
Lineage-Specific Genes and Family Expansions in Dictyostelid Genomes Display Expression Bias and Evolutionary Diversification during Development


Abstract
:1. Introduction
2. Materials and Methods
2.1. Gene Families and Lineage-Specific Genes
2.2. Sequence Analysis
2.3. Transcriptional Analysis
2.4. Gene Ontology Analysis and Visualizations
3. Results
3.1. Gene Family Distribution and Lineage-Specific Genes
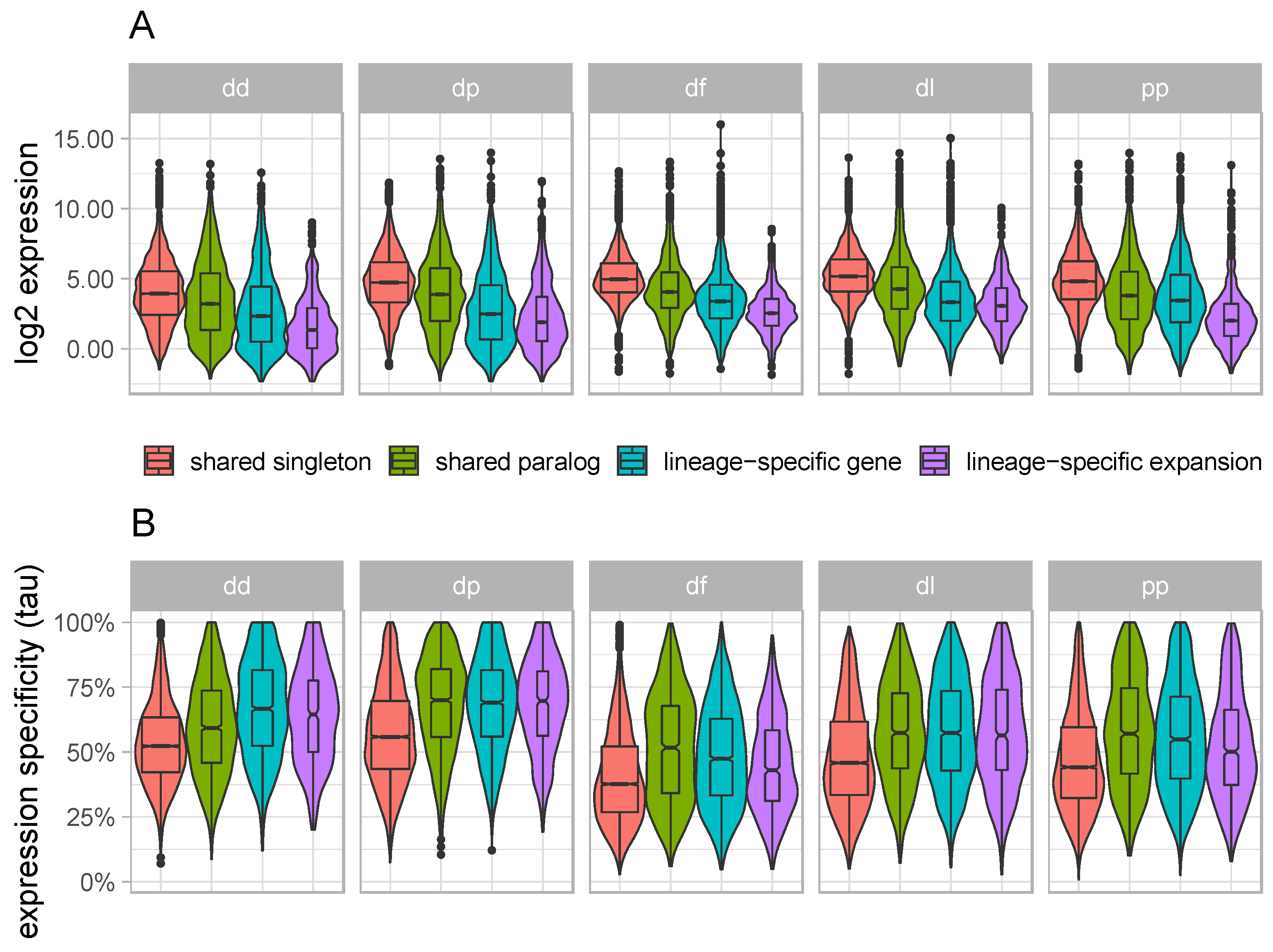
3.2. Lineage-Specific Genes and Expansions Tend to Have Low and Narrow Expression
3.3. Biased Genes Are Enriched during the Early and Late Stages of Development
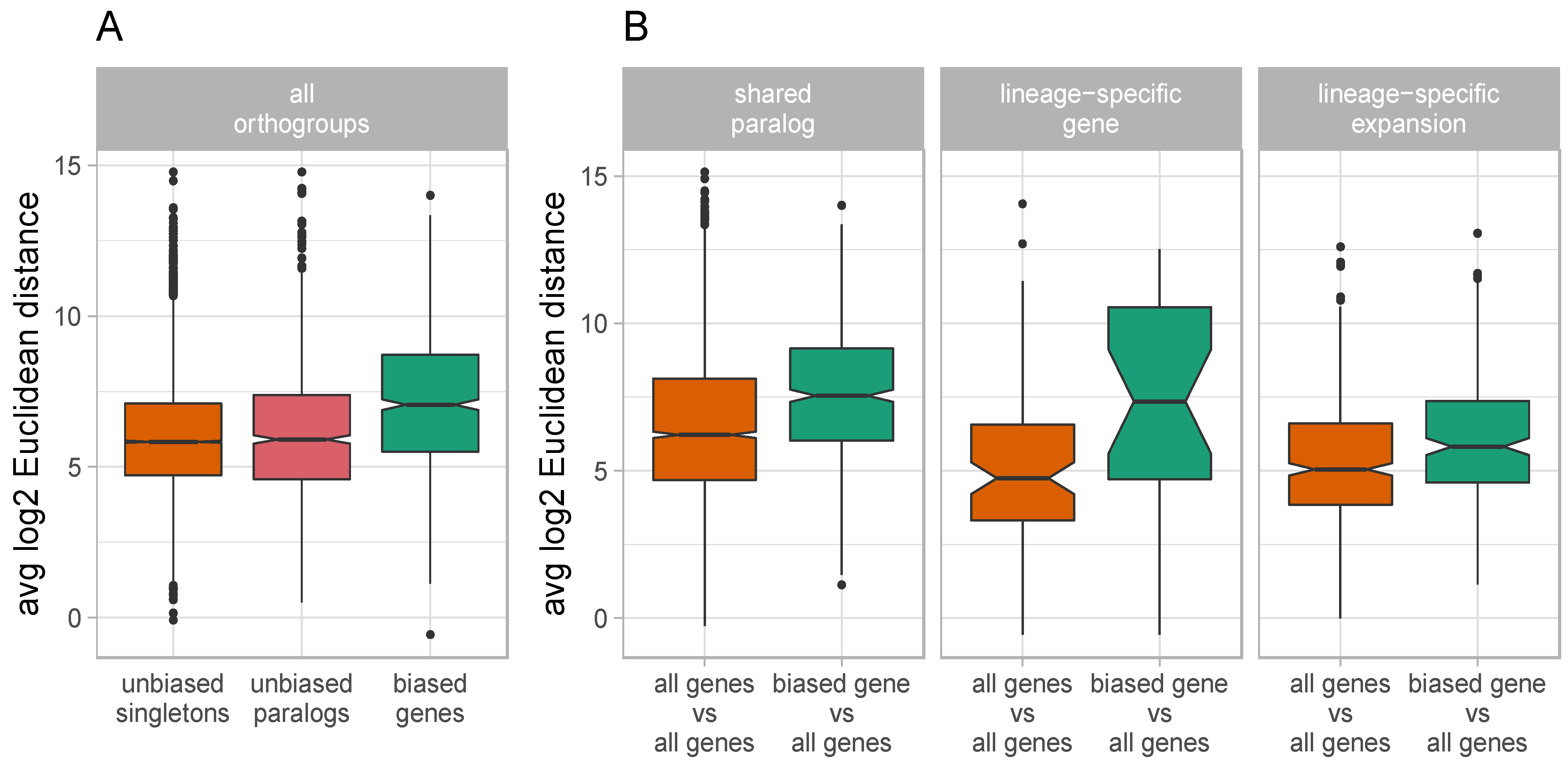
3.4. Biased Duplicate Genes Display High Levels of Expression Divergence
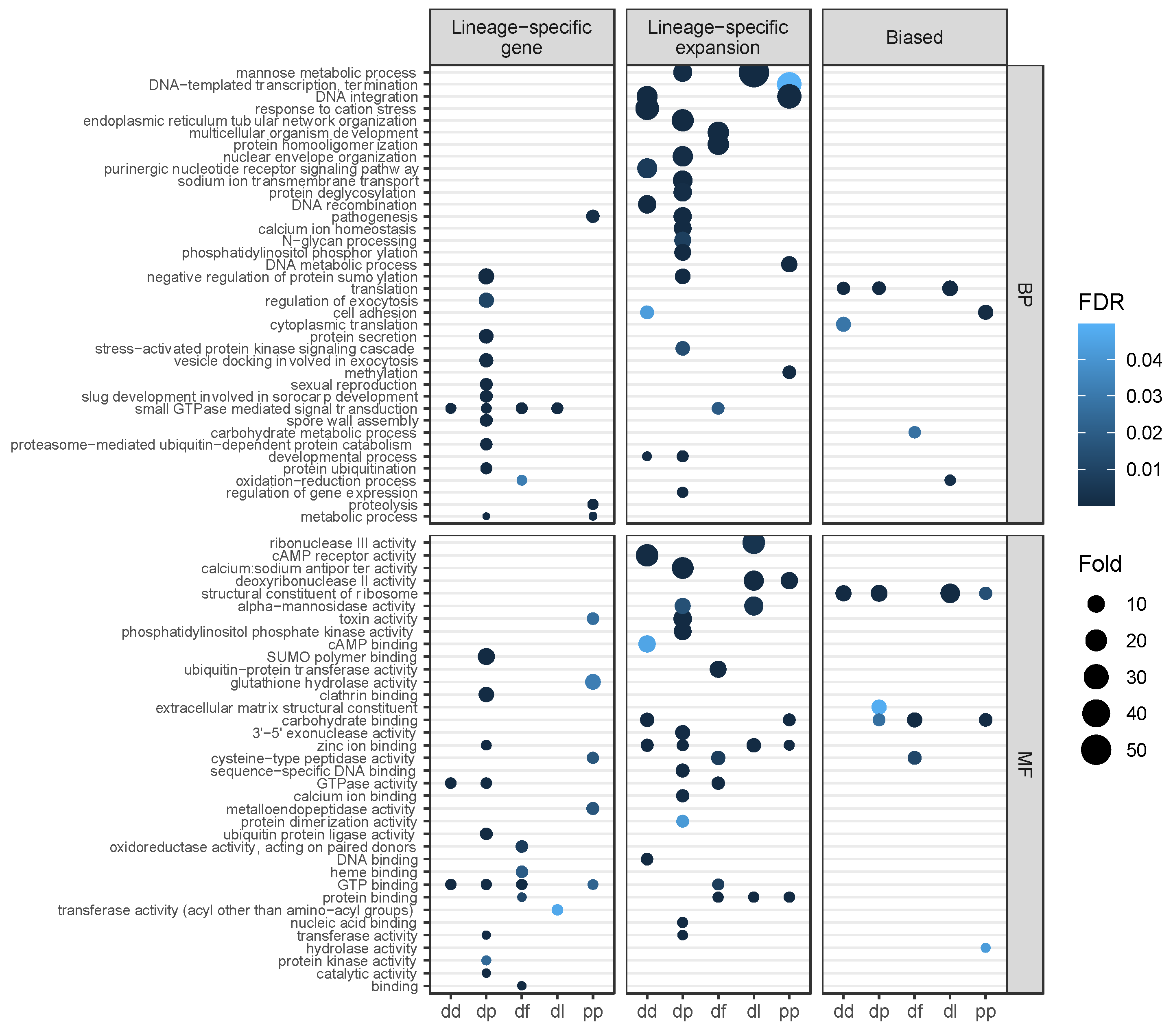
3.5. Gene Function and Positive Selection among Lineage-Specific Expansions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ohno, S. Evolution by Gene Duplication; Springer: Berlin/Heidelberg, Germany, 1970; ISBN 978-3-642-86661-6. [Google Scholar]
- Hurles, M. Gene Duplication: The Genomic Trade in Spare Parts. PLoS Biol. 2004, 2, e206. [Google Scholar] [CrossRef] [PubMed]
- Reams, D.; Roth, J.R. Mechanisms of Gene Duplication and Amplification. Cold Spring Harb. Perspect. Biol. 2015, 7, a016592. [Google Scholar] [CrossRef] [Green Version]
- Fortna, A.; Kim, Y.; MacLaren, E.J.; E Marshall, K.; Hahn, G.; Meltesen, L.; Brenton, M.; Hink, R.; Burgers, S.; Hernandez-Boussard, T.; et al. Lineage-Specific Gene Duplication and Loss in Human and Great Ape Evolution. PLoS Biol. 2004, 2, e207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynch, M. The Evolutionary Fate and Consequences of Duplicate Genes. Science 2000, 290, 1151–1155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Force, A.; Lynch, M.; Pickett, F.B.; Amores, A.; Yan, Y.-L.; Postlethwait, J. Preservation of Duplicate Genes by Complementary, Degenerative Mutations. Genetics 1999, 151, 1531–1545. [Google Scholar] [CrossRef] [PubMed]
- Li, W.-H.; Yang, J.; Gu, X. Expression divergence between duplicate genes. Trends Genet. 2005, 21, 602–607. [Google Scholar] [CrossRef]
