 2020, 49. [Google Scholar] [CrossRef] [PubMed]
- Butler, J.M.; Kline, M.C.; Coble, M.D. NIST Interlaboratory Studies Involving DNA Mixtures (MIX05 and MIX13): Varia-tion Observed and Lessons Learned. Forensic Sci. Int. Genet. 2018, 37, 81–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrio, P.A.; Crespillo, M.; Luque, J.; Aler, M.; Baeza-Richer, C.; Baldassarri, L.; Carnevali, E.; Coufalova, P.; Flores, I.; García, O.; et al. GHEP-ISFG collaborative exercise on mixture profiles (GHEP-MIX06). Reporting conclusions: Results and evaluation. Forensic Sci. Int. Genet. 2018, 35, 156–163. [Google Scholar] [CrossRef]
- Prieto, L.; Haned, H.; Mosquera, A.; Crespillo, M.; Alemañ, M.; Aler, M.; Álvarez, F.; Baeza-Richer, C.; Dominguez, A.; Doutremepuich, C.; et al. Euroforgen-NoE collaborative exercise on LRmix to demonstrate standardization of the interpretation of complex DNA profiles. Forensic Sci. Int. Genet. 2014, 9, 47–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buckleton, J.S.; Bright, J.-A.; Cheng, K.; Budowle, B.; Coble, M.D. NIST interlaboratory studies involving DNA mixtures (MIX13): A modern analysis. Forensic Sci. Int. Genet. 2018, 37, 172–179. [Google Scholar] [CrossRef]
- Bille, T.; Bright, J.-A.; Buckleton, J. Application of Random Match Probability Calculations to Mixed STR Profiles. J. Forensic Sci. 2013, 58, 474–485. [Google Scholar] [CrossRef]
- Alladio, E.; Omedei, M.; Cisana, S.; D’Amico, G.; Caneparo, D.; Vincenti, M.; Garofano, P. DNA mixtures interpretation—A proof-of-concept multi-software comparison highlighting different proba-bilistic methods’ performances on challenging samples. Forensic Sci. Int. Genet. 2018, 37, 143–150. [Google Scholar] [CrossRef]
- Iyer, H.K. Validation Principles, Practices, Parameters, Performance Evaluations, and Protocols Reliability Assessment of LR Systems: General Concepts. In Proceedings of the ISHI 2020 Validation Workshop, Baltimore, MD, USA, 18 September 2020; Available online: https://strbase.nist.gov/pub_pres/5_W10-Hari.pdf (accessed on 28 September 2021).
- Garofano, P.; Caneparo, D.; D’Amico, G.; Vincenti, M.; Alladio, E. An alternative application of the consensus method to DNA typing interpretation for Low Tem-plate-DNA mixtures. FSI: Genet. Suppl. Ser. 2015, 5, e422–e424. [Google Scholar]
- Cheng, K.; Bleka, Ø.; Gill, P.; Curran, J.; Bright, J.; Taylor, D.; Buckleton, J. A comparison of likelihood ratios obtained from EuroForMix and STRmix™. J. Forensic Sci. 2021. [Google Scholar] [CrossRef]
- Taylor, D.A.; Buckleton, J.S.; Bright, J.-A. Comment on “DNA mixtures interpretation—A proof-of-concept multi-software comparison highlighting different probabilistic methods’ performances on challenging samples” by Alladio et al. Forensic Sci. Int. Genet. 2019, 40, e248–e251. [Google Scholar] [CrossRef]
- Swaminathan, H.; Qureshi, M.O.; Grgicak, C.M.; Duffy, K.; Lun, D.S. Four model variants within a continuous forensic DNA mixture interpretation framework: Effects on evidential inference and reporting. PLoS ONE 2018, 13, e0207599. [Google Scholar] [CrossRef]
- Zweig, M.H.; Campbell, A.G. Receiver-operating characteristic (ROC) plots: A fundamental evaluation tool in clinical medi-cine. Clin. Chem. 1993, 39, 561–577. [Google Scholar] [CrossRef]
- Bleka, Ø.; Benschop, C.C.; Storvik, G.O.; Gill, P. A comparative study of qualitative and quantitative models used to interpret complex STR DNA profiles. Forensic Sci. Int. Genet. 2016, 25, 85–96. [Google Scholar] [CrossRef] [Green Version]
- You, Y.; Balding, D. A comparison of software for the evaluation of complex DNA profiles. Forensic Sci. Int. Genet. 2019, 40, 114–119. [Google Scholar] [CrossRef]
- Manabe, S.; Morimoto, C.; Hamano, Y.; Fujimoto, S.; Tamaki, K. Development and validation of open-source software for DNA mixture interpretation based on a quantita-tive continuous model. PLoS ONE 2017, 12, e0188183. [Google Scholar] [CrossRef] [Green Version]
- Bright, J.-A.; Cheng, K.; Kerr, Z.; McGovern, C.; Kelly, H.; Moretti, T.R.; Smith, M.A.; Bieber, F.R.; Budowle, B.; Coble, M.D.; et al. STRmix™ collaborative exercise on DNA mixture interpretation. Forensic Sci. Int. Genet. 2019, 40, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Bright, J.-A.; Richards, R.; Kruijver, M.; Kelly, H.; McGovern, C.; Magee, A.; McWhorter, A.; Ciecko, A.; Peck, B.; Baumgartner, C.; et al. Internal validation of STRmix™ – A multi laboratory response to PCAST, Forensic Sci. Int. Genet. 2019, 34, 11–24. Forensic Sci. Int. Genet. 2019, 41, e14–e17. [Google Scholar]
- Ramos, D.; Gonzalez-Rodriguez, J. Reliable support: Measuring calibration of likelihood ratios. Forensic Sci. Int. 2013, 230, 156–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buckleton, J.S.; Pugh, S.N.; Bright, J.-A.; Taylor, D.A.; Curran, J.M.; Kruijver, M.; Gill, P.; Budowle, B.; Cheng, K. Are low LRs reliable? Forensic Sci. Int. Genet. 2020, 49, 102350. [Google Scholar] [CrossRef] [PubMed]
- Hannig, J.; Riman, S.; Iyer, H.; Vallone, P.M. Are reported likelihood ratios well calibrated? Forensic Sci. Int. Genet. Suppl. Ser. 2019, 7, 572–574. [Google Scholar] [CrossRef]
- Bright, J.-A.; Dukes, M.J.; Pugh, S.N.; Evett, I.W.; Buckleton, J.S. Applying calibration to LRs produced by a DNA interpretation software. Aust. J. Forensic Sci. 2019, 53, 147–153. [Google Scholar] [CrossRef]
- Taylor, D.; Buckleton, J.; Evett, I. Testing likelihood ratios produced from complex DNA profiles. Forensic Sci. Int. Genet. 2015, 16, 165–171. [Google Scholar] [CrossRef]
- Taylor, D.; Curran, J.M.; Buckleton, J. Importance sampling allows Hd true tests of highly discriminating DNA profiles. Forensic Sci. Int. Genet. 2017, 27, 74–81. [Google Scholar] [CrossRef]
- Butler, J.M. Advanced Topics in Forensic DNA Typing: Interpretation; Academic Press: Cambridge, MA, USA, 2014. [Google Scholar] [CrossRef]
- Gill, P.; Bleka, Ø.; Hansson, O.; Benschop, C.; Haned, H. Forensic Practitioner’s Guide to the Interpretation of Complex DNA Profiles; Academic Press: London, UK, 2020. [Google Scholar] [CrossRef]
- SWGDAM Interpretation Guidelines for Autosomal STR Typing by Forensic DNA Testing Laboratories. 2010. Available online: http://www.fbi.gov/about-us/lab/codis/swgdam-interpretation-guidelines (accessed on 28 September 2021).
