
MOSES: A New Approach to Integrate Interactome Topology and Functional Features for Disease Gene Prediction

 , , , and
, , , and
Abstract
:1. Introduction
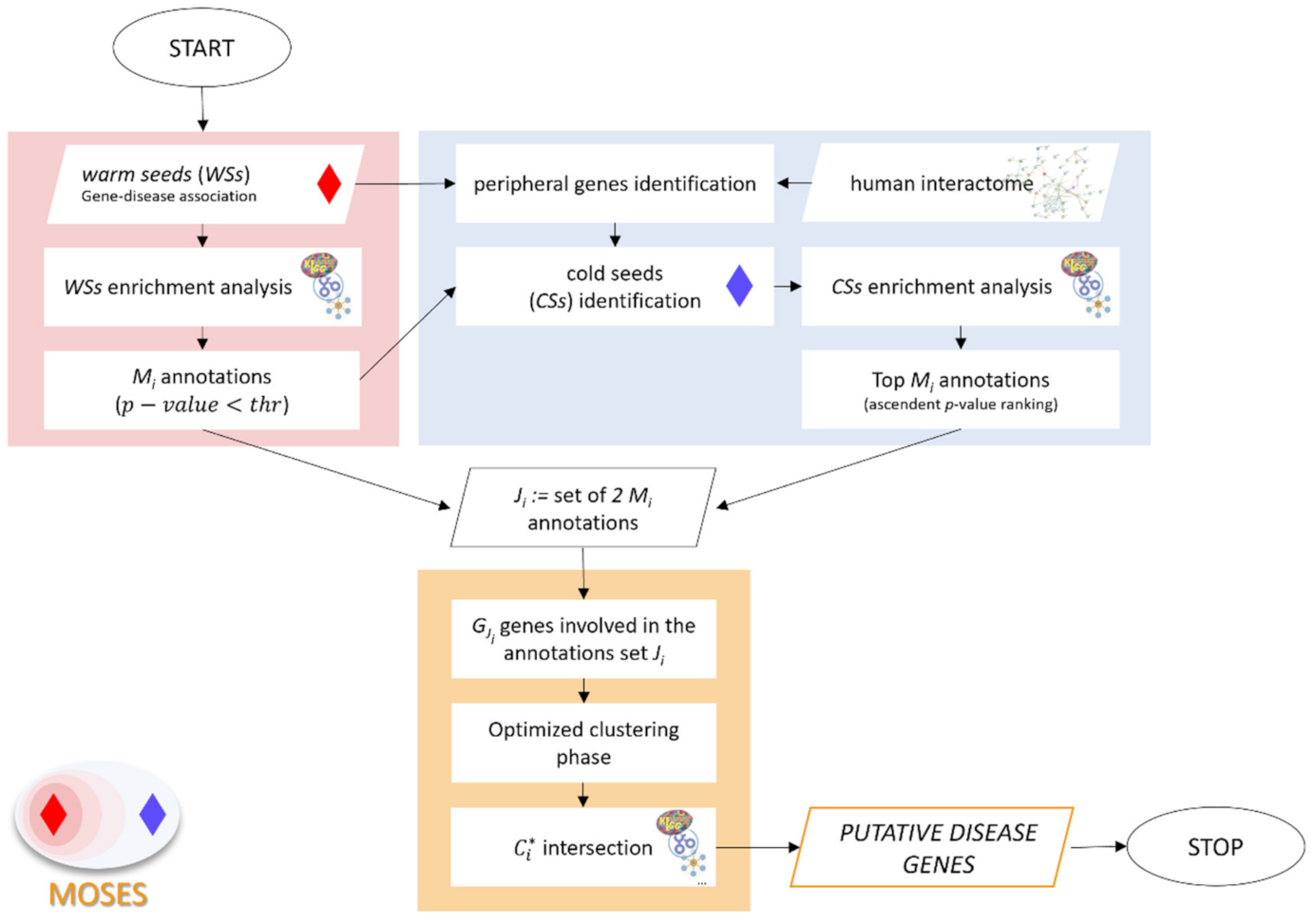
2. Methods
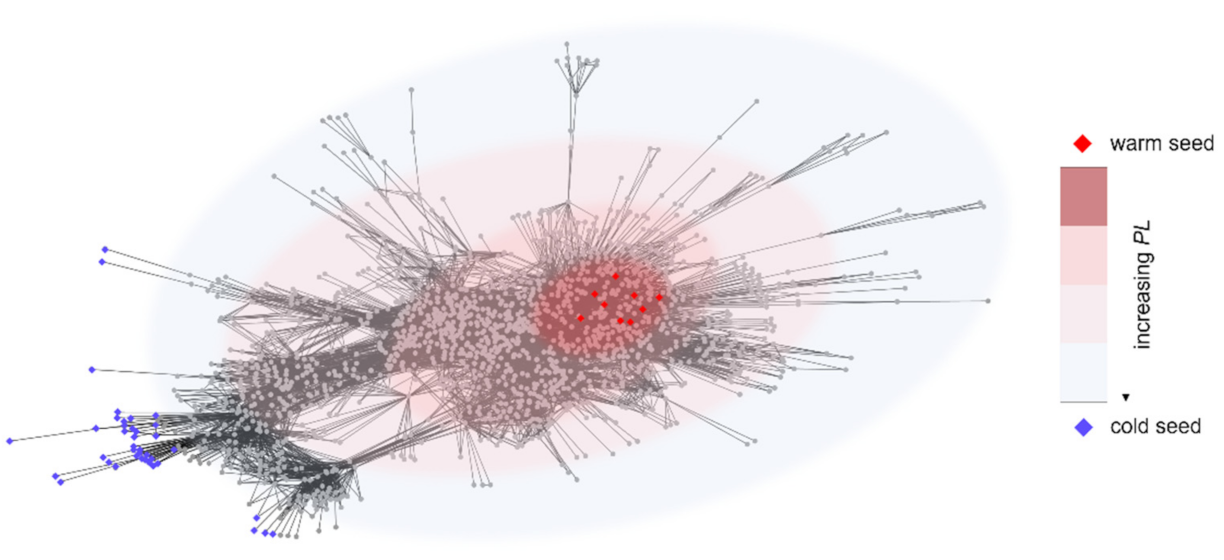
2.1. Functional Characterization of the Warm Seeds
2.2. Identification and Enrichment Analysis of the Cold Seeds
- identification of the non-seeds set NSi. At the first iteration, NS1 is the difference set between the interactome and the disease genes:
