
Interrogating Mitochondrial Biology and Disease Using CRISPR/Cas9 Gene Editing

{kind=link}
Abstract
:1. Introduction
2. Gene Editing in Mitochondrial Research
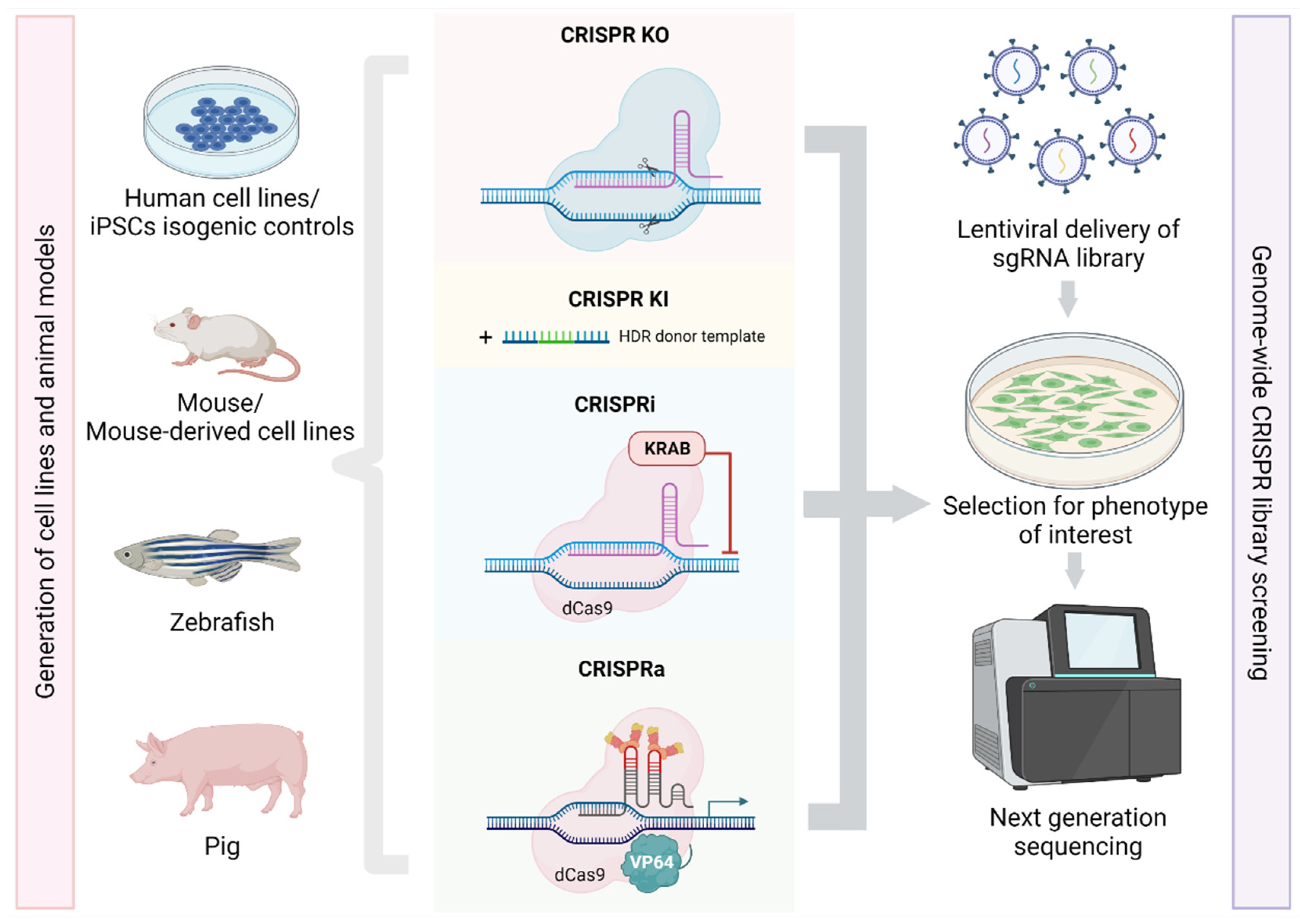
3. Application of CRISPR/Cas9 in Mitochondrial Research
3.1. Generation of Cell Lines and Animal Models
- Delineating specific gene functions to understand basic mitochondrial molecular mechanisms;
- Generating cell lines and animal models to establish and examine gene causality for mitochondrial dysfunction;
- Generating isogenic control cell lines (i) to provide reliable controls minimising experimental variability and (ii) to verify pathogenicity of mitochondrial disease-causing candidates
3.1.1. Delineating Gene Functions in Mitochondrial Biology
3.1.2. Generation of Cell Lines and Animal Models to Examine Gene Causality for Mitochondrial Dysfunction
3.1.3. Generating Isogenic Control Cell Lines to Verify Pathogenicity of Mitochondrial Proteins
3.2. High-Throughput CRISPR/Cas9 Library Screening
4. The Use of CRISPR/Cas9 System for Targeting the Mitochondrial Genome
5. Concluding Remarks and Future Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Doudna, J.A. CRISPR-Cas9 Structures and Mechanisms. Annu. Rev. Biophys. 2017, 46, 505–529. [Google Scholar] [CrossRef] [Green Version]
- Bétermier, M.; Bertrand, P.; Lopez, B.S. Is non-homologous end-joining really an inherently error-prone process? PLoS Genet. 2014, 10, e1004086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardo, B.; Gómez-González, B.; Aguilera, A. DNA repair in mammalian cells: DNA double-strand break repair: How to fix a broken relationship. Cell Mol. Life Sci. 2009, 66, 1039–1056. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [Green Version]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larson, M.H.; Gilbert, L.A.; Wang, X.; Lim, W.A.; Weissman, J.S.; Qi, L.S. CRISPR interference (CRISPRi) for sequence-specific control of gene expression. Nat. Protoc. 2013, 8, 2180–2196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, L.A.; Larson, M.H.; Morsut, L.; Liu, Z.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A.; et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 2013, 154, 442–451. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, L.A.; Horlbeck, M.A.; Adamson, B.; Villalta, J.E.; Chen, Y.; Whitehead, E.H.; Guimaraes, C.; Panning, B.; Ploegh, H.L.; Bassik, M.C.; et al. