
Nephronophthisis-Pathobiology and Molecular Pathogenesis of a Rare Kidney Genetic Disease


Abstract
:1. Introduction
2. Epidemiology
3. Pathophysiology and Clinical Findings
3.1. Kidneys
3.2. Eyes
3.3. Central Nervous System
3.4. Liver
3.5. Musculoskeletal System
3.6. Reproductive System
3.7. Cardiovascular System
4. Genetics
5. NPHP Proteins
5.1. NPHP1
5.2. NPHP2
5.3. NPHP3
5.4. NPHP4
5.5. NPHP5
5.6. NPHP6
5.7. NPHP7
5.8. NPHP8
5.9. NPHP9
5.10. NPHP10-20 and Other Putative NPHP Genes
6. Biochemical Pathways
6.1. Mechanosensation in Renal Cilia
6.2. Vasopressin, cAMP and Ca2+ Signalling in Renal Cilia
6.3. PI3K/Akt/mTOR Signalling in Renal Cilia
6.4. Jak/Stat and Inflammatory Signalling in Renal Cilia
6.5. Wnt Signalling and Planar Cell Polarity Defects in Renal Cilia
6.6. Hedgehog Signalling in Renal Cilia
6.7. Hippo Signalling
6.8. DNA Damage Response (DDR)
6.9. Cilia Dependent Cyst Activation (CDCA)
7. Diagnosis, Screening and Prevention
- (1)
- Positive family history of either cystic kidneys, CKD, need for renal replacement therapy or renal transplant, or consanguineous families;
- (2)
- Childhood onset CKD, often associated with hyperechoic kidneys prenatally;
- (3)
- Clinically concentration defect, polyuria, polydipsia, and hyponatremia;
- (4)
- Ultrasound findings confirming bilateral cystic kidneys;
- (5)
- Associated syndromic features, especially if involving eyes (e.g., retinitis pigmentosa); brain (e.g., in Joubert syndrome), musculoskeletal system (as in BBS), developmental delays, autism, cardiac features (especially situs inversus), and short stature etc.
8. Management
9. Quality of Life
10. Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wilson, P.D. Polycystic kidney disease. N. Engl. J. Med. 2004, 350, 151–164. [Google Scholar] [CrossRef]
- Hildebrandt, F. Genetic kidney diseases. Lancet 2010, 375, 1287–1295. [Google Scholar] [CrossRef] [Green Version]
- Braun, D.A.; Hildebrandt, F. Ciliopathies. Cold Spring Harb. Perspect. Biol. 2017, 9. [Google Scholar] [CrossRef]
- Smith, C.H.; Graham, J.B. Congenital medullary cysts of the kidneys with severe refractory anemia. Am. J. Dis. Child 1945, 69, 369–377. [Google Scholar] [CrossRef]
- Fanconi, G.; Hanhart, E.; Albertini, A.; Uhlinger, E.; Dolivo, G.; Prader, A. Die familiäre juvenile Nephronophthise. Helv. Paediatr. Acta 1951, 6, 1–49. [Google Scholar] [PubMed]
- Hildebrandt, F. Chapter 25—Nephronophthisis. In Genetic Diseases of the Kidney; Lifton, R.P., Somlo, S., Giebisch, G.H., Seldin, D.W., Eds.; Academic Press: Cambridge, MA, USA, 2009; pp. 425–446. [Google Scholar]
- Waldherr, R.; Lennert, T.; Weber, H.P.; Fodisch, H.J.; Scharer, K. The nephronophthisis complex. A clinicopathologic study in children. Virchows Arch. A Pathol. Anat. Histol. 1982, 394, 235–254. [Google Scholar] [CrossRef]
- Simms, R.J.; Hynes, A.M.; Eley, L.; Sayer, J.A. Nephronophthisis: A genetically diverse ciliopathy. Int. J. Nephrol. 2011, 2011, 527137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallett, A.; Patel, C.; Salisbury, A.; Wang, Z.; Healy, H.; Hoy, W. The prevalence and epidemiology of genetic renal disease amongst adults with chronic kidney disease in Australia. Orphanet. J. Rare Dis. 2014, 9, 98. [Google Scholar] [CrossRef] [Green Version]
- Pistor, K.; Scharer, K.; Olbing, H.; Tamminen-Mobius, T. Children with chronic renal failure in the Federal Republic of Germany: II. Primary renal diseases, age and intervals from early renal failure to renal death. Arbeitsgemeinschaft fur Padiatrische Nephrologie. Clin. Nephrol. 1985, 23, 278–284. [Google Scholar]