- Buckleton, J.S.; Curran, J.M.; Gill, P. Towards understanding the effect of uncertainty in the number of contributors to DNA stains. Forensic Sci. Int. Genet. 2007, 1, 20–28. [Google Scholar] [CrossRef]
- Paoletti, D.R.; Doom, T.E.; Krane, C.M.; Raymer, M.L.; Krane, D.E. Empirical Analysis of the STR Profiles Resulting from Conceptual Mixtures. J. Forensic Sci. 2005, 50, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Norsworthy, S.; Lun, D.S.; Grgicak, C.M. Determining the number of contributors to DNA mixtures in the low-template regime: Exploring the impacts of sampling and detection effects. Leg. Med. 2018, 32, 1–8. [Google Scholar] [CrossRef]
- Weir, B.S.; Triggs, C.M.; Starling, L.; Stowell, L.I.; Walsh, K.A.; Buckleton, J. Interpreting DNA mixtures. J. Forensic Sci. 1997, 42, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Benschop, C.C.; Haned, H.; Jeurissen, L.; Gill, P.D.; Sijen, T. The effect of varying the number of contributors on likelihood ratios for complex DNA mixtures. Forensic Sci. Int. Genet. 2015, 19, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Haned, H.; Benschop, C.C.; Gill, P.D.; Sijen, T. Complex DNA mixture analysis in a forensic context: Evaluating the probative value using a likelihood ratio model. Forensic Sci. Int. Genet. 2015, 16, 17–25. [Google Scholar] [CrossRef]
- Bright, J.A.; Curran, J.M.; Buckleton, J.S. The effect of the uncertainty in the number of contributors to mixed DNA pro-files on profile interpretation. Forensic Sci. Int. Genet. 2014, 12, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Buckleton, J.S.; Bright, J.-A.; Cheng, K.; Kelly, H.; Taylor, D.A. The effect of varying the number of contributors in the prosecution and alternate propositions. Forensic Sci. Int. Genet. 2018, 38, 225–231. [Google Scholar] [CrossRef] [Green Version]
- Bille, T.; Weitz, S.; Buckleton, J.S.; Bright, J.-A. Interpreting a major component from a mixed DNA profile with an unknown number of minor contributors. Forensic Sci. Int. Genet. 2019, 40, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Benschop, C.C.; Nijveld, A.; Duijs, F.E.; Sijen, T. An assessment of the performance of the probabilistic genotyping software EuroForMix: Trends in likelihood ratios and analysis of Type I & II errors. Forensic Sci. Int. Genet. 2019, 42, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Slooten, K. A top-down approach to DNA mixtures. Forensic Sci. Int. Genet. 2020, 46, 102250. [Google Scholar] [CrossRef] [PubMed]
- Coble, M.D.; Bright, J.-A.; Buckleton, J.S.; Curran, J.M. Uncertainty in the number of contributors in the proposed new CODIS set. Forensic Sci. Int. Genet. 2015, 19, 207–211. [Google Scholar] [CrossRef]
- Curran, J.M.; Buckleton, J. Uncertainty in the number of contributors for the European Standard Set of loci. Forensic Sci. Int. Genet. 2014, 11, 205–206. [Google Scholar] [CrossRef]
- Dembinski, G.M.; Sobieralski, C.; Picard, C.J. Estimation of the number of contributors of theoretical mixture profiles based on allele counting: Does increasing the number of loci increase success rate of estimates? Forensic Sci. Int. Genet. 2018, 33, 24–32. [Google Scholar] [CrossRef] [Green Version]
- Young, B.A.; Gettings, K.B.; McCord, B.; Vallone, P.M. Estimating number of contributors in massively parallel sequencing data of STR loci. Forensic Sci. Int. Genet. 2019, 38, 15–22. [Google Scholar] [CrossRef]
- Haned, H.; Pène, L.; Lobry, J.R.; Dufour, A.B.; Pontier, D. Estimating the Number of Contributors to Forensic DNA Mixtures: Does Maximum Likelihood Perform Better Than Maximum Allele Count? J. Forensic Sci. 2011, 56, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Haned, H.; Pène, L.; Sauvage, F.; Pontier, D. The predictive value of the maximum likelihood estimator of the number of contributors to a DNA mixture. Forensic Sci. Int. Genet. 2011, 5, 281–284. [Google Scholar] [CrossRef]
- Biedermann, A.; Bozza, S.; Konis, K.; Taroni, F. Inference about the number of contributors to a DNA mixture: Comparative analyses of a Bayesian network approach and the maximum allele count method. Forensic Sci. Int. Genet. 2012, 6, 689–696. [Google Scholar] [CrossRef]
- Tvedebrink, T. On the exact distribution of the numbers of alleles in DNA mixtures. Int. J. Leg. Med. 2013, 128, 427–437. [Google Scholar] [CrossRef]
- Benschop, C.; Haned, H.; Sijen, T. Consensus and pool profiles to assist in the analysis and interpretation of complex low template DNA mixtures. Int. J. Leg. Med. 2011, 127, 11–23. [Google Scholar] [CrossRef] [Green Version]
- Paoletti, D.R.; Krane, D.E.; Raymer, M.L.; Doom, T.E. Inferring the Number of Contributors to Mixed DNA Profiles. IEEE/ACM Trans. Comput. Biol. Bioinform. 2011, 9, 113–122. [Google Scholar] [CrossRef]
- Perez, J.; Mitchell, A.A.; Ducasse, N.; Tamariz, J.; Caragine, T. Estimating the number of contributors to two-, three-, and four-person mixtures containing DNA in high template and low template amounts. Croat. Med. J. 2011, 52, 314–326. [Google Scholar] [CrossRef] [Green Version]
- Benschop, C.C.; van der Beek, C.P.; Meiland, H.C.; van Gorp, A.G.; Westen, A.A.; Sijen, T. Low template STR typing: Effect of replicate number and consensus method on genotyping reliability and DNA database search results. Forensic Sci. Int. Genet. 2011, 5, 316–328. [Google Scholar] [CrossRef] [PubMed]