- Gu, X.; Zhang, Z.; Huang, W. Rapid evolution of expression and regulatory divergences after yeast gene duplication. Proc. Natl. Acad. Sci. USA 2005, 102, 707–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duarte, J.M.; Cui, L.; Wall, P.K.; Zhang, Q.; Zhang, X.; Leebens-Mack, J.; Ma, H.; Altman, N.; Depamphilis, C.W. Expression Pattern Shifts Following Duplication Indicative of Subfunctionalization and Neofunctionalization in Regulatory Genes of Arabidopsis. Mol. Biol. Evol. 2005, 23, 469–478. [Google Scholar] [CrossRef]
- Hanada, K.; Kuromori, T.; Myouga, F.; Toyoda, T.; Shinozaki, K. Increased Expression and Protein Divergence in Duplicate Genes Is Associated with Morphological Diversification. PLoS Genet. 2009, 5, e1000781. [Google Scholar] [CrossRef] [Green Version]
- Chain, F.J.; Dushoff, J.; Evans, B.J. The odds of duplicate gene persistence after polyploidization. BMC Genom. 2011, 12, 599. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Ma, H.; Lin, J. Angiosperm-Wide and Family-Level Analyses of AP2/ERF Genes Reveal Differential Retention and Sequence Divergence after Whole-Genome Duplication. Front. Plant Sci. 2019, 10, 196. [Google Scholar] [CrossRef] [Green Version]
- Carvunis, A.; Rolland, T.; Wapinski, I.; Calderwood, M.A.; Yildirim, M.A.; Simonis, N.; Charloteaux, B.; Hidalgo, C.A.; Barbette, J.; Santhanam, B.; et al. Proto-gene and de novo gene brith. Nature 2012, 487, 370–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitz, J.F.; Chain, F.J.J.; Bornberg-Bauer, E. Evolution of novel genes in three-spined stickleback populations. Heredity 2020, 125, 50–59. [Google Scholar] [CrossRef]
- Assis, R.; Bachtrog, D. Neofunctionalization of young duplicate genes in Drosophila. Proc. Natl. Acad. Sci. USA 2013, 110, 17409–17414. [Google Scholar] [CrossRef] [Green Version]
- Assis, R.; Bachtrog, D. Rapid divergence and diversification of mammalian duplicate gene functions. BMC Evol. Biol. 2015, 15, 138. [Google Scholar] [CrossRef] [Green Version]