- Ala-Mello, S.; Kivivuori, S.M.; Ronnholm, K.A.; Koskimies, O.; Siimes, M.A. Mechanism underlying early anaemia in children with familial juvenile nephronophthisis. Pediatr. Nephrol. 1996, 10, 578–581. [Google Scholar] [CrossRef]
- Potter, D.E.; Holliday, M.A.; Piel, C.F.; Feduska, N.J.; Belzer, F.O.; Salvatierra, O., Jr. Treatment of end-stage renal disease in children: A 15-year experience. Kidney Int. 1980, 18, 103–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hildebrandt, F.; Zhou, W. Nephronophthisis-associated ciliopathies. J. Am. Soc. Nephrol. 2007, 18, 1855–1871. [Google Scholar] [CrossRef] [Green Version]
- Bollee, G.; Fakhouri, F.; Karras, A.; Noel, L.H.; Salomon, R.; Servais, A.; Lesavre, P.; Moriniere, V.; Antignac, C.; Hummel, A. Nephronophthisis related to homozygous NPHP1 gene deletion as a cause of chronic renal failure in adults. Nephrol. Dial. Transplant. 2006, 21, 2660–2663. [Google Scholar] [CrossRef] [PubMed]
- Hoefele, J.; Nayir, A.; Chaki, M.; Imm, A.; Allen, S.J.; Otto, E.A.; Hildebrandt, F. Pseudodominant inheritance of nephronophthisis caused by a homozygous NPHP1 deletion. Pediatr. Nephrol. 2011, 26, 967–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudson, R.; Patel, C.; Hawley, C.M.; O’Shea, S.; Snelling, P.; Ho, G.; Holman, K.; Bennetts, B.; Crawford, J.; Francis, L.; et al. Adult-Diagnosed Nonsyndromic Nephronophthisis in Australian Families Caused by Biallelic NPHP4 Variants. Am. J. Kidney Dis. 2020, 76, 282–287. [Google Scholar] [CrossRef] [Green Version]
- Salomon, R.; Saunier, S.; Niaudet, P. Nephronophthisis. Pediatr. Nephrol. 2009, 24, 2333–2344. [Google Scholar] [CrossRef] [Green Version]
- Saunier, S.; Salomon, R.; Antignac, C. Nephronophthisis. Curr. Opin. Genet. Dev. 2005, 15, 324–331. [Google Scholar] [CrossRef]
- Cuppage, F.E.; Huseman, R.A.; Chapman, A.; Grantham, J.J. Ultrastructure and function of cysts from human adult polycystic kidneys. Kidney Int. 1980, 17, 372–381. [Google Scholar] [CrossRef] [Green Version]
- Evan, A.P.; Gardner, K.D.; Bernstein, J. Polypoid and papillary epithelial hyperplasia: A potential cause of ductal obstruction in adult polycystic disease. Kidney Int. 1979, 16, 743–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullivan, L.P.; Wallace, D.P.; Grantham, J.J. Epithelial transport in polycystic kidney disease. Physiol. Rev. 1998, 78, 1165–1191. [Google Scholar] [CrossRef]
- Verani, R.R.; Silva, F.G. Histogenesis of the renal cysts in adult (autosomal dominant) polycystic kidney disease: A histochemical study. Mod. Pathol. Off. J. United States Can. Acad. Pathol. 1988, 1, 457–463. [Google Scholar]
- Grantham, J.J.; Geiser, J.L.; Evan, A.P. Cyst formation and growth in autosomal dominant polycystic kidney disease. Kidney Int. 1987, 31, 1145–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terryn, S.; Ho, A.; Beauwens, R.; Devuyst, O. Fluid transport and cystogenesis in autosomal dominant polycystic kidney disease. Biochim. Biophys. Acta 2011, 1812, 1314–1321. [Google Scholar] [CrossRef]
- Hildebrandt, F.; Attanasio, M.; Otto, E. Nephronophthisis: Disease mechanisms of a ciliopathy. J. Am. Soc. Nephrol. 2009, 20, 23–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seeman, T.; Dusek, J.; Vondrák, K.; Bláhová, K.; Simková, E.; Kreisinger, J.; Dvorák, P.; Kyncl, M.; Hríbal, Z.; Janda, J. Renal concentrating capacity is linked to blood pressure in children with autosomal dominant polycystic kidney disease. Physiol. Res. 2004, 53, 629–634. [Google Scholar] [PubMed]