- Alfonse, L.; Tejada, G.; Swaminathan, H.; Lun, D.S.; Grgicak, C.M. Inferring the Number of Contributors to Complex DNA Mixtures Using Three Methods: Exploring the Limits of Low-Template DNA Interpretation. J. Forensic Sci. 2016, 62, 308–316. [Google Scholar] [CrossRef]
- Swaminathan, H.; Grgicak, C.M.; Medard, M.; Lun, D.S. NOCIt: A computational method to infer the number of contributors to DNA samples analyzed by STR genotyping. Forensic Sci. Int. Genet. 2015, 16, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Benschop, C.; Backx, A.; Sijen, T. Automated estimation of the number of contributors in autosomal STR profiles. Forensic Sci. Int. Genet. Suppl. Ser. 2019, 7, 7–8. [Google Scholar] [CrossRef]
- Marciano, M.A.; Adelman, J.D. PACE: Probabilistic Assessment for Contributor Estimation—A machine learning-based assessment of the number of contributors in DNA mixtures. Forensic Sci. Int. Genet. 2017, 27, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Benschop, C.C.; van der Linden, J.; Hoogenboom, J.; Ypma, R.; Haned, H. Automated estimation of the number of contributors in autosomal short tandem repeat profiles using a machine learning approach. Forensic Sci. Int. Genet. 2019, 43, 102150. [Google Scholar] [CrossRef] [PubMed]
- Kruijver, M.; Kelly, H.; Cheng, K.; Lin, M.-H.; Morawitz, J.; Russell, L.; Buckleton, J.; Bright, J.-A. Estimating the number of contributors to a DNA profile using decision trees. Forensic Sci. Int. Genet. 2020, 50, 102407. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.; Bright, J.A.; Buckleton, J. Interpreting forensic DNA profiling evidence without specifying the number of con-tribuors. Forensic Sci. Int. Genet. 2014, 13, 269–280. [Google Scholar] [CrossRef]
- Slooten, K.; Caliebe, A. Contributors are a nuisance (parameter) for DNA mixture evidence evaluation. Forensic Sci. Int. Genet. 2018, 37, 116–125. [Google Scholar] [CrossRef]
- Buckleton, J.; Taylor, D.; Bright, J.A.; Hicks, T.; Curran, J. When evaluating DNA evidence within a likelihood ratio framework, should the propositions be exhaustive? Forensic Sci. Int. Genet. 2021, 50, 102406. [Google Scholar] [CrossRef]
- Hicks, T.; Kerr, Z.; Pugh, S.; Bright, J.-A.; Curran, J.; Taylor, D.; Buckleton, J. Comparing multiple POI to DNA mixtures. Forensic Sci. Int. Genet. 2021, 52, 102481. [Google Scholar] [CrossRef]
- Kelly, H. The effect of user defined number of contributors within the LR assignment. Aust. J. Forensic Sci. 2021, 1–14. [Google Scholar] [CrossRef]
- McGovern, C.; Cheng, K.; Kelly, H.; Ciecko, A.; Taylor, D.; Buckleton, J.S.; Bright, J.A. Performance of a method for weighting a range in the number of contributors in probabilistic genotyping. Forensic Sci. Int. Genet. 2020, 48, 102352. [Google Scholar] [CrossRef]
- Akaike, H. A new look at the statistical model identification. IEEE Trans. Autom. Control. 1974, 19, 716–723. [Google Scholar] [CrossRef]
- Coble, M.D.; Bright, J.-A. Probabilistic genotyping software: An overview. Forensic Sci. Int. Genet. 2018, 38, 219–224. [Google Scholar] [CrossRef]
- Cook, R.; Evett, I.; Jackson, G.; Jones, P.; Lambert, J. A hierarchy of propositions: Deciding which level to address in casework. Sci. Justice 1998, 38, 231–239. [Google Scholar] [CrossRef]
- Evett, I.W.; Jackson, G.; Lambert, J.A. More on the hierarchy of propositions: Exploring the distinction between explana-tions and propositions. Sci. Justice 2000, 40, 3–10. [Google Scholar] [CrossRef]
- Gittelson, S.; Kalafut, T.; Myers, S.; Taylor, D.; Hicks, T.; Taroni, F.; Buckleton, J. A Practical Guide for the Formulation of Propositions in the Bayesian Approach to DNA Evidence Inter-pretation in an Adversarial Environment. J. Forensic Sci. 2016, 61, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.A.; Bright, J.-A.; Buckleton, J.S. The ‘factor of two’ issue in mixed DNA profiles. J. Theor. Biol. 2014, 363, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Evett, I. On meaningful questions: A two-trace transfer problem. J. Forensic Sci. Soc. 1987, 27, 375–381. [Google Scholar] [CrossRef]
- Gill, P. Analysis and implications of the miscarriages of justice of Amanda Knox and Raffaele Sollecito. Forensic Sci. Int. Genet. 2016, 23, 9–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foreman, L.; Smith, A.F.M.; Evett, I.W.; Aitken, C.G.G.; Taroni, A.F. Comment on Foreman L., Smith A.F.M., Evett I.W., Bayesian analysis of DNA profiling data in forensic identification applications. J. R. Stat. Soc. 1997, 160, 463. [Google Scholar] [CrossRef]
- Scientific Working Group on DNA Analysis Methods (SWGDAM): Guidelines for the Validation of Probabilistic Genotyping Systems. 2015. Available online: https://1ecb9588-ea6f-4feb-971a-73265dbf079c.filesusr.com/ugd/4344b0_22776006b67c4a32a5ffc04fe3b56515.pdf (accessed on 25 September 2021).
- Coble, M.; Buckleton, J.; Butler, J.; Egeland, T.; Fimmers, R.; Gill, P.; Gusmão, L.; Guttman, B.; Krawczak, M.; Morling, N.; et al. DNA Commission of the International Society for Forensic Genetics: Recommendations on the validation of software programs performing biostatistical calculations for forensic genetics applications. Forensic Sci. Int. Genet. 2016, 25, 191–197. [Google Scholar] [CrossRef]
- ANSI/ASB Standard 018, First Edition. 2020: Standard for Validation of Probabilistic Genotyping Systems. Available online: http://www.asbstandardsboard.org/wp-content/uploads/2020/07/018_Std_e1.pdf (accessed on 28 September 2021).