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell 2014, 159, 647–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konermann, S.; Brigham, M.D.; Trevino, A.; Hsu, P.D.; Heidenreich, M.; Cong, L.; Platt, R.J.; Scott, D.A.; Church, G.M.; Zhang, F. Optical control of mammalian endogenous transcription and epigenetic states. Nature 2013, 500, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Maeder, M.L.; Linder, S.J.; Cascio, V.M.; Fu, Y.; Ho, Q.H.; Joung, J.K. CRISPR RNA-guided activation of endogenous human genes. Nat. Methods 2013, 10, 977–979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Pinera, P.; Kocak, D.D.; Vockley, C.M.; Adler, A.F.; Kabadi, A.M.; Polstein, L.R.; Thakore, P.I.; Glass, K.A.; Ousterout, D.G.; Leong, K.W.; et al. RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nat. Methods 2013, 10, 973–976. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Xu, X.; Nguyen, C.M.; Liu, Y.; Gao, Y.; Lin, X.; Daley, T.; Kipniss, N.H.; La Russa, M.; Qi, L.S. CRISPR-Mediated Programmable 3D Genome Positioning and Nuclear Organization. Cell 2018, 175, 1405–1417.e1414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nuñez, J.K.; Chen, J.; Pommier, G.C.; Cogan, J.Z.; Replogle, J.M.; Adriaens, C.; Ramadoss, G.N.; Shi, Q.; Hung, K.L.; Samelson, A.J.; et al. Genome-wide programmable transcriptional memory by CRISPR-based epigenome editing. Cell 2021, 184, 2503–2519.e2517. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Gilbert, L.A.; Cimini, B.A.; Schnitzbauer, J.; Zhang, W.; Li, G.W.; Park, J.; Blackburn, E.H.; Weissman, J.S.; Qi, L.S.; et al. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell 2013, 155, 1479–1491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelles, D.A.; Fang, M.Y.; O’Connell, M.R.; Xu, J.L.; Markmiller, S.J.; Doudna, J.A.; Yeo, G.W. Programmable RNA Tracking in Live Cells with CRISPR/Cas9. Cell 2016, 165, 488–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Nakamura, M.; Abbott, T.R.; Zhao, D.; Luo, K.; Yu, C.; Nguyen, C.M.; Lo, A.; Daley, T.P.; La Russa, M.; et al. CRISPR-mediated live imaging of genome editing and transcription. Science 2019, 365, 1301–1305. [Google Scholar] [CrossRef]
- Wang, T.; Wei, J.J.; Sabatini, D.M.; Lander, E.S. Genetic screens in human cells using the CRISPR-Cas9 system. Science 2014, 343, 80–84. [Google Scholar] [CrossRef] [Green Version]
- Shalem, O.; Sanjana, N.E.; Hartenian, E.; Shi, X.; Scott, D.A.; Mikkelson, T.; Heckl, D.; Ebert, B.L.; Root, D.E.; Doench, J.G.; et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 2014, 343, 84–87. [Google Scholar] [CrossRef] [Green Version]
- Konermann, S.; Brigham, M.D.; Trevino, A.E.; Joung, J.; Abudayyeh, O.O.; Barcena, C.; Hsu, P.D.; Habib, N.; Gootenberg, J.S.; Nishimasu, H.; et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 2015, 517, 583–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joung, J.; Konermann, S.; Gootenberg, J.S.; Abudayyeh, O.O.; Platt, R.J.; Brigham, M.D.; Sanjana, N.E.; Zhang, F. Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nat. Protoc. 