- identification of the first neighbors of genes in Si (set FNi)
- update of the sets Si and NSi:
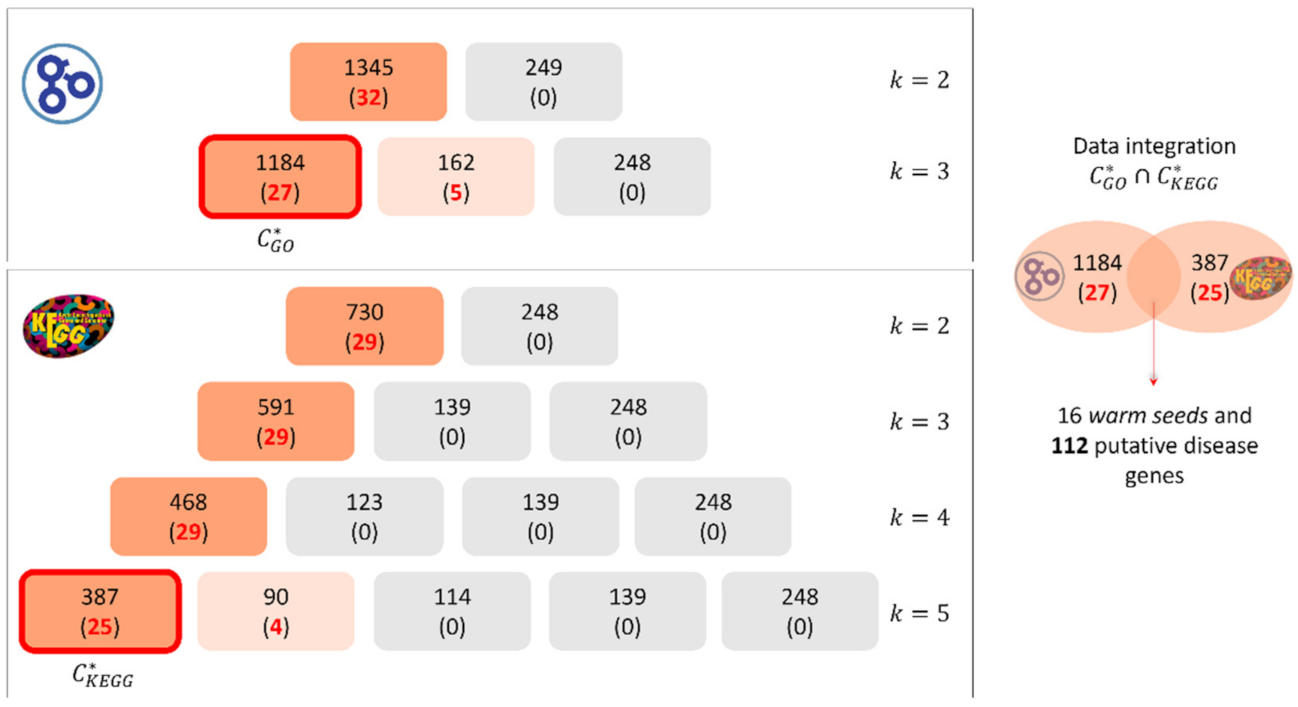
2.3. Optimized Clustering Phase and Selection of Putative Disease Genes
3. Data and Preprocessing
- annotations labeled with evidence code IPI (Inferred from Physical Interaction) were excluded to avoid circularity;
- annotations not associated with the gene products (evidence code “NOT”) were excluded.