- Chain, F.J. Sex-Biased Expression of Young Genes in Silurana (Xenopus) tropicalis. Cytogenet. Genome Res. 2015, 145, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Guschanski, K.; Warnefors, M.; Kaessmann, H. The evolution of duplicate gene expression in mammalian organs. Genome Res. 2017, 27, 1461–1474. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Assis, R. Rapid functional divergence after small-scale gene duplication in grasses. BMC Evol. Biol. 2019, 19, 97. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Ren, Y.; Yang, T.; Li, G.; Chen, J.; Gschwend, A.; Yu, Y.; Hou, G.; Zi, J.; Zhou, R.; et al. Rapid evolution of protein diversity by de novo origination in Oryza. Nat. Ecol. Evol. 2019, 3, 679–690. [Google Scholar] [CrossRef]
- Dowling, D.; Schmitz, J.F.; Bornberg-Bauer, E. Stochastic Gain and Loss of Novel Transcribed Open Reading Frames in the Human Lineage. Genome Biol. Evol. 2020, 12, 2183–2195. [Google Scholar] [CrossRef]
- Blevins, W.R.; Ruiz-Orera, J.; Messeguer, X.; Blasco-Moreno, B.; Villanueva-Cañas, J.L.; Espinar, L.; Díez, J.; Carey, L.B.; Albà, M.M. Uncovering de novo gene birth in yeast using deep transcriptomics. Nat. Commun. 2021, 12, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Long, M.; Betran, E.; Thornton, K.; Wang, W. The origin of new genes: Glimpses from the young and old. Nat. Rev. Genet. 2003, 4, 865–875. [Google Scholar] [CrossRef] [PubMed]
- Lespinet, O.; Wolf, Y.; Koonin, E.V.; Aravind, L. The Role of Lineage-Specific Gene Family Expansion in the Evolution of Eukaryotes. Genome Res. 2002, 12, 1048–1059. [Google Scholar] [CrossRef] [Green Version]
- Jordan, I.K.; Makarova, K.S.; Spouge, J.L.; Wolf, Y.I.; Koonin, E.V. Lineage-Specific Gene Expansions in Bacterial and Archaeal Genomes. Genome Res. 2001, 11, 555–565. [Google Scholar] [CrossRef] [Green Version]
- Cortesi, F.; Musilová, Z.; Stieb, S.; Hart, N.; Siebeck, U.; Malmstrøm, M.; Tørresen, O.K.; Jentoft, S.; Cheney, K.; Marshall, J.; et al. Ancestral duplications and highly dynamic opsin gene evolution in percomorph fishes. Proc. Natl. Acad. Sci. USA 2015, 112, 1493–1498. [Google Scholar] [CrossRef] [Green Version]
- Niimura, Y.; Nei, M. Evolutionary dynamics of olfactory and other chemosensory receptor genes in vertebrates. J. Hum. Genet. 2006, 51, 505–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anantharaman, V.; Iyer, L.M.; Aravind, L. Comparative Genomics of Protists: New Insights into the Evolution of Eukaryotic Signal Transduction and Gene Regulation. Annu. Rev. Microbiol. 2007, 61, 453–475. [Google Scholar] [CrossRef]