- Ballim, A.; Wilks, A.Y. Beliefs, stereotypes and dynamic agent modeling. User Model. User-Adapt. Interact. 1991, 1, 33–65. [Google Scholar] [CrossRef]
- Bright, J.-A.; Evett, I.W.; Taylor, D.; Curran, J.M.; Buckleton, J. A series of recommended tests when validating probabilistic DNA profile interpretation software. Forensic Sci. Int. Genet. 2015, 14, 125–131. [Google Scholar] [CrossRef]
- Haned, H.; Gill, P.; Lohmueller, K.; Inman, K.; Rudin, N. Validation of probabilistic genotyping software for use in forensic DNA casework: Definitions and illustra-tions. Sci. Justice 2016, 56, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Ropero-Miller, J.; Melton, P.; Ferrara, L.; Hall, J. Landscape Study of DNA Mixture Interpretation Software. National Institute of Justice, Forensic Technology Centre of Excellence. 2015. Available online: https://nij.ojp.gov/library/publications/landscape-study-dna-mixture-interpretation-software (accessed on 28 September 2021).
- European Network of Forensic Science Institutes (ENFSI). Best Practice Manual for the Internal Validation of Probabilistic Software to Undertake DNA Mixture Interpretation ENFSI-BPM-DNA-01 issue 001. 17 May 2017. Available online: https://enfsi.eu/wp-content/uploads/2017/09/Best-Practice-Manual-for-the-internal-validation-of-probabilistic-software-to-undertake-DNA-mixture-interpretation-v1.docx.pdf (accessed on 28 September 2021).
- Forensic Science Regulator, Software Validation for DNA Mixture Interpretation, FSR-G-223 (2). 2018. Available online: https://www.gov.uk/government/publications/software-validation-for-dna-mixture-interpretation-fsr-g-223 (accessed on 28 September 2021).
- PCAST, Forensic Science in Criminal Courts: Ensuring Scientific Validity of Feature Comparison Methods, US President’s Council of Advisors on Science and Technology. 2016. Available online: https://obamawhitehouse.archives.gov/sites/default/files/microsites/ostp/PCAST/pcast_forensic_science_report_final.pdf (accessed on 28 September 2021).
- Box, G.E.P.; Draper, N.R. Empirical Model-Building and Response Surfaces; Wiley: New York, NY, USA, 1987. [Google Scholar]
- Rykiel, E.J., Jr. Testing ecological models: The meaning of validation. Ecol. Model. 1996, 90, 229–244. [Google Scholar] [CrossRef]
- Gill, P.; Kirkham, A.; Curran, J. LoComatioN: A software tool for the analysis of low copy number DNA profiles. Forensic Sci. Int. 2007, 166, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Haned, H. Forensim: An open-source initiative for the evaluation of statistical methods in forensic genetics. Forensic Sci. Int. Genet. 2011, 5, 265–268. [Google Scholar] [CrossRef]
- Haned, H.; Gill, P. Analysis of complex DNA mixtures using the Forensim package. Forensic Sci. Int. Genet. Suppl. Ser. 2011, 3, e79–e80. [Google Scholar] [CrossRef]
- Evett, I.W.; Gill, P.D.; Lambert, J.A. Taking account of peak areas when interpreting mixed DNA profiles. J. Forensic Sci. 1998, 43, 62–69. [Google Scholar] [CrossRef]
- Cowell, R.G.; Lauritzen, S.L.; Mortera, J. A γ model for {DNA} mixture analyses. Bayesian Anal. 2007, 2, 333–348. [Google Scholar] [CrossRef]
- Graversen, T.; Lauritzen, S. Estimation of parameters in DNA mixture analysis. J. Appl. Stat. 2013, 40, 2423–2436. [Google Scholar] [CrossRef] [Green Version]
- Graversen, T.; Lauritzen, S. Computational aspects of DNA mixture analysis. Stat. Comput. 2014, 25, 527–541. [Google Scholar] [CrossRef] [Green Version]
- Haldemann, B.; Dornseifer, S.; Heylen, T.; Aelbrecht, C.; Bleka, O.; Larsen, H.J.; Neuhaus-Steinmetz, U. eDNA—An expert software system for comparison and evaluation of DNA profiles in forensic case-work. Forensic Sci. Int. Genet. Suppl. Ser. 2015, 5, e400–e402. [Google Scholar] [CrossRef]
- Benschop, C.C.; Hoogenboom, J.; Hovers, P.; Slagter, M.; Kruise, D.; Parag, R.; Steensma, K.; Slooten, K.; Nagel, J.H.; Dieltjes, P.; et al. DNAxs/DNAStatistX: Development and validation of a software suite for the data management and probabilistic interpretation of DNA profiles. Forensic Sci. Int. Genet. 2019, 42, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Veldhuis, M. Explainable artificial intelligence in forensics: Realistic explanations for number of contributor predictions of DNA profiles. Forensic Sci. Int. Genet. 2021, submitted. [Google Scholar]
- Cowell, R.G.; Graversen, T.; Lauritzen, S.L.; Mortera, J. Analysis of forensic DNA mixtures with artefacts. J. R. Stat. Soc. Ser. C Appl. Stat. 2015, 64, 1–48. [Google Scholar] [CrossRef] [Green Version]
- Duijs, F.E.; Hoogenboom, J.; Sijen, T.; Benschop, C. Performance of EuroForMix deconvolution on PowerPlex® Fusion 6C profiles. Forensic Sci. Int. Genet. Suppl. Ser. 2019, 7, 5–6. [Google Scholar] [CrossRef]
- Taylor, D.; Buckleton, J. Do low template DNA profiles have useful quantitative data? Forensic Sci. Int. Genet. 2015, 16, 13–16. [Google Scholar] [CrossRef]
- Bruijns, B.; Tiggelaar, R.M.; Gardeniers, J. Massively parallel sequencing techniques for forensics: A review. Electrophoresis 2018, 39, 2642–2654. [Google Scholar] [CrossRef] [Green Version]
- Just, R.S.; Irwin, J.A. Use of the LUS in sequence allele designations to facilitate probabilistic genotyping of NGS-based STR typing results. Forensic Sci. Int. Genet. 2018, 34, 197–205. [Google Scholar] [CrossRef] [Green Version]
- Just, R.S.; Le, J.; Irwin, J.A. LUS+: Extension of the LUS designator concept to differentiate most sequence alleles for 27 STR loci. Forensic Sci. Int. Rep. 2020, 2, 100059. [Google Scholar] [CrossRef]
- Vilsen, S.B.; Tvedebrink, T.; Eriksen, P.S.; Bøsting, C.; Hussing, C.; Mogensen, H.S.; Morling, N. Stutter analysis of complex STR MPS data. Forensic Sci. Int. Genet. 2018, 35, 107–112. [Google Scholar] [CrossRef]
- Bleka, Ø.; Just, R.; Le, J.; Gill, P. Automation of high volume MPS mixture interpretation using CaseSolver. Forensic Sci. Int. Genet. Suppl. Ser. 2019, 7, 14–15. [Google Scholar] [CrossRef]