4. Results and Discussion
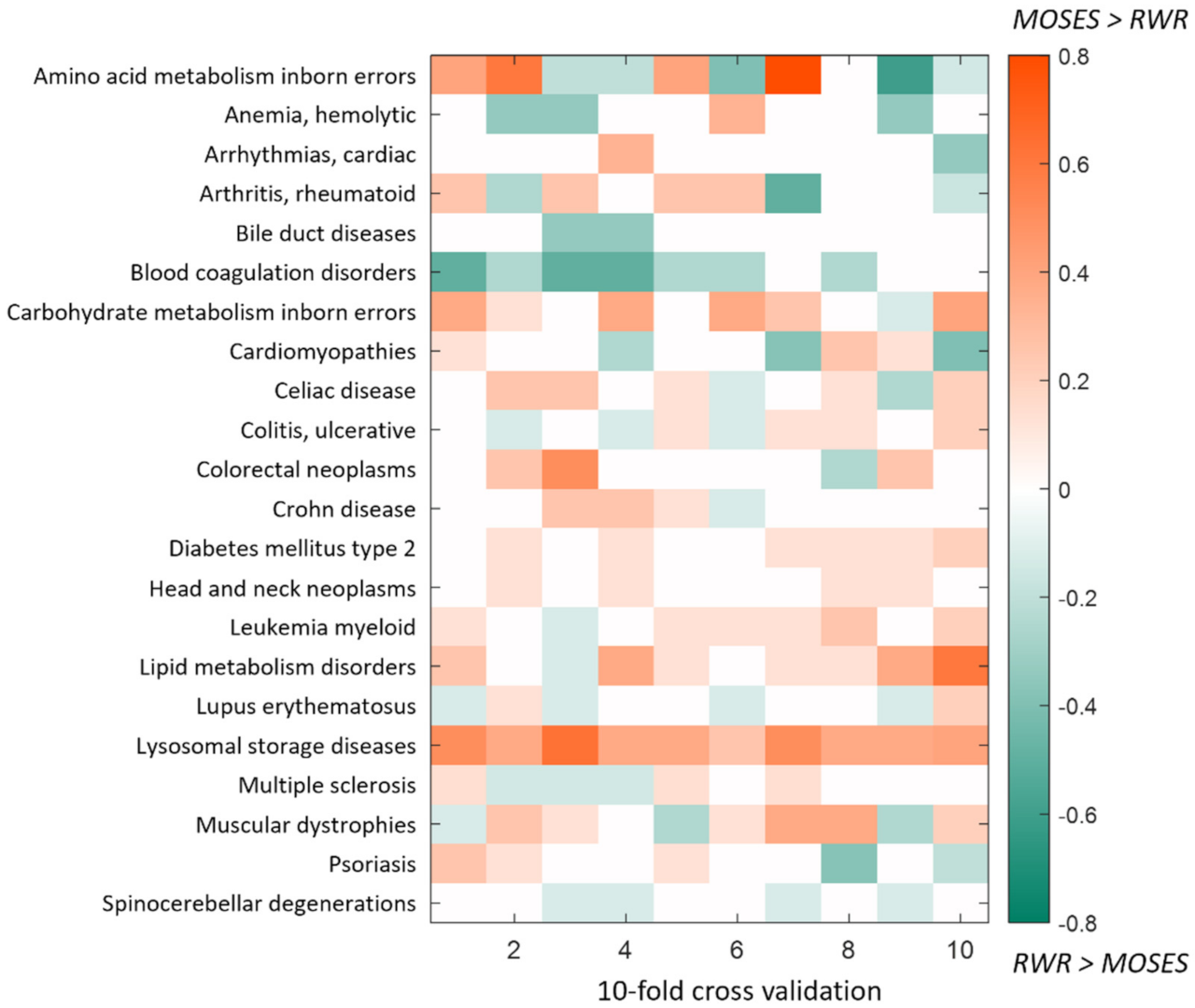
4.1. Computational Cross-Validation and Comparison with Random Walk with Restart
4.2. Enrichment Analysis of Putative Disease Genes
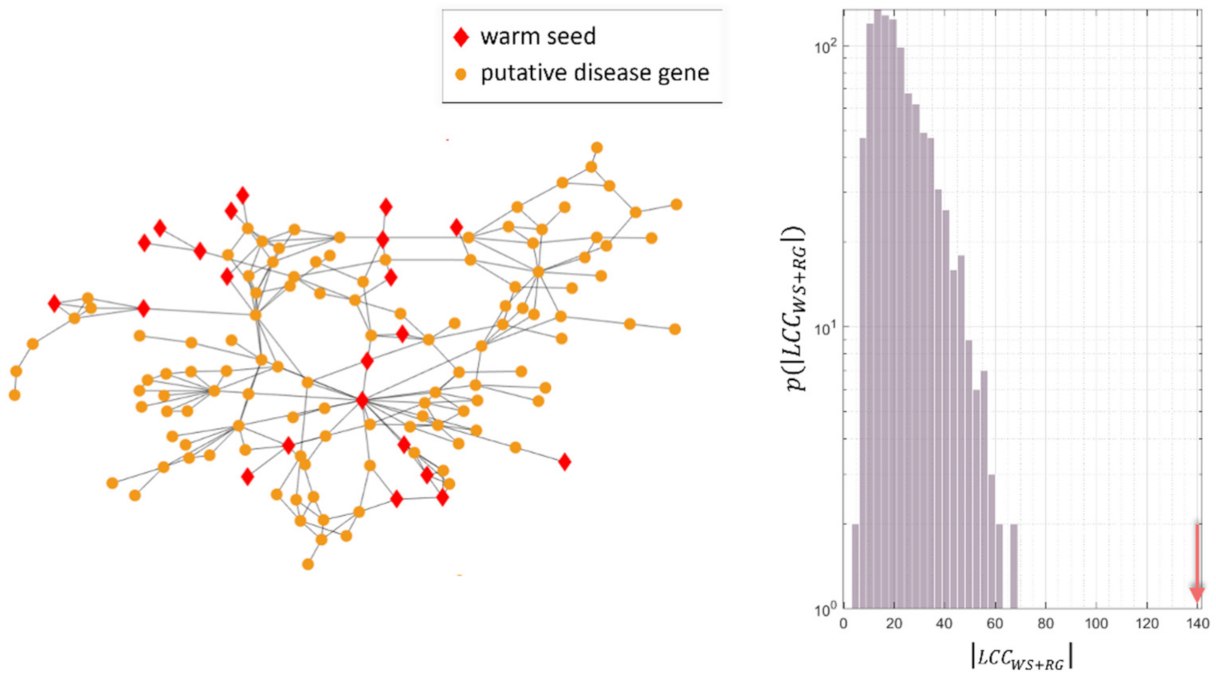
4.3. Study of the Predicted Disease Module
4.4. Case Studies on Colorectal Neoplasms and Rheumatoid Arthritis
4.4.1. Rheumatoid Arthritis
4.4.2. Colorectal Neoplasms
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- What Is Precision Medicine? MedlinePlus Genetics. Available online: https://medlineplus.gov/genetics/understanding/precisionmedicine/definition/ (accessed on 21 October 2021).
- Hamosh, A.; Scott, A.F.; Amberger, J.S.; Bocchini, C.A.; Valle, D.; McKusick, V.A. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 2002, 30, 52–55. [Google Scholar] [CrossRef]
- Ramos, E.M.; Hoffman, D.; Junkins, H.A.; Maglott, D.; Phan, L.; Sherry, S.T.; Feolo, M.; Hindorff, L.A. Phenotype-Genotype Integrator (PheGenI): Synthesizing genome-wide association study (GWAS) data with existing genomic resources. Eur. J. Hum. Genet. 2014, 22, 144–147. [Google Scholar] [CrossRef] [PubMed]
- Piñero, J.; Bravo, À.; Queralt-Rosinach, N.; Gutiérrez-Sacristán, A.; Deu-Pons, J.; Centeno, E.; García-García, J.; Sanz, F.; Furlong, L.I. DisGeNET: A comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Res. 2017, 45, D833–D839. [Google Scholar] [CrossRef]
- Babbi, G.; Martelli, P.L.; Profiti, G.; Bovo, S.; Savojardo, C.; Casadio, R. eDGAR: A database of Disease-Gene Associations with annotated Relationships among genes. BMC Genom. 2017, 18, 554. [Google Scholar] [CrossRef]
- Bromberg, Y. Chapter 15: Disease Gene Prioritization. PLoS Comput. Biol. 2013, 9, e1002902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreau, Y.; Tranchevent, L.-C. Computational tools for prioritizing candidate genes: Boosting disease gene discovery. Nat. Rev. Genet. 2012, 13, 523–536. [Google Scholar] [CrossRef] [PubMed]
- Piro, R.M.; di Cunto, F. Computational approaches to disease-gene prediction: Rationale, classification and successes. FEBS J. 2012, 279, 678–696. [Google Scholar] [CrossRef] [PubMed]