- Lucas, E.R.; Miles, A.; Harding, N.; Clarkson, C.S.; Lawniczak, M.K.; Kwiatkowski, D.P.; Weetman, D.; Donnelly, M. Whole-genome sequencing reveals high complexity of copy number variation at insecticide resistance loci in malaria mosquitoes. Genome Res. 2019, 29, 1250–1261. [Google Scholar] [CrossRef] [Green Version]
- Sucgang, R.; Kuo, A.; Tian, X.; Salerno, W.; Parikh, A.; Feasley, C.L.; Dalin, E.; Tu, H.; Huang, E.; Barry, K.; et al. Comparative genomics of the social amoebae Dictyostelium discoideum and Dictyostelium purpureum. Genome Biol. 2011, 12, R20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kin, K.; Schaap, P. Evolution of Multicellular Complexity in the Dictyostelid Social Amoebas. Genes 2021, 12, 487. [Google Scholar] [CrossRef]
- Parikh, A.; Miranda, E.R.; Katoh-Kurasawa, M.; Fuller, D.; Rot, G.; Zagar, L.; Curk, T.; Sucgang, R.; Chen, R.; Zupan, B.; et al. Conserved developmental transcriptomes in evolutionarily divergent species. Genome Biol. 2010, 11, R35. [Google Scholar] [CrossRef] [Green Version]
- Schilde, C.; Lawal, H.M.; Noegel, A.A.; Eichinger, L.; Schaap, P.; Glöckner, G. A set of genes conserved in sequence and expression traces back the establishment of multicellularity in social amoebae. BMC Genom. 2016, 17, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Glöckner, G.; Lawal, H.M.; Felder, M.; Singh, R.; Singer, G.; Weijer, C.J.; Schaap, P. The multicellularity genes of dictyostelid social amoebas. Nat. Commun. 2016, 7, 12085. [Google Scholar] [CrossRef] [Green Version]
- Howe, K.L.; Contreras-Moreira, B.; De Silva, N.; Maslen, G.; Akanni, W.; Allen, J.; Alvarez-Jarreta, J.; Barba, M.; Bolser, D.M.; Cambell, L.; et al. Ensembl Genomes 2020—enabling non-vertebrate genomic research. Nucleic Acids Res. 2020, 48, D689–D695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felsenstein, J. PHYLIP—Phylogeny Inference Package (Version 3.2). Cladistics 1989, 5, 164–166. [Google Scholar]