2017, 12, 828–863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinnery, P.F.; Hudson, G. Mitochondrial genetics. Br. Med. Bull. 2013, 106, 135–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rath, S.; Sharma, R.; Gupta, R.; Ast, T.; Chan, C.; Durham, T.J.; Goodman, R.P.; Grabarek, Z.; Haas, M.E.; Hung, W.H.W.; et al. MitoCarta3.0, an updated mitochondrial proteome now with sub-organelle localization and pathway annotations. Nucleic Acids Res. 2021, 49, D1541–D1547. [Google Scholar] [CrossRef] [PubMed]
- Floyd, B.J.; Wilkerson, E.M.; Veling, M.T.; Minogue, C.E.; Xia, C.; Beebe, E.T.; Wrobel, R.L.; Cho, H.; Kremer, L.S.; Alston, C.L.; et al. Mitochondrial Protein Interaction Mapping Identifies Regulators of Respiratory Chain Function. Mol. Cell. 2016, 63, 621–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Primers 2016, 2, 16080. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.W.; Turnbull, D.M. Mitochondrial DNA mutations in human disease. Nat. Rev. Genet. 2005, 6, 389–402. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, S.; Moraes, C.T. Manipulating mitochondrial DNA heteroplasmy by a mitochondrially targeted restriction endonuclease. Hum. Mol. Genet. 2001, 10, 3093–3099. [Google Scholar] [CrossRef] [Green Version]
- Peeva, V.; Blei, D.; Trombly, G.; Corsi, S.; Szukszto, M.J.; Rebelo-Guiomar, P.; Gammage, P.A.; Kudin, A.P.; Becker, C.; Altmüller, J.; et al. Linear mitochondrial DNA is rapidly degraded by components of the replication machinery. Nat. Commun. 2018, 9, 1727. [Google Scholar] [CrossRef] [Green Version]
- Minczuk, M.; Papworth, M.A.; Kolasinska, P.; Murphy, M.P.; Klug, A. Sequence-specific modification of mitochondrial DNA using a chimeric zinc finger methylase. Proc. Natl. Acad. Sci. USA 2006, 103, 19689–19694. [Google Scholar] [CrossRef] [Green Version]
- Gammage, P.A.; Rorbach, J.; Vincent, A.I.; Rebar, E.J.; Minczuk, M. Mitochondrially targeted ZFNs for selective degradation of pathogenic mitochondrial genomes bearing large-scale deletions or point mutations. EMBO Mol. Med. 2014, 6, 458–466. [Google Scholar] [CrossRef]
- Bacman, S.R.; Williams, S.L.; Pinto, M.; Peralta, S.; Moraes, C.T. Specific elimination of mutant mitochondrial genomes in patient-derived cells by mitoTALENs. Nat. Med. 2013, 19, 1111–1113. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, M.; Bacman, S.R.; Peralta, S.; Falk, M.J.; Chomyn, A.; Chan, D.C.; Williams, S.L.; Moraes, C.T. MitoTALEN: A General Approach to Reduce Mutant mtDNA Loads and Restore Oxidative Phosphorylation Function in Mitochondrial Diseases. Mol. Ther. 2015, 23, 1592–1599. [Google Scholar] [CrossRef] [Green Version]
- Bacman, S.R.; Kauppila, J.H.K.; Pereira, C.V.; Nissanka, N.; Miranda, M.; Pinto, M.; Williams, S.L.; Larsson, N.G.; Stewart, J.B.; Moraes, C.T. MitoTALEN reduces mutant mtDNA load and restores tRNA(Ala) levels in a mouse model of heteroplasmic mtDNA mutation. Nat. Med. 2018, 24, 1696–1700. [Google Scholar] [CrossRef]