- Kaushal, P.; Singh, S. Network-based disease gene prioritization based on Protein–Protein Interaction Networks. Netw. Modeling Anal. Health Inform. Bioinform. 2020, 9, 55. [Google Scholar] [CrossRef]
- van Driel, M.A.; Bruggeman, J.; Vriend, G.; Brunner, H.G.; Leunissen, J.A.M. A text-mining analysis of the human phenome. Eur. J. Hum. Genet. 2006, 14, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Freudenberg, J.; Propping, P. A similarity-based method for genome-wide prediction of disease-relevant human genes. Bioinformatics 2002, 18, S110–S115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silverman, E.K.; Schmidt, H.H.H.W.; Anastasiadou, E.; Altucci, L.; Angelini, M.; Badimon, L.; Balligand, J.; Benincasa, G.; Capasso, G.; Conte, F.; et al. Molecular networks in Network Medicine: Development and applications. Wiley Interdiscip. Rev. Syst. Biol. Med. 2020, 12, e1489. [Google Scholar] [CrossRef] [PubMed]
- Barabási, A.-L.; Gulbahce, N.; Loscalzo, J. Network Medicine: A Network-based Approach to Human Disease. Nat. Rev. Genet. 2011, 12, 56–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tieri, P.; Farina, L.; Petti, M.; Astolfi, L.; Paci, P.; Castiglione, F. Network Inference and Reconstruction in Bioinformatics. In Encyclopedia of Bioinformatics and Computational Biology; Ranganathan, S., Gribskov, M., Nakai, K., Schönbach, C., Eds.; Academic Press: Oxford, UK, 2019; pp. 805–813. Available online: http://www.sciencedirect.com/science/article/pii/B9780128096338202902 (accessed on 26 April 2019).
- Bassett, D.S.; Sporns, O. Network neuroscience. Nat. Neurosci. 2017, 20, 353–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toppi, J.; Petti, M.; Fallani, F.D.V.; Vecchiato, G.; Maglione, A.G.; Cincotti, F.; Salinari, S.; Mattia, D.; Babiloni, F.; Astolfi, L. Describing relevant indices from the resting state electrophysiological networks. In Proceedings of the 2012 Annual International Conference of the IEEE Engineering in Medicine and Biology Society, San Diego, CA, USA, 28 August–1 September 2012; pp. 2547–2550. Available online: http://ieeexplore.ieee.org/lpdocs/epic03/wrapper.htm?arnumber=6346483 (accessed on 7 June 2015).