- Suyama, M.; Torrents, D.; Bork, P. PAL2NAL: Robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006, 34, W609–W612. [Google Scholar] [CrossRef] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Cepas, J.; Serra, F.; Bork, P. ETE 3: Reconstruction, Analysis, and Visualization of Phylogenomic Data. Mol. Biol. Evol. 2016, 33, 1635–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Nielsen, R.; Yang, Z. Evaluation of an Improved Branch-Site Likelihood Method for Detecting Positive Selection at the Molecular Level. Mol. Biol. Evol. 2005, 22, 2472–2479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z. PAML 4: Phylogenetic Analysis by Maximum Likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [Green Version]
- Stajdohar, M.; Rosengarten, R.D.; Kokosar, J.; Jeran, L.; Blenkus, D.; Shaulsky, G.; Zupan, B. dictyExpress: A web-based platform for sequence data management and analytics in Dictyostelium and beyond. BMC Bioinform. 2017, 18, 291. [Google Scholar] [CrossRef] [Green Version]
- Yanai, I.; Benjamin, H.; Shmoish, M.; Chalifa-Caspi, V.; Shklar, M.; Ophir, R.; Bar-Even, A.; Horn-Saban, S.; Safran, M.; Domany, E.; et al. Genome-wide midrange transcription profiles reveal expression level relationships in human tissue specification. Bioinformatics 2004, 21, 650–659. [Google Scholar] [CrossRef] [Green Version]
- Alexa, A.; Rahnenfuehrer, J.; Lengauer, T. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics 2006, 22, 1600–1607. [Google Scholar] [CrossRef] [Green Version]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; ISBN 978-3-319-24277-4. Available online: https://ggplot2.tidyverse.org (accessed on 20 August 2021).
- R Core Team. R: A Language and Environment for Statistical Computing; Team RC: Vienna, Austria, 2017. [Google Scholar]
- Cai, H.; Gu, J.; Wang, Y. Core genome components and lineage specific expansions in malaria parasites Plasmodium. BMC Genom. 2010, 11, S13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.E.; Landback, P.; Vibranovski, M.; Long, M. New genes expressed in human brains: Implications for annotating evolving genomes. BioEssays 2012, 34, 982–991. [Google Scholar] [CrossRef] [PubMed]