- Bleka, Ø.; Just, R.; Le, J.; Gill, P. An examination of STR nomenclatures, filters and models for MPS mixture interpretation. Forensic Sci. Int. Genet. 2020, 48, 102319. [Google Scholar] [CrossRef]
- Gill, P.; Just, R.; Parson, W.; Phillips, C.; Bleka, Ø. Interpretation of complex DNA profiles generated by Massively Parallel Sequencing. In Forensic Practitioner’s Guide to the Interpretation of Complex DNA Profiles; Gill, P., Bleka, O., Hansson, O., Benschop, C., Haned, H., Eds.; Academic Press: Cambridge, MA, USA; Elsevier: Amsterdam, The Netherlands, 2020; pp. 419–451. [Google Scholar] [CrossRef]
- van der Gaag, K.J.; de Leeuw, R.H.; Hoogenboom, J.; Patel, J.; Storts, D.R.; Laros, J.F.; de Knijff, P. Massively parallel sequencing of short tandem re-peats-Population data and mixture analysis results for the PowerSeq system. Forensic Sci. Int. Genet. 2016, 24, 86–96. [Google Scholar] [CrossRef] [Green Version]
- Hoogenboom, J.; van der Gaag, K.J.; de Leeuw, R.H.; Sijen, T.; de Knijff, P.; Laros, J.F. FDSTools: A software package for analysis of massively parallel sequencing data with the ability to recognise and correct STR stutter and other PCR or sequencing noise. Forensic Sci. Int. Genet. 2016, 27, 27–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benschop, C.C.; van der Gaag, K.J.; de Vreede, J.; Backx, A.J.; de Leeuw, R.H.; Zuñiga, S.; Hoogenboom, J.; de Knijff, P.; Sijen, T. Application of a probabilistic genotyping software to MPS mixture STR data is supported by similar trends in LRs compared with CE data. Forensic Sci. Int. Genet. 2021, 52, 102489. [Google Scholar] [CrossRef] [PubMed]
- Bleka, Ø.; Eduardoff, M.; Santos, C.; Phillips, C.; Parson, W.; Gill, P. Open source software EuroForMix can be used to analyse complex SNP mixtures. Forensic Sci. Int. Genet. 2017, 31, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Bleka, Ø.; Eduardoff, M.; Santos, C.; Phillips, C.; Parson, W.; Gill, P. Using EuroForMix to analyse complex SNP mixtures, up to six contributors. Forensic Sci. Int. Genet. Suppl. Ser. 2017, 6, e277–e279. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.-W.; Li, Y.-H.; Chou, C.-F.; Lai, F.-P.; Chien, Y.-H.; Yin, H.-I.; Lee, T.-T.; Hwa, H.-L. DNA mixture interpretation using linear regression and neural networks on massively parallel sequencing data of single nucleotide polymorphisms. Aust. J. Forensic Sci. 2021, 1–13. [Google Scholar] [CrossRef]
- Benschop, C.C.; Haned, H.; Yoo, S.Y.; Sijen, T. Evaluation of samples comprising minute amounts of DNA. Sci. Justice 2015, 55, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Benschop, C.C.; Yoo, S.Y.; Sijen, T. Split DNA over replicates or perform one amplification? Forensic Sci. Int. Genet. Suppl. Ser. 2015, 5, e532–e533. [Google Scholar] [CrossRef] [Green Version]
- Benschop, C.C.; Graaf, E.S.; Sijen, T. Is an increased drop-in rate appropriate with enhanced DNA profiling? Forensic Sci. Int. Genet. Suppl. Ser. 2015, 5, e71–e72. [Google Scholar] [CrossRef]
- Slagter, M.; Kruise, D.; van Ommen, L.; Hoogenboom, J.; Steensma, K.; de Jong, J.; Hovers, P.; Parag, R.; van der Linden, J.; Kneppers, A.L.; et al. The DNAxs software suite: A three-year retrospective study on the development, architecture, testing and implementation in forensic casework. Forensic Sci. Int. Rep. 2021, 3, 100212. [Google Scholar] [CrossRef]
- Beecham, G.W.; Weir, B.S. Confidence interval of the likelihood ratio associated with mixed stain DNA evidence. J. Forensic Sci. 2010, 56, S166–S171. [Google Scholar] [CrossRef]
- Curran, J.M.; Triggs, C.M.; Buckleton, J.; Weir, B.S. Interpreting DNA mixtures in structured populations. J. Forensic Sci. 1999, 44, 12028J. [Google Scholar] [CrossRef]
- Slooten, K. The analogy between DNA kinship and DNA mixture evaluation, with applications for the interpretation of like-lihood ratios produced by possibly imperfect models. Forensic Sci. Int. Genet. 2021, 52, 102449. [Google Scholar] [CrossRef]
- Good, I.J. Probability and the Weighing of Evidence; Charles Griffin & Company Limited: London, UK, 1950. [Google Scholar]
- Bright, J.A.; Taylor, D.; Curran, J.M.; Buckleton, J.S. Degradation of forensic DNA profiles. Aust. J. Forensic Sci. 2013, 45, 445–449. [Google Scholar] [CrossRef]
- Taylor, D.; Bright, J.-A.; Kelly, H.; Lin, M.-H.; Buckleton, J. A fully continuous system of DNA profile evidence evaluation that can utilise STR profile data produced under different conditions within a single analysis. Forensic Sci. Int. Genet. 2017, 31, 149–154. [Google Scholar] [CrossRef] [Green Version]
- Bright, J.-A.; Taylor, D.; Curran, J.M.; Buckleton, J.S. Developing allelic and stutter peak height models for a continuous method of DNA interpretation. Forensic Sci. Int. Genet. 2013, 7, 296–304. [Google Scholar] [CrossRef]
- Taylor, D.; Bright, J.-A.; McGovern, C.; Hefford, C.; Kalafut, T.; Buckleton, J. Validating multiplexes for use in conjunction with modern interpretation strategies. Forensic Sci. Int. Genet. 2016, 20, 6–19. [Google Scholar] [CrossRef] [Green Version]
- Bright, J.-A.; Buckleton, J.S.; Taylor, D.; Fernando, M.A.C.S.S.; Curran, J.M. Modeling forward stutter: Toward increased objectivity in forensic DNA interpretation. Electrophoresis 2014, 35, 3152–3157. [Google Scholar] [CrossRef]
- Bright, J.-A.; Taylor, D.; Kerr, Z.; Buckleton, J.; Kruijver, M. The efficacy of DNA mixture to mixture matching. Forensic Sci. Int. Genet. 2019, 41, 64–71. [Google Scholar] [CrossRef] [Green Version]
- Cheng, K.; Bright, J.-A.; Kerr, Z.; Taylor, D.; Ciecko, A.; Curran, J.; Buckleton, J. Examining the additivity of peak heights in forensic DNA profiles. Aust. J. Forensic Sci. 2020, 1–15. [Google Scholar] [CrossRef]