- Barabási, A.-L.; Oltvai, Z.N. Network biology: Understanding the cell’s functional organization. Nat. Rev. Genet. 2004, 5, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Köhler, S.; Bauer, S.; Horn, D.; Robinson, P.N. Walking the Interactome for Prioritization of Candidate Disease Genes. Am. J. Hum. Genet. 2008, 82, 949–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navlakha, S.; Kingsford, C. The power of protein interaction networks for associating genes with diseases. Bioinformatics 2010, 26, 1057–1063. [Google Scholar] [CrossRef] [PubMed]
- Petti, M.; Bizzarri, D.; Verrienti, A.; Falcone, R.; Farina, L. Connectivity Significance for Disease Gene Prioritization in an Expanding Universe. IEEE/ACM Trans. Comput. Biol. Bioinform. 2020, 17, 2155–2161. [Google Scholar] [CrossRef] [PubMed]
- Ghiassian, S.D.; Menche, J.; Barabási, A.-L. A DIseAse MOdule Detection (DIAMOnD) Algorithm Derived from a Systematic Analysis of Connectivity Patterns of Disease Proteins in the Human Interactome. PLoS Comput. Biol. 2015, 11, e1004120. [Google Scholar] [CrossRef] [PubMed]
- Selim, S.Z.; Ismail, M.A. K-Means-Type Algorithms: A Generalized Convergence Theorem and Characterization of Local Optimality. IEEE Trans. Pattern Anal. Mach. Intell. 1984, 1, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Vaquero, M.L.; Gama-Carvalho, M.; Rivas, J.D.L.; Pinto, F.R. Searching the overlap between network modules with specific betweeness (S2B) and its application to cross-disease analysis. Sci. Rep. 2018, 8, 11555. [Google Scholar] [CrossRef]
- Cáceres, J.J.; Paccanaro, A. Disease gene prediction for molecularly uncharacterized diseases. PLoS Comput. Biol. 2019, 15, e1007078. [Google Scholar] [CrossRef] [PubMed]
- Maiorino, E.; Baek, S.H.; Guo, F.; Zhou, X.; Kothari, P.H.; Silverman, E.K.; Barabási, A.L.; Weiss, S.T.; Raby, B.A.; Sharma, A. Discovering the genes mediating the interactions between chronic respiratory diseases in the human interactome. Nat. Commun. 2020, 11, 811. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, F.; Kovács, I.A.; Barabási, A.-L. Network-based prediction of drug combinations. Nat. Commun. 2019, 10, 1197. [Google Scholar] [CrossRef]