- Minczuk, M.; Papworth, M.A.; Miller, J.C.; Murphy, M.P.; Klug, A. Development of a single-chain, quasi-dimeric zinc-finger nuclease for the selective degradation of mutated human mitochondrial DNA. Nucleic Acids Res. 2008, 36, 3926–3938. [Google Scholar] [CrossRef] [Green Version]
- Zekonyte, U.; Bacman, S.R.; Smith, J.; Shoop, W.; Pereira, C.V.; Tomberlin, G.; Stewart, J.; Jantz, D.; Moraes, C.T. Mitochondrial targeted meganuclease as a platform to eliminate mutant mtDNA in vivo. Nat. Commun. 2021, 12, 3210. [Google Scholar] [CrossRef] [PubMed]
- Gammage, P.A.; Moraes, C.T.; Minczuk, M. Mitochondrial Genome Engineering: The Revolution May Not Be CRISPR-Ized. Trends Genet. 2018, 34, 101–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.B.; Komor, A.C.; Levy, J.M.; Packer, M.S.; Zhao, K.T.; Liu, D.R. Increasing the genome-targeting scope and precision of base editing with engineered Cas9-cytidine deaminase fusions. Nat. Biotechnol. 2017, 35, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Mok, B.Y.; de Moraes, M.H.; Zeng, J.; Bosch, D.E.; Kotrys, A.V.; Raguram, A.; Hsu, F.; Radey, M.C.; Peterson, S.B.; Mootha, V.K.; et al. A bacterial cytidine deaminase toxin enables CRISPR-free mitochondrial base editing. Nature 2020, 583, 631–637. [Google Scholar] [CrossRef]
- Van Haute, L.; Lee, S.Y.; McCann, B.J.; Powell, C.A.; Bansal, D.; Vasiliauskaitė, L.; Garone, C.; Shin, S.; Kim, J.S.; Frye, M.; et al. NSUN2 introduces 5-methylcytosines in mammalian mitochondrial tRNAs. Nucleic Acids Res. 2019, 47, 8720–8733. [Google Scholar] [CrossRef]
- Brzezicha, B.; Schmidt, M.; Makalowska, I.; Jarmolowski, A.; Pienkowska, J.; Szweykowska-Kulinska, Z. Identification of human tRNA:m5C methyltransferase catalysing intron-dependent m5C formation in the first position of the anticodon of the pre-tRNA Leu (CAA). Nucleic Acids Res. 2006, 34, 6034–6043. [Google Scholar] [CrossRef] [PubMed]
- Tuorto, F.; Liebers, R.; Musch, T.; Schaefer, M.; Hofmann, S.; Kellner, S.; Frye, M.; Helm, M.; Stoecklin, G.; Lyko, F. RNA cytosine methylation by Dnmt2 and NSun2 promotes tRNA stability and protein synthesis. Nat. Struct. Mol. Biol. 2012, 19, 900–905. [Google Scholar] [CrossRef] [PubMed]
- Shinoda, S.; Kitagawa, S.; Nakagawa, S.; Wei, F.Y.; Tomizawa, K.; Araki, K.; Araki, M.; Suzuki, T.; Suzuki, T. Mammalian NSUN2 introduces 5-methylcytidines into mitochondrial tRNAs. Nucleic Acids Res. 2019, 47, 8734–8745. [Google Scholar] [CrossRef] [Green Version]