- Hansson, O.; Egeland, T.; Gill, P. Characterization of degradation and heterozygote balance by simulation of the forensic DNA analysis process. Int. J. Leg. Med. 2016, 131, 303–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bright, J.-A.; Curran, J.M.; Buckleton, J.S. Investigation into the performance of different models for predicting stutter. Forensic Sci. Int. Genet. 2013, 7, 422–427. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D. Using continuous DNA interpretation methods to revisit likelihood ratio behaviour. Forensic Sci. Int. Genet. 2014, 11, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.A.; Bright, J.-A.; Buckleton, J.S. The effect of varying the number of contributors in the prosecution and alternate propositions. Forensic Sci. Int. Genet. 2019, 13, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, M. Computing the Bayes Factor from a Markov Chain Monte Carlo Simulation of the Posterior Distribution. Bayesian Anal. 2012, 7, 737–770. [Google Scholar] [CrossRef]
- Weinberg, M.D.; Yoon, I.; Katz, N. A remarkably simple and accurate method for computing the Bayes Factor from a Markov chain Monte Carlo Simulation of the Posterior Distribution in high dimension. arXiv 2013, arXiv:1301.3156v1. [Google Scholar]
- Buckleton, J.; Bright, J.-A.; Taylor, D.; Evett, I.; Hicks, T.; Jackson, G.; Curran, J.M. Helping formulate propositions in forensic DNA analysis. Sci. Justice 2014, 54, 258–261. [Google Scholar] [CrossRef]
- Taylor, D.; Bright, J.-A.; Buckleton, J. Considering relatives when assessing the evidential strength of mixed DNA profiles. Forensic Sci. Int. Genet. 2014, 13, 259–263. [Google Scholar] [CrossRef]
- Balding, D.J. Weight-of-Evidence for Forensic DNA Profiles; John Wiley and Sons: Chichester, UK, 2005. [Google Scholar]
- Buckleton, J.; Triggs, C. Relatedness and DNA: Are we taking it seriously enough? Forensic Sci. Int. 2005, 152, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.; Bright, J.-A.; Buckleton, J.; Curran, J. An illustration of the effect of various sources of uncertainty on DNA likelihood ratio calculations. Forensic Sci. Int. Genet. 2014, 11, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Balding, D.; Nichols, R.A. DNA profile match probability calculation: How to allow for population stratification, relatedness, database selection and single bands. Forensic Sci. Int. 1994, 64, 125–140. [Google Scholar] [CrossRef]
- Balding, D.J. Estimating products in forensic identification using DNA profiles. J. Am. Stat. Assoc. 1995, 90, 839–844. [Google Scholar] [CrossRef]
- Curran, J.M.; Buckleton, J.S. An investigation into the performance of methods for adjusting for sampling uncertainty in DNA likelihood ratio calculations. Forensic Sci. Int. Genet. 2011, 5, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Triggs, C.; Harbison, S.; Buckleton, J. The calculation of DNA match probabilities in mixed race populations. Sci. Justice 2000, 40, 33–38. [Google Scholar] [CrossRef]
- Curran, J.M. An introduction to Bayesian credible intervals for sampling error in DNA profiles. Law Probab. Risk 2005, 4, 115–126. [Google Scholar] [CrossRef] [Green Version]
- Morrison, G.S. Special issue on measuring and reporting the precision of forensic likelihood ratios: Introduction to the debate. Sci. Justice 2016, 56, 371–373. [Google Scholar] [CrossRef] [PubMed]
- Morrison, G.; Enzinger, E. What should a forensic practitioner’s likelihood ratio be? Sci. Justice 2016, 5, 374–379. [Google Scholar] [CrossRef]
- Curran, J.M. Admitting to uncertainty in the LR. Sci. Justice 2016, 56, 380–382. [Google Scholar] [CrossRef]
- Ommen, D.M.; Saunders, C.P.; Neumann, C. An argument against presenting interval quantifications as a surrogate for the value of evidence. Sci. Justice 2016, 56, 383–387. [Google Scholar] [CrossRef]
- Berger, C.E.; Slooten, K. The LR does not exist. Sci. Justice 2016, 56, 388–391. [Google Scholar] [CrossRef] [PubMed]
- Biedermann, A.; Bozza, S.; Taroni, F.; Aitken, C. Reframing the debate: A question of probability, not of likelihood ratio. Sci. Justice 2016, 56, 392–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hout, A.v.d.; Alberink, I. Posterior distribution for likelihood ratios in forensic science. Sci. Justice 2016, 5, 397–401. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.; Hicks, T.; Champod, C. Using sensitivity analyses in Bayesian networks to highlight the impact of data pauci-ty and direct future analyses: A contribution to the debate on measuring and reporting the precision of likelihood ratios. Sci. Justice 2016, 56, 402–410. [Google Scholar] [CrossRef] [PubMed]
- Bright, J.-A. Testing Methods for Quantifying Monte Carlo Variation for Categorical Variables in Probabilistic Genotyping; Report; Institute of Environmental Science and Research: Wellington, New Zealand, 2020. [Google Scholar] [CrossRef]
- Bright, J.-A. Revisiting the STRmix™ Likelihood Ratio Probability Interval Coverage Considering Multiple Factors; Report; Institute of Environmental Science and Research: Wellington, New Zealand, 2021. [Google Scholar]
- Buckleton, J.; Curran, J.; Goudet, J.; Taylor, D.; Thiery, A.; Weir, B.S. Population-specific FST values for forensic STR markers: A worldwide survey. Forensic Sci. Int. Genet. 2016, 23, 126–133. [Google Scholar]
- Steele, C.D.; Court, D.S.; Balding, D.J. Worldwide F(ST) estimates relative to five continental-scale populations. Ann. Hum. Genet. 2014, 78, 468–477. [Google Scholar] [CrossRef] [Green Version]
- Weir, B.S.; Cockerham, C.C. Estimating F-statistics for the analysis of population structure. Evolution 1984, 38, 1358–1370. [Google Scholar]
- Bright, J.-A.; Taylor, D.; Curran, J.; Buckleton, J. Searching mixed DNA profiles directly against profile databases. Forensic Sci. Int. Genet. 2014, 9, 102–110. [Google Scholar] [CrossRef]
- Abarno, D.; Sobieraj, T.; Summers, C.; Taylor, D. The first Australian conviction resulting from a familial search. Aust. J. Forensic Sci. 2019, 51, S56–S59. [Google Scholar] [CrossRef]