- Goh, K.-I.; Cusick, M.E.; Valle, D.; Childs, B.; Vidal, M.; Barabási, A.-L. The human disease network. Proc. Natl. Acad. Sci. USA 2007, 104, 8685–8690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caldera, M.; Buphamalai, P.; Müller, F.; Menche, J. Interactome-based approaches to human disease. Curr. Opin. Syst. Biol. 2017, 3, 88–94. [Google Scholar] [CrossRef]
- Paci, P.; Fiscon, G.; Conte, F.; Wang, R.-S.; Farina, L.; Loscalzo, J. Gene co-expression in the interactome: Moving from correlation toward causation via an integrated approach to disease module discovery. NPJ Syst. Biol. Appl. 2021, 7, 1–11. [Google Scholar] [CrossRef]
- Wang, R.-S.; Loscalzo, J. Network-Based Disease Module Discovery by a Novel Seed Connector Algorithm with Pathobiological Implications. J. Mol. Biol. 2018, 430, 2939–2950. [Google Scholar] [CrossRef] [PubMed]
- Keewan, E.; Naser, S.A. The Role of Notch Signaling in Macrophages during Inflammation and Infection: Implication in Rheumatoid Arthritis? Cells 2020, 9, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.; Kim, S.; Kim, K.; Jin, C.; Choi, K.Y.; Jang, J.; Choi, Y.; Gwon, A.; Baik, S.; Yun, U.J.; et al. Inhibition of notch signalling ameliorates experimental inflammatory arthritis. Ann. Rheum. Dis. 2015, 74, 267–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- da Rosa Franchi Santos, L.F.; Costa, N.T.; Maes, M.; Simão, A.N.C.; Dichi, I. Influence of treatments on cell adhesion molecules in patients with systemic lupus erythematosus and rheumatoid arthritis: A review. Inflammopharmacology 2020, 28, 363–384. [Google Scholar] [CrossRef]
- Walker, J.G.; Smith, M.D. The Jak-STAT pathway in rheumatoid arthritis. J. Rheumatol. 2005, 32, 1650–1653. [Google Scholar] [PubMed]
- Vieira Borba, V.; Shoenfeld, N.; Perricone, C.; Shoenfeld, Y. Chapter 27-Smell and Autoimmunity—State of the Art. In Mosaic of Autoimmunity; Perricone, C., Shoenfeld, Y., Eds.; Academic Press: Oxford, UK, 2019; pp. 269–277. Available online: https://www.sciencedirect.com/science/article/pii/B978012814307000027X (accessed on 21 May 2021).