- Romeralo, M.; Skiba, A.; Gonzalez-Voyer, A.; Schilde, C.; Lawal, H.; Kedziora, S.; Cavender, J.C.; Glöckner, G.; Urushihara, H.; Schaap, P. Analysis of phenotypic evolution in Dictyostelia highlights developmental plasticity as a likely consequence of colonial multicellularity. Proc. R. Soc. B: Boil. Sci. 2013, 280, 20130976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivero, F.; Xiong, H. Chapter Two—Rho Signaling in Dictyostelium discoideum. In International Review of Cell and Molecular Biology; Jeon, K.W., Ed.; Academic Press: Cambridge, MA, USA, 2016; Volume 322, pp. 61–181. [Google Scholar]
- Storz, J.F.; Opazo, J.C.; Hoffmann, F. Gene duplication, genome duplication, and the functional diversification of vertebrate globins. Mol. Phylogenetics Evol. 2013, 66, 469–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loomis, W.F. Cell signaling during development of Dictyostelium. Dev. Biol. 2014, 391, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kjellin, J.; Avesson, L.; Reimegård, J.; Liao, Z.; Eichinger, L.; Noegel, A.; Glöckner, G.; Schaap, P.; Söderbom, F. Abundantly expressed class of noncoding RNAs conserved through the multicellular evolution of dictyostelid social amoebas. Genome Res. 2021, 31, 436–447. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Ding, Q.; Guo, Z.; Zhao, Z.; Wei, L.; Li, Y.; Song, S.; Zheng, H.-L. Identification, characterization and expression analysis of lineage-specific genes within mangrove species Aegiceras corniculatum. Mol. Genet. Genom. 2021, 1–13. [Google Scholar] [CrossRef]
- Huber, R.J.; O’Day, D.H. Proteomic profiling of the extracellular matrix (slime sheath) of Dictyostelium discoideum. Proteomics 2015, 15, 3315–3319. [Google Scholar] [CrossRef] [PubMed]
- Forbes, G.; Chen, Z.-H.; Kin, K.; Lawal, H.M.; Schilde, C.; Yamada, Y.; Schaap, P. Phylogeny-wide conservation and change in developmental expression, cell-type specificity and functional domains of the transcriptional regulators of social amoebas. BMC Genom. 2019, 20, 890. [Google Scholar] [CrossRef] [PubMed]
- Rosengarten, R.D.; Santhanam, B.; Kokosar, J.; Shaulsky, G. The Long Noncoding RNA Transcriptome of Dictyostelium discoideum Development. G3 Genes Genomes Genet. 2017, 7, 387–398. [Google Scholar] [CrossRef] [Green Version]
- Gaiti, F.; Calcino, A.D.; Tanurdžić, M.; Degnan, B.M. Origin and evolution of the metazoan non-coding regulatory genome. Dev. Biol. 2017, 427, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Kaessmann, H. Origins, evolution, and phenotypic impact of new genes. Genome Res. 2010, 20, 1313–1326. [Google Scholar] [CrossRef] [Green Version]