- Selistre, L.; de Souza, V.; Ranchin, B.; Hadj-Aissa, A.; Cochat, P.; Dubourg, L. Early renal abnormalities in children with postnatally diagnosed autosomal dominant polycystic kidney disease. Paediatr. Nephrol. 2012, 27, 1589–1593. [Google Scholar] [CrossRef] [PubMed]
- Helal, I.; Reed, B.; McFann, K.; Yan, X.-D.; Fick-Brosnahan, G.M.; Cadnapaphornchai, M.; Schrier, R.W. Glomerular hyperfiltration and renal progression in children with autosomal dominant polycystic kidney disease. Clin. J. Am. Soc. Nephrol. CJASN 2011, 6, 2439–2443. [Google Scholar] [CrossRef] [Green Version]
- Georges, B.; Cosyns, J.P.; Dahan, K.; Snyers, B.; Carlier, B.; Loute, G.; Pirson, Y. Late-onset renal failure in Senior-Loken syndrome. Am. J. Kidney Dis. 2000, 36, 1271–1275. [Google Scholar] [CrossRef]
- Brancati, F.; Camerota, L.; Colao, E.; Vega-Warner, V.; Zhao, X.; Zhang, R.; Bottillo, I.; Castori, M.; Caglioti, A.; Sangiuolo, F.; et al. Biallelic variants in the ciliary gene TMEM67 cause RHYNS syndrome. Eur. J. Hum. Genet. 2018, 26, 1266–1271. [Google Scholar] [CrossRef]
- Sayer, J.A.; Otto, E.A.; O’Toole, J.F.; Nurnberg, G.; Kennedy, M.A.; Becker, C.; Hennies, H.C.; Helou, J.; Attanasio, M.; Fausett, B.V.; et al. The centrosomal protein nephrocystin-6 is mutated in Joubert syndrome and activates transcription factor ATF4. Nat. Genet. 2006, 38, 674–681. [Google Scholar] [CrossRef]
- McGraw, P. The molar tooth sign. Radiology 2003, 229, 671–672. [Google Scholar] [CrossRef]
- Betz, R.; Rensing, C.; Otto, E.; Mincheva, A.; Zehnder, D.; Lichter, P.; Hildebrandt, F. Chilren with ocular motor apraxia type Cogan carry deletions in the gene (NPHP1) for juvenile nephronopthisis. J. Pediatr. 2000, 136, 828–831. [Google Scholar] [CrossRef]
- Boichis, H.; Passwell, J.; David, R.; Miller, H. Congenital Hepatic Fibrosis and Nephronophthisis: A Family Study. QJM 1973, 42, 221–226. [Google Scholar] [PubMed]
- Doherty, D.; Parisi, M.A.; Finn, L.S.; Gunay-Aygun, M.; Al-Mateen, M.; Bates, D.; Clericuzio, C.; Demir, H.; Dorschner, M.; van Essen, A.J.; et al. Mutations in 3 genes (MKS3, CC2D2A and RPGRIP1L) cause COACH syndrome (Joubert syndrome with congenital hepatic fibrosis). J. Med. Genet. 2010, 47, 8–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrault, I.; Saunier, S.; Hanein, S.; Filhol, E.; Bizet, A.A.; Collins, F.; Salih, M.A.M.; Gerber, S.; Delphin, N.; Bigot, K.; et al. Mainzer-Saldino Syndrome is a ciliopathy caused by IFT140 mutations. Am. J. Hum. Genet. 2012, 90, 864–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, S. Clinical and Genetic Epidemiology of Bardet–Biedl Syndrome in Newfoundland: A 22-Year Prospective, Population-Based, Cohort Study. Am. J. Med. Genet. A 2005, 132A, 352–360. [Google Scholar] [CrossRef] [Green Version]
- de Vries, J.; Yntema, J.L.; van Die, C.E.; Crama, N.; Cornelissen, E.A.; Hamel, B.C. Jeune syndrome: Description of 13 cases and a proposal for follow-up protocol. Eur. J. Pediatr. 2010, 169, 177–188. [Google Scholar] [CrossRef] [Green Version]
- Walczak-Sztulpa, J.; Eggenschwiler, J.; Osborn, D.; Brown, D.A.; Emma, F.; Klingenberg, C.; Hennekam, R.C.; Torre, G.; Garshasbi, M.; Tzschach, A.; et al. Cranioectodermal Dysplasia, Sensenbrenner syndrome, is a ciliopathy caused by mutations in the IFT122 gene. Am. J. Hum. Genet. 2010, 86, 949–956. [Google Scholar] [CrossRef] [Green Version]