- Perricone, C.; Shoenfeld, N.; Agmon-Levin, N.; de Carolis, C.; Perricone, R.; Shoenfeld, Y. Smell and Autoimmunity: A Comprehensive Review. Clin. Rev. Allergy Immunol. 2013, 45, 87–96. [Google Scholar] [CrossRef]
- Moscavitch, S.-D.; Szyper-Kravitz, M.; Shoenfeld, Y. Autoimmune pathology accounts for common manifestations in a wide range of neuro-psychiatric disorders: The olfactory and immune system interrelationship. Clin. Immunol. 2009, 130, 235–243. [Google Scholar] [CrossRef]
- Li, Y.; Leung, E.L.; Pan, H.; Yao, X.; Huang, Q.; Wu, M.; Xu, T.; Wang, Y.; Cai, J.; Li, R.; et al. Identification of potential genetic causal variants for rheumatoid arthritis by whole-exome sequencing. Oncotarget 2017, 8, 111119–111129. [Google Scholar] [CrossRef]
- Makarov, S.S. NF-κB in rheumatoid arthritis: A pivotal regulator of inflammation, hyperplasia, and tissue destruction. Arthritis Res. Ther. 2001, 3, 200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer. J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Lasry, A.; Zinger, A.; Ben-Neriah, A.L.A.Z.Y. Inflammatory networks underlying colorectal cancer. Nat. Immunol. 2016, 17, 230–240. [Google Scholar] [CrossRef] [PubMed]
- West, N.R.; McCuaig, S.; Franchini, F.; Powrie, F. Emerging cytokine networks in colorectal cancer. Nat. Rev. Immunol. 2015, 15, 615–629. [Google Scholar] [CrossRef] [PubMed]
- Akao, Y.; Nakagawa, Y.; Naoe, T. MicroRNA-143 and -145 in Colon Cancer. DNA Cell Biol. 2007, 26, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Wang, F.; Jiang, H.; Xu, J.; Jiang, Y.; Wang, Z. MicroRNA-145 suppresses cell migration and invasion by targeting paxillin in human colorectal cancer cells. Int. J. Clin. Exp. Pathol. 2015, 8, 1328. [Google Scholar] [PubMed]
- Wang, Z.; Zhang, X.; Yang, Z.; Du, H.; Wu, Z.; Gong, J.; Yan, J.; Zheng, Q. MiR-145 regulates PAK4 via the MAPK pathway and exhibits an antitumor effect in human colon cells. Biochem. Biophys. Res. Commun. 2012, 427, 444–449. [Google Scholar] [CrossRef] [PubMed]
- Slaby, O.; Svoboda, M.; Fabian, P.; Smerdova, T.; Knoflickova, D.; Bednarikova, M.; Nenutil, R.; Vyzula, R. Altered Expression of miR-21, miR-31, miR-143 and miR-145 Is Related to Clinicopathologic Features of Colorectal Cancer. Oncology 2007, 72, 397–402. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | Warm Seeds | Peripheral Genes | Cold Seeds |
|---|---|---|---|
| Amino acid metabolism inborn errors | 52 | 119 | 113 |
| Anemia, hemolytic | 29 | 155 | 143 |
| Arrhythmias, cardiac | 30 | 171 | 163 |
| Arthritis, rheumatoid | 42 | 87 | 77 |
| Asthma | 37 | 91 | 85 |
| Bile duct diseases | 31 | 109 | 103 |
| Blood coagulation disorders | 40 | 142 | 129 |
| Blood platelet disorders | 26 | 193 | 170 |
| Carbohydrate metabolism inborn errors | 77 | 81 | 79 |
| Cardiomyopathies | 50 | 70 | 63 |
| Celiac disease | 36 | 137 | 120 |
| Colitis, ulcerative | 56 | 90 | 72 |
| Colorectal neoplasms | 42 | 79 | 68 |
| Crohn disease | 72 | 65 | 56 |
| Diabetes mellitus, type 2 | 73 | 77 | 75 |
| Head and neck neoplasms | 35 | 87 | 80 |
| Leukemia, myeloid | 43 | 97 | 93 |
| Lipid metabolism disorders | 50 | 93 | 83 |
| Lung diseases, obstructive | 40 | 88 | 82 |
| Lupus erythematosus | 75 | 51 | 48 |
| Lysosomal storage diseases | 45 | 152 | 150 |
| Multiple sclerosis | 69 | 71 | 62 |
| Muscular dystrophies | 36 | 113 | 107 |
| Psoriasis | 54 | 86 | 76 |
| Renal tubular transport inborn errors | 34 | 229 | 211 |
| Spinocerebellar ataxias | 28 | 147 | 132 |
| Spinocerebellar degenerations | 30 | 147 | 137 |
| Disease | #PG | DisGeNET Disease | #Validated | Adjusted p-Value |
|---|---|---|---|---|
| Amino acid metabolism, inborn errors | 122 | Amino Acid Metabolism, Inborn Errors | 2 | 1.68 × 10−02 |
| Anemia, hemolytic | 50 | Anemia, Hemolytic | 2 | 7.52 × 10−02 |
| Arrhythmias, cardiac | 59 | Cardiac Arrhythmia | 5 | 7.08 × 10−04 |