- Kalinka, A.T.; Varga, K.M.; Gerrard, D.; Preibisch, S.; Corcoran, D.; Jarrells, J.; Ohler, U.; Bergman, C.; Tomancak, P. Gene expression divergence recapitulates the developmental hourglass model. Nat. Cell Biol. 2010, 468, 811–814. [Google Scholar] [CrossRef]
- Liu, J.; Frochaux, M.; Gardeux, V.; Deplancke, B.; Robinson-Rechavi, M. Inter-embryo gene expression variability recapitulates the hourglass pattern of evo-devo. BMC Biol. 2020, 18, 1–12. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Genes | Shared Singletons 1 | Shared Paralogs 2 | Lineage-Specific Genes 3 | Lineage-Specific Expansion Genes 4 | Lineage-Specific Expansion Families 5 |
|---|---|---|---|---|---|---|
| dd: D. discoideum | 13,243 | 6918 | 2318 | 3304 | 703 | 52 |
| dp: D. purpureum | 12,398 | 6782 | 2078 | 2791 | 747 | 56 |
| df: D. (C.) fasciculata | 12,165 | 6247 | 1483 | 3358 | 1077 | 78 |
| dl: D. (T.) lacteum | 10,224 | 6508 | 1222 | 1852 | 642 | 53 |
| pp: P. (H.) pallidum | 12,367 | 6255 | 1659 | 3246 | 1207 | 79 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luna, S.K.; Chain, F.J.J. Lineage-Specific Genes and Family Expansions in Dictyostelid Genomes Display Expression Bias and Evolutionary Diversification during Development. Genes 2021, 12, 1628. https://doi.org/10.3390/genes12101628
Luna SK, Chain FJJ. Lineage-Specific Genes and Family Expansions in Dictyostelid Genomes Display Expression Bias and Evolutionary Diversification during Development. Genes. 2021; 12(10):1628. https://doi.org/10.3390/genes12101628
Chicago/Turabian StyleLuna, Saara K., and Frédéric J. J. Chain. 2021. "Lineage-Specific Genes and Family Expansions in Dictyostelid Genomes Display Expression Bias and Evolutionary Diversification during Development" Genes 12, no. 10: 1628. https://doi.org/10.3390/genes12101628
APA StyleLuna, S. K., & Chain, F. J. J. (2021). Lineage-Specific Genes and Family Expansions in Dictyostelid Genomes Display Expression Bias and Evolutionary Diversification during Development. Genes, 12(10), 1628. https://doi.org/10.3390/genes12101628