- Koscinski, I.; Mark, M.; Messaddeq, N.; Braun, J.J.; Celebi, C.; Muller, J.; Zinetti-Bertschy, A.; Goetz, N.; Dollfus, H.; Rossignol, S. Reproduction Function in Male Patients With Bardet Biedl Syndrome. J. Clin. Endocrinol. Metab. 2020, 105, e4417–e4429. [Google Scholar] [CrossRef]
- Guran, T.; Ekinci, G.; Atay, Z.; Turan, S.; Akcay, T.; Bereket, A. Radiologic and hormonal evaluation of pituitary abnormalities in patients with Bardet-Biedl syndrome. Clin. Dysmorphol. 2011, 20, 26–31. [Google Scholar] [CrossRef]
- Marlais, M.; Cuthell, O.; Langan, D.; Jan, D.; Sinha, M.D.; Winyard, P.J.D. Hypertension in autosomal dominant polycystic kidney disease: A meta-analysis. Arch. Dis. Child. 2016, 101, 1142–1147. [Google Scholar] [CrossRef]
- Seeman, T. Hypertension in Children with Cystic Kidney Diseases. Curr. Hypertens. Rev. 2006, 2, 167–177. [Google Scholar] [CrossRef]
- Otto, E.A.; Schermer, B.; Obara, T.; O’Toole, J.F.; Hiller, K.S.; Mueller, A.M.; Ruf, R.G.; Hoefele, J.; Beekmann, F.; Landau, D.; et al. Mutations in INVS encoding inversin cause nephronophthisis type 2, linking renal cystic disease to the function of primary cilia and left-right axis determination. Nat. Genet. 2003, 34, 413–420. [Google Scholar] [CrossRef]
- Saunier, S.; Calado, J.; Heilig, R.; Silbermann, F.; Benessy, F.; Morin, G.; Konrad, M.; Broyer, M.; Gubler, M.C.; Weissenbach, J.; et al. A novel gene that encodes a protein with a putative src homology 3 domain is a candidate gene for familial juvenile nephronophthisis. Hum. Mol. Genet. 1997, 6, 2317–2323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hildebrandt, F.; Otto, E.; Rensing, C.; Nothwang, H.G.; Vollmer, M.; Adolphs, J.; Hanusch, H.; Brandis, M. A novel gene encoding an SH3 domain protein is mutated in nephronophthisis type 1. Nat. Genet 1997, 17, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Otto, E.A.; Trapp, M.L.; Schultheiss, U.T.; Helou, J.; Quarmby, L.M.; Hildebrandt, F. NEK8 mutations affect ciliary and centrosomal localization and may cause nephronophthisis. J. Am. Soc. Nephrol. 2008, 19, 587–592. [Google Scholar] [CrossRef] [Green Version]
- Delous, M.; Baala, L.; Salomon, R.; Laclef, C.; Vierkotten, J.; Tory, K.; Golzio, C.; Lacoste, T.; Besse, L.; Ozilou, C.; et al. The ciliary gene RPGRIP1L is mutated in cerebello-oculo-renal syndrome (Joubert syndrome type B) and Meckel syndrome. Nat. Genet. 2007, 39, 875–881. [Google Scholar] [CrossRef] [PubMed]
- Attanasio, M.; Uhlenhaut, N.H.; Sousa, V.H.; O’Toole, J.F.; Otto, E.; Anlag, K.; Klugmann, C.; Treier, A.C.; Helou, J.; Sayer, J.A.; et al. Loss of GLIS2 causes nephronophthisis in humans and mice by increased apoptosis and fibrosis. Nat. Genet. 2007, 39, 1018–1024. [Google Scholar] [CrossRef]
- Valente, E.M.; Silhavy, J.L.; Brancati, F.; Barrano, G.; Krishnaswami, S.R.; Castori, M.; Lancaster, M.A.; Boltshauser, E.; Boccone, L.; Al-Gazali, L.; et al. Mutations in CEP290, which encodes a centrosomal protein, cause pleiotropic forms of Joubert syndrome. Nat. Genet. 2006, 38, 623–625. [Google Scholar] [CrossRef] [PubMed]
- Otto, E.A.; Loeys, B.; Khanna, H.; Hellemans, J.; Sudbrak, R.; Fan, S.; Muerb, U.; O’Toole, J.F.; Helou, J.; Attanasio, M.; et al. Nephrocystin-5, a ciliary IQ domain protein, is mutated in Senior-Loken syndrome and interacts with RPGR and calmodulin. Nat. Genet. 2005, 37, 282–288. [Google Scholar] [CrossRef]