| Arthritis, rheumatoid | 447 | Rheumatoid Arthritis | 156 | 7.92 × 10−49 |
| Bile duct diseases | 55 | Bile Duct Diseases | 1 | 3.71 × 10−02 |
| Blood coagulation disorders | 104 | Blood Coagulation Disorders | 13 | 9.73 × 10−10 |
| Carbohydrate metabolism inborn errors | 256 | - | - | - |
| Cardiomyopathies | 32 | Cardiomyopathies | 22 | 1.04 × 10−04 |
| Celiac disease | 112 | Celiac Disease | 16 | 4.16 × 10−10 |
| Colitis, ulcerative | 165 | Ulcerative Colitis | 68 | 7.88 × 10−45 |
| Colorectal neoplasms | 1160 | Colorectal Carcinoma | 433 | 2.13 × 10−84 |
| Crohn disease | 162 | Crohn Disease | 58 | 3.50 × 10−34 |
| Diabetes mellitus, type 2 | 52 | Diabetes Mellitus, Non-Insulin-Dependent | 29 | 4.68 × 10−15 |
| Head and neck neoplasms | 412 | Malignant Head and Neck Neoplasm | 52 | 1.21 × 10−21 |
| Leukemia, myeloid | 184 | Myeloid Leukemia | 22 | 3.32 × 10−08 |
| Lipid metabolism disorders | 43 | - | - | - |
| Lupus erythematosus | 248 | Lupus Erythematosus, Systemic | 103 | 1.19 × 10−59 |
| Lysosomal storage diseases | 112 | Lysosomal Storage Diseases | 5 | 3.18 × 10−03 |
| Multiple sclerosis | 396 | Multiple Sclerosis | 101 | 1.31 × 10−37 |
| Muscular dystrophies | 122 | Muscular Dystrophy, Duchenne | 6 | 6.63 × 10−03 |
| Psoriasis | 421 | Psoriasis | 77 | 3.51 × 10−27 |
| Spinocerebellar degenerations | 38 | Ataxia, Spinocerebellar | 2 | 3.52 × 10−02 |
| Disease | #WSs | #PGs | ||||
|---|---|---|---|---|---|---|
| Amino acid metabolism inborn errors | 52 | 122 | 11 | 42 | 14 | 27 |
| Anemia, hemolytic | 29 | 50 | 11 | 55 | 12 | 16 |
| Arrhythmias, cardiac | 30 | 59 | 2 | 36 | 6 | 16 |
| Arthritis, rheumatoid | 42 | 447 | 6 | 306 | 31 | 201 |
| Bile duct diseases | 31 | 55 | 3 | 35 | 7 | 12 |
| Blood coagulation disorders | 40 | 104 | 22 | 98 | 34 | 37 |
| Carbohydrate metabolism inborn errors | 77 | 256 | 9 | 168 | 39 | 96 |
| Cardiomyopathies | 50 | 32 | 27 | 42 | 32 | 33 |
| Celiac disease | 36 | 112 | 2 | 57 | 7 | 15 |
| Colitis, ulcerative | 56 | 165 | 5 | 140 | 22 | 44 |
| Colorectal neoplasms | 42 | 1160 | 18 | 992 | 35 | 771 |
| crohn disease | 72 | 162 | 10 | 150 | 27 | 57 |
| Diabetes mellitus type 2 | 73 | 52 | 7 | 19 | 9 | 16 |
| Head and neck neoplasms | 35 | 412 | 6 | 320 | 25 | 172 |
| Leukemia myeloid | 43 | 184 | 16 | 136 | 32 | 69 |
| Lipid metabolism disorders | 50 | 43 | 11 | 37 | 19 | 17 |
| Lupus erythematosus | 75 | 248 | 5 | 180 | 39 | 92 |
| Lysosomal storage diseases | 45 | 112 | 8 | 13 | 5 | 20 |
| Multiple sclerosis | 69 | 396 | 11 | 287 | 40 | 185 |
| Muscular dystrophies | 36 | 122 | 12 | 84 | 24 | 31 |
| Psoriasis | 54 | 421 | 6 | 309 | 36 | 194 |
| Spinocerebellar degenerations | 30 | 38 | 2 | 37 | 9 | 12 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petti, M.; Farina, L.; Francone, F.; Lucidi, S.; Macali, A.; Palagi, L.; De Santis, M. MOSES: A New Approach to Integrate Interactome Topology and Functional Features for Disease Gene Prediction. Genes 2021, 12, 1713. https://doi.org/10.3390/genes12111713
Petti M, Farina L, Francone F, Lucidi S, Macali A, Palagi L, De Santis M. MOSES: A New Approach to Integrate Interactome Topology and Functional Features for Disease Gene Prediction. Genes. 2021; 12(11):1713. https://doi.org/10.3390/genes12111713
Chicago/Turabian StylePetti, Manuela, Lorenzo Farina, Federico Francone, Stefano Lucidi, Amalia Macali, Laura Palagi, and Marianna De Santis. 2021. "MOSES: A New Approach to Integrate Interactome Topology and Functional Features for Disease Gene Prediction" Genes 12, no. 11: 1713. https://doi.org/10.3390/genes12111713
APA StylePetti, M., Farina, L., Francone, F., Lucidi, S., Macali, A., Palagi, L., & De Santis, M. (2021). MOSES: A New Approach to Integrate Interactome Topology and Functional Features for Disease Gene Prediction. Genes, 12(11), 1713. https://doi.org/10.3390/genes12111713