- Taylor, D.; Abarno, D.; Rowe, E.; Rask-Nielsen, L. Observations of DNA transfer within an operational Forensic Biology Laboratory. Forensic Int. Genet. 2016, 23, 33–49. [Google Scholar] [CrossRef] [PubMed]
- Kruijver, M.; Meester, R.; Slooten, K. p-Values should not be used for evaluating the strength of DNA evidence. Forensic Sci. Int. Genet. 2015, 16, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Budowle, B.; Onorato, A.J.; Callaghan, T.F.; Della Manna, A.; Gross, A.M.; Guerrieri, R.A.; Luttman, J.C.; McClure, D.L. Mixture Interpretation: Defining the Relevant Features for Guidelines for the Assessment of Mixed DNA Profiles in Forensic Casework. J. Forensic Sci. 2009, 54, 810–821. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.; Bright, J.-A.; Scandrett, L.; Abarno, D.; Lee, S.-I.; Wivell, R.; Kelly, H.; Buckleton, J. Validation of a top-down DNA profile analysis for database searching using a fully continuous probabilistic genotyping model. Forensic Sci. Int. Genet. 2021, 52, 102479. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.; Buckleton, J.; Bright, J.-A. Factors affecting peak height variability for short tandem repeat data. Forensic Sci. Int. Genet. 2015, 21, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Bright, J.-A.; Curran, J.M. Investigation into stutter ratio variability between different laboratories. Forensic Sci. Int. Genet. 2014, 13, 79–81. [Google Scholar] [CrossRef]
- Kelly, H.; Bright, J.-A.; Kruijver, M.; Cooper, S.; Taylor, D.; Duke, K.; Strong, M.; Beamer, V.; Buettner, C.; Buckleton, J. A sensitivity analysis to determine the robustness of STRmix™ with respect to laboratory calibration. Forensic Sci. Int. Genet. 2018, 35, 113–122. [Google Scholar] [CrossRef]
- Kelly, H. Developmental validation of a software implementation of a flexible framework for the assignment of likeli-hood ratios for forensic investigations. Forensic Sci. Int. Rep. 2021, 100231. [Google Scholar] [CrossRef]
- Kruijver, M.; Bright, J.-A.; Kelly, H.; Buckleton, J. Exploring the probative value of mixed DNA profiles. Forensic Sci. Int. Genet. 2019, 41, 1–10. [Google Scholar] [CrossRef]
- Taylor, D.; Kruijver, M. Combining evidence across multiple mixed DNA profiles for improved resolution of a donor when a common contributor can be assumed. Forensic Sci. Int. Genet. 2020, 49. [Google Scholar] [CrossRef]
- Kruijver, M.; Taylor, D.; Bright, J.-A. Evaluating DNA evidence possibly involving multiple (mixed) samples, common donors and related contributors. Forensic Sci. Int. Genet. 2021, 54, 102532. [Google Scholar] [CrossRef]
- Allen, P.S.; Pugh, S.N.; Bright, J.-A.; Taylor, D.A.; Curran, J.M.; Kerr, Z.; Buckleton, J.S. Relaxing the assumption of unrelatedness in the numerator and denominator of likelihood ratios for DNA mixtures. Forensic Sci. Int. Genet. 2020, 51, 102434. [Google Scholar] [CrossRef] [PubMed]
- Regulator, F.S. The Forensic Science Regulator Guidance on DNA Mixture Interpretation FSR-G-222; The Forensic Science Regulator: Birmingham, UK, 2018; pp. 1–63. [Google Scholar]
- Bright, J.-A.; Taylor, D.; McGovern, C.; Cooper, S.; Russell, L.; Abarno, D.; Buckleton, J. Developmental validation of STRmix™, expert software for the interpretation of forensic DNA profiles. Forensic Sci. Int. Genet. 2016, 23, 226–239. [Google Scholar] [CrossRef] [PubMed]
- Brookes, C.; Bright, J.-A.; Harbison, S.; Buckleton, J. Characterising stutter in forensic STR multiplexes. Forensic Sci. Int. Genet. 2012, 6, 58–63. [Google Scholar] [CrossRef]
- Kelly, H.; Bright, J.-A.; Buckleton, J.S.; Curran, J.M. Identifying and modelling the drivers of stutter in forensic DNA profiles. Aust. J. Forensic Sci. 2013, 46, 194–203. [Google Scholar] [CrossRef]
- Triggs, C.; Curran, J. The sensitivity of the Bayesian HPD method to the choice of prior. Sci. Justice 2006, 46, 169–178. [Google Scholar] [CrossRef]
- Bright, J.-A.; Stevenson, K.; Curran, J.M.; Buckleton, J.S. The variability in likelihood ratios due to different mechanisms. Forensic Sci. Int. Genet. 2015, 14, 187–190. [Google Scholar] [CrossRef]
- Taylor, D.; Buckleton, J.; Bright, J.-A. Does the use of probabilistic genotyping change the way we should view sub-threshold data? Aust. J. Forensic Sci. 2017, 49, 78–92. [Google Scholar] [CrossRef]
- Russell, L.; Cooper, S.; Wivell, R.; Kerr, Z.; Taylor, D.; Buckleton, J.; Bright, J. A guide to results and diagnostics within a STRmix™ report. Wiley Interdiscip. Rev. Forensic Sci. 2019, 1, e1354. [Google Scholar] [CrossRef]
- Moretti, T.; Just, R.S.; Kehl, S.C.; Willis, L.E.; Buckleton, J.S.; Bright, J.-A.; Taylor, D..; Onorato, A.J. Internal validation of STRmix for the interpetation of single source and mixed DNA profiles. Forensic Sci. Int. Genet. 2017, 29, 126–144. [Google Scholar] [CrossRef]
- Noël, S.; Noël, J.; Granger, D.; Lefebvre, J.-F.; Séguin, D. STRmix™ put to the test: 300,000 non-contributor profiles compared to four-contributor DNA mixtures and the impact of replicates. Forensic Sci. Int. Genet. 2019, 41, 24–31. [Google Scholar] [CrossRef]
- Duke, K.R.; Myers, S.P. Systematic evaluation of STRmix™ performance on degraded DNA profile data. Forensic Sci. Int. Genet. 2019, 44, 102174. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.-H.; Bright, J.-A.; Pugh, S.N.; Buckleton, J.S. The interpretation of mixed DNA profiles from a mother, father, and child trio. Forensic Sci. Int. Genet. 2019, 44, 102175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buckleton, J.S.; Bright, J.-A.; Gittelson, S.; Moretti, T.R.; Onorato, A.J.; Bieber, F.R.; Budowle, B.; Taylor, D.A. The Probabilistic Genotyping SoftwareSTRmix: Utility and Evidence for its Validity. J. Forensic Sci. 2018, 64, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Riman, S.; Iyer, H.; Vallone, P. Exploring DNA interpretation software using the PROVEDIt dataset. Forensic Sci. Int. Genet. Suppl. Ser. 2019, 7, 724–726. [Google Scholar] [CrossRef]