- Jin, X.; Zhang, Z.; Nie, Z.; Wang, C.; Meng, F.; Yi, Q.; Chen, M.; Sun, J.; Zou, J.; Jiang, P.; et al. An animal model for mitochondrial tyrosyl-tRNA synthetase deficiency reveals links between oxidative phosphorylation and retinal function. J. Biol. Chem. 2021, 296, 100437. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Huang, Y.P.; Wu, H.; Song, K.; Wan, C.; Chi, A.N.; Xiao, Y.M.; Zhao, X.Y. Mitochondrial complex I deficiency leads to the retardation of early embryonic development in Ndufs4 knockout mice. PeerJ 2017, 5, e3339. [Google Scholar] [CrossRef] [PubMed]
- Zeharia, A.; Shaag, A.; Pappo, O.; Mager-Heckel, A.M.; Saada, A.; Beinat, M.; Karicheva, O.; Mandel, H.; Ofek, N.; Segel, R.; et al. Acute infantile liver failure due to mutations in the TRMU gene. Am. J. Hum. Genet. 2009, 85, 401–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, M.X.; Yan, Q.; Li, X.; Bykhovskaya, Y.; Gallo-Teran, J.; Hajek, P.; Umeda, N.; Zhao, H.; Garrido, G.; Mengesha, E.; et al. Mutation in TRMU related to transfer RNA modification modulates the phenotypic expression of the deafness-associated mitochondrial 12S ribosomal RNA mutations. Am. J. Hum. Genet. 2006, 79, 291–302. [Google Scholar] [CrossRef] [Green Version]
- Meng, F.; Cang, X.; Peng, Y.; Li, R.; Zhang, Z.; Li, F.; Fan, Q.; Guan, A.S.; Fischel-Ghosian, N.; Zhao, X.; et al. Biochemical Evidence for a Nuclear Modifier Allele (A10S) in TRMU (Methylaminomethyl-2-thiouridylate-methyltransferase) Related to Mitochondrial tRNA Modification in the Phenotypic Manifestation of Deafness-associated 12S rRNA Mutation. J. Biol. Chem. 2017, 292, 2881–2892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Zhang, L.; Chen, D.; He, X.; Yao, S.; Zhang, Z.; Chen, Y.; Guan, M.X. Deletion of Mtu1 (Trmu) in zebrafish revealed the essential role of tRNA modification in mitochondrial biogenesis and hearing function. Nucleic Acids Res. 2018, 46, 10930–10945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dell’agnello, C.; Leo, S.; Agostino, A.; Szabadkai, G.; Tiveron, C.; Zulian, A.; Prelle, A.; Roubertoux, P.; Rizzuto, R.; Zeviani, M. Increased longevity and refractoriness to Ca(2+)-dependent neurodegeneration in Surf1 knockout mice. Hum. Mol. Genet. 2007, 16, 431–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baden, K.N.; Murray, J.; Capaldi, R.A.; Guillemin, K. Early developmental pathology due to cytochrome c oxidase deficiency is revealed by a new zebrafish model. J. Biol. Chem. 2007, 282, 34839–34849. [Google Scholar] [CrossRef] [Green Version]
- Da-Rè, C.; von Stockum, S.; Biscontin, A.; Millino, C.; Cisotto, P.; Zordan, M.A.; Zeviani, M.; Bernardi, P.; De Pittà, C.; Costa, R. Leigh syndrome in Drosophila melanogaster: Morphological and biochemical characterization of Surf1 post-transcriptional silencing. J. Biol. Chem. 2014, 289, 29235–29246. [Google Scholar] [CrossRef] [Green Version]