- Otto, E.; Hoefele, J.; Ruf, R.; Mueller, A.M.; Hiller, K.S.; Wolf, M.T.; Schuermann, M.J.; Becker, A.; Birkenhager, R.; Sudbrak, R.; et al. A gene mutated in nephronophthisis and retinitis pigmentosa encodes a novel protein, nephroretinin, conserved in evolution. Am. J. Hum. Genet. 2002, 71, 1161–1167. [Google Scholar] [CrossRef] [Green Version]
- Olbrich, H.; Fliegauf, M.; Hoefele, J.; Kispert, A.; Otto, E.; Volz, A.; Wolf, M.T.; Sasmaz, G.; Trauer, U.; Reinhardt, R.; et al. Mutations in a novel gene, NPHP3, cause adolescent nephronophthisis, tapeto-retinal degeneration and hepatic fibrosis. Nat. Genet. 2003, 34, 455–459. [Google Scholar] [CrossRef]
- Srivastava, S.; Molinari, E.; Raman, S.; Sayer, J.A. Many Genes-One Disease? Genetics of Nephronophthisis (NPHP) and NPHP-Associated Disorders. Front. Pediatr. 2017, 5, 287. [Google Scholar] [CrossRef]
- Luo, F.; Tao, Y.H. Nephronophthisis: A review of genotype-phenotype correlation. Nephrology (Carlton) 2018, 23, 904–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halbritter, J.; Porath, J.D.; Diaz, K.A.; Braun, D.A.; Kohl, S.; Chaki, M.; Allen, S.J.; Soliman, N.A.; Hildebrandt, F.; Otto, E.A.; et al. Identification of 99 novel mutations in a worldwide cohort of 1,056 patients with a nephronophthisis-related ciliopathy. Hum. Genet. 2013, 132, 865–884. [Google Scholar] [CrossRef] [Green Version]
- Wolf, M.T. Nephronophthisis and related syndromes. Curr. Opin. Pediatr. 2015, 27, 201–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sang, L.; Miller, J.J.; Corbit, K.C.; Giles, R.H.; Brauer, M.J.; Otto, E.A.; Baye, L.M.; Wen, X.; Scales, S.J.; Kwong, M.; et al. Mapping the NPHP-JBTS-MKS protein network reveals ciliopathy disease genes and pathways. Cell 2011, 145, 513–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoff, S.; Halbritter, J.; Epting, D.; Frank, V.; Nguyen, T.M.; van Reeuwijk, J.; Boehlke, C.; Schell, C.; Yasunaga, T.; Helmstadter, M.; et al. ANKS6 is a central component of a nephronophthisis module linking NEK8 to INVS and NPHP3. Nat. Genet. 2013, 45, 951–956. [Google Scholar] [CrossRef] [PubMed]
- Avasthi, P.; Maser, R.L.; Tran, P.V. Primary Cilia in Cystic Kidney Disease. Results Probl. Cell Differ. 2017, 60, 281–321. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, J.; Pelletier, L. The Ciliary Transition Zone: Finding the Pieces and Assembling the Gate. Mol. Cells 2017, 40, 243–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anvarian, Z.; Mykytyn, K.; Mukhopadhyay, S.; Pedersen, L.B.; Christensen, S.T. Cellular signalling by primary cilia in development, organ function and disease. Nat. Rev. Nephrol. 2019, 15, 199–219. [Google Scholar] [CrossRef] [PubMed]
- Chaki, M.; Hoefele, J.; Allen, S.J.; Ramaswami, G.; Janssen, S.; Bergmann, C.; Heckenlively, J.R.; Otto, E.A.; Hildebrandt, F. Genotype-phenotype correlation in 440 patients with NPHP-related ciliopathies. Kidney Int. 2011, 80, 1239–1245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoefele, J.; Wolf, M.T.; O’Toole, J.F.; Otto, E.A.; Schultheiss, U.; Deschenes, G.; Attanasio, M.; Utsch, B.; Antignac, C.; Hildebrandt, F. Evidence of oligogenic inheritance in nephronophthisis. J. Am. Soc. Nephrol. 2007, 18, 2789–2795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsanis, N.; Ansley, S.J.; Badano, J.L.; Eichers, E.R.; Lewis, R.A