- Takano, M.H. R.4368—Justice in Forensic ALGORITHMS Act of 2019; 2019. [Google Scholar]
- Access to STRmix™ Software by Defence Legal Teams (“Access Policy”); 2020. [CrossRef]
- Adams, N.; Koppl, R.; Krane, D.; Thompson, W.; Zabell, S. Letter to the Editor-Appropriate Standards for Verification and Validation of Probabilistic Genotyping Systems. J. Forensic Sci. 2018, 63, 339–340. [Google Scholar] [CrossRef] [PubMed]
- Software and Systems Engineering Standards Committee of the IEEE Computer Society. IEEE Standard for System, Software, and Hardware Verification and Validation IEEE Std 1012™-2016; IEEE: New York, NY, USA, 2017. [Google Scholar]
- The Queen v. Clinton James Tuite. S CR 2014 007, 23 October 2017.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Algorithms, Scientific Principles and Methods | Version Introduced | Reference |
|---|---|---|
| Allele and stutter peak height variability as separate constants within the MCMC | V2.0 | [15] |
| Peak height variability as random variables within the MCMC | V2.3 | [196] |
| Model for calibrating laboratory peak height variability | V2.0 | [196] |
| Application of a Gaussian random walk to the MCMC process | V2.3 | [205] |
| Modelling of back stutter by regressing stutter ratio against allelic designation | V2.0 | [156,197,206,207] |
| Modelling of back stutter by regressing stutter ratio against LUS | V2.3 | [156,162,206,207] |
| Modelling of forward stutter | V2.4 | [157] |
| Modelling of allelic drop-in using a simple exponential or uniform distribution | V2.0 | [15] |
| Modelling of allelic drop-in using a γ distribution | V2.3 | [13] |
| Modelling of degradation and dropout | V2.0 | [154] |
| Modelling of the uncertainties in the allele frequencies using the HPD | V2.0 | [208] |
| Modelling of the uncertainties in the MCMC | V2.3 | [171,208,209] |
| Database searching of mixed DNA profiles | V2.0 | [190] |
| Familial searching of mixed DNA profiles | V2.3 | [168] |
| Relatives as alternate contributors under the defence proposition | V2.3 | [168] |
| Modelling expected stutter peak heights in saturated data | V2.3 | [157] |
| Taking into account the ‘factor of two’ in LR calculations | V2.3 | [104] |
| Model for incorporating prior beliefs in mixture proportions | V2.3 | [210] |
| Combining DNA profiles produced under different conditions into a single analysis | V2.5 | [155] |
| Assigning a range for the number of contributors to a DNA profile | V2.6 | [164] |
| Mixture-to-mixture comparison to identify common DNA donors | V2.7 | [20] |
| A top-down DNA search approach | V2.8 | [74] |
| The diagnostic outputs of STRmix™ | V2.3 | [211] |
| Focus of Validation | Reference |
|---|---|
| Ability of STRmix™ to deconvolute profiles and assign LRs that comport to manual interpretation and human expectation | [15] |
| Ability of STRmix™ to discriminate between donors and non-donors in database searches | [190] |
| Behaviour of STRmix™ to assign LRs when dealing with multiple replicates, different number of contributors, and assumed contributors | [163] |
| Sensitivity of LR produced by STRmix™ to different factors of uncertainty such as theta, relatedness of alternate DNA source and length of MCMC analysis | [171] |
| Tests to be performed when validating probabilistic genotyping, using STRmix™ as an example | [112] |
| Ability of individuals from different laboratories to standardise evaluations when using STRmix™ | [33,53] |
| Ability of STRmix™ to reliably use peak height information in very low intensity profiles | [56,132,210] |
| Ability of STRmix™ to discriminate between donors and non-donors in large-scale Hd true tests, or using importance sampling | [59,60,190,200,212,213] |
| Sensitivity of STRmix™ model parameters to laboratory factors | [196,198] |
| Ability of STRmix™ to utilise information from profiles produced under different laboratory conditions within a single analysis | [155] |
| Effect of mixture complexity, allele sharing and contributor proportions on the ability STRmix™ to distinguish contributors from non-contributors | [54] |
| The ability of STRmix™ to identify common DNA donors in mixed samples | [25,159] |
| The sensitivity of LRs produced in STRmix™ to the choice of the number of contributors | [71,72,97] |
| Ability to use STRmix™ to resolve major components of mixtures | [72] |
| Testing the assumption of additivity of peak heights in STRmix™ models | [159,160] |
| Performance of the degradation model within STRmix™ | [214] |
| The effect of relatedness of contributors to the STRmix™ analysis | [203,215] |
| Testing the calibration of LRs produced in STRmix™ | [58] |
| Validation overviews of STRmix™ | [205,216] |
| Comparison of STRmix™ to other probabilistic genotyping software | [41,43,112,217] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gill, P.; Benschop, C.; Buckleton, J.; Bleka, Ø.; Taylor, D. A Review of Probabilistic Genotyping Systems: EuroForMix, DNAStatistX and STRmix™. Genes 2021, 12, 1559. https://doi.org/10.3390/genes12101559
Gill P, Benschop C, Buckleton J, Bleka Ø, Taylor D. A Review of Probabilistic Genotyping Systems: EuroForMix, DNAStatistX and STRmix™. Genes. 2021; 12(10):1559. https://doi.org/10.3390/genes12101559
Chicago/Turabian StyleGill, Peter, Corina Benschop, John Buckleton, Øyvind Bleka, and Duncan Taylor. 2021. "A Review of Probabilistic Genotyping Systems: EuroForMix, DNAStatistX and STRmix™" Genes 12, no. 10: 1559. https://doi.org/10.3390/genes12101559
APA StyleGill, P., Benschop, C., Buckleton, J., Bleka, Ø., & Taylor, D. (2021). A Review of Probabilistic Genotyping Systems: EuroForMix, DNAStatistX and STRmix™. Genes, 12(10), 1559. https://doi.org/10.3390/genes12101559