- Quadalti, C.; Brunetti, D.; Lagutina, I.; Duchi, R.; Perota, A.; Lazzari, G.; Cerutti, R.; Di Meo, I.; Johnson, M.; Bottani, E.; et al. SURF1 knockout cloned pigs: Early onset of a severe lethal phenotype. Biochim. Biophys. Acta Mol. Basis. Dis. 2018, 1864, 2131–2142. [Google Scholar] [CrossRef] [PubMed]
- Perks, K.L.; Rossetti, G.; Kuznetsova, I.; Hughes, L.A.; Ermer, J.A.; Ferreira, N.; Busch, J.D.; Rudler, D.L.; Spahr, H.; Schöndorf, T.; et al. PTCD1 Is Required for 16S rRNA Maturation Complex Stability and Mitochondrial Ribosome Assembly. Cell Rep. 2018, 23, 127–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKnight, C.L.; Low, Y.C.; Elliott, D.A.; Thorburn, D.R.; Frazier, A.E. Modelling Mitochondrial Disease in Human Pluripotent Stem Cells: What Have We Learned? Int. J. Mol. Sci. 2021, 22, 7730. [Google Scholar] [CrossRef] [PubMed]
- Van Lent, J.; Verstraelen, P.; Asselbergh, B.; Adriaenssens, E.; Mateiu, L.; Verbist, C.; De Winter, V.; Eggermont, K.; Van Den Bosch, L.; De Vos, W.H.; et al. Induced pluripotent stem cell-derived motor neurons of CMT type 2 patients reveal progressive mitochondrial dysfunction. Brain 2021, 144, 2471–2485. [Google Scholar] [CrossRef]
- Romero-Moya, D.; Castaño, J.; Santos-Ocaña, C.; Navas, P.; Menendez, P. Generation, genome edition and characterization of iPSC lines from a patient with coenzyme Q(10) deficiency harboring a heterozygous mutation in COQ4 gene. Stem Cell Res. 2017, 24, 144–147. [Google Scholar] [CrossRef]
- Arroyo, J.D.; Jourdain, A.A.; Calvo, S.E.; Ballarano, C.A.; Doench, J.G.; Root, D.E.; Mootha, V.K. A Genome-wide CRISPR Death Screen Identifies Genes Essential for Oxidative Phosphorylation. Cell Metab. 2016, 24, 875–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendelsohn, B.A.; Bennett, N.K.; Darch, M.A.; Yu, K.; Nguyen, M.K.; Pucciarelli, D.; Nelson, M.; Horlbeck, M.A.; Gilbert, L.A.; Hyun, W.; et al. A high-throughput screen of real-time ATP levels in individual cells reveals mechanisms of energy failure. PLoS Biol. 2018, 16, e2004624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoshino, A.; Wang, W.J.; Wada, S.; McDermott-Roe, C.; Evans, C.S.; Gosis, B.; Morley, M.P.; Rathi, K.S.; Li, J.; Li, K.; et al. The ADP/ATP translocase drives mitophagy independent of nucleotide exchange. Nature 2019, 575, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The integrated stress response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Aviles, G.; Liu, Y.; Tian, R.; Unger, B.A.; Lin, Y.T.; Wiita, A.P.; Xu, K.; Correia, M.A.; Kampmann, M. Mitochondrial stress is relayed to the cytosol by an OMA1-DELE1-HRI pathway. Nature 2020, 579, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Jain, I.H.; Zazzeron, L.; Goli, R.; Alexa, K.; Schatzman-Bone, S.; Dhillon, H.; Goldberger, O.; Peng, J.; Shalem, O.; Sanjana, N.E.; et al. Hypoxia as a therapy for mitochondrial disease. Science 2016, 352, 54–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrow, J.J.; Balsa, E.; Verdeguer, F.; Tavares, C.D.; Soustek, M.S.; Hollingsworth, L.R.T.; Jedrychowski, M.; Vogel, R.; Paulo, J.A.; Smeitink, J.; et al. Bromodomain Inhibitors Correct Bioenergetic Deficiency Caused by Mitochondrial Disease Complex I Mutations. Mol. Cell. 2016, 64, 163–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gammage, P.A.; Van Haute, L.; Minczuk, M. Engineered mtZFNs for Manipulation of Human Mitochondrial DNA Heteroplasmy. Methods Mol. Biol. 2016, 1351, 145–162. [Google Scholar] [PubMed]
- Moretton, A.; Morel, F.; Macao, B.; Lachaume, P.; Ishak, L.; Lefebvre, M.; Garreau-Balandier, I.; Vernet, P.; Falkenberg, M.; Farge, G. Selective mitochondrial DNA degradation following double-strand breaks. PLoS ONE 2017, 12, e0176795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahal, S.; Dubey, S.; Raghavan, S.C. Homologous recombination-mediated repair of DNA double-strand breaks operates in mammalian mitochondria. Cell Mol. Life Sci. 2018, 75, 1641–1655. [Google Scholar] [CrossRef]
- Jo, A.; Ham, S.; Lee, G.H.; Lee, Y.I.; Kim, S.; Lee, Y.S.; Shin, J.H.; Lee, Y. Efficient Mitochondrial Genome Editing by CRISPR/Cas9. Biomed. Res. Int. 2015, 2015, 305716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bian, W.P.; Chen, Y.L.; Luo, J.J.; Wang, C.; Xie, S.L.; Pei, D.S. Knock-In Strategy for Editing Human and Zebrafish Mitochondrial DNA Using Mito-CRISPR/Cas9 System. ACS Synth. Biol. 2019, 8, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Gammage, P.A.; Viscomi, C.; Simard, M.-L.; Costa, A.S.H.; Gaude, E.; Powell, C.A.; Van Haute, L.; McCann, B.J.; Rebelo-Guiomar, P.; Cerutti, R.; et al. Genome editing in mitochondria corrects a pathogenic mtDNA mutation in vivo. Nat. Med. 2018, 24, 1691–1695. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, J.-X.; Pyle, A.; Taylor, R.W.; Oláhová, M. Interrogating Mitochondrial Biology and Disease Using CRISPR/Cas9 Gene Editing. Genes 2021, 12, 1604. https://doi.org/10.3390/genes12101604
Tang J-X, Pyle A, Taylor RW, Oláhová M. Interrogating Mitochondrial Biology and Disease Using CRISPR/Cas9 Gene Editing. Genes. 2021; 12(10):1604. https://doi.org/10.3390/genes12101604
Chicago/Turabian StyleTang, Jia-Xin, Angela Pyle, Robert W. Taylor, and Monika Oláhová. 2021. "Interrogating Mitochondrial Biology and Disease Using CRISPR/Cas9 Gene Editing" Genes 12, no. 10: 1604. https://doi.org/10.3390/genes12101604
APA StyleTang, J. -X., Pyle, A., Taylor, R. W., & Oláhová, M. (2021). Interrogating Mitochondrial Biology and Disease Using CRISPR/Cas9 Gene Editing. Genes, 12(10), 1604. https://doi.org/10.3390/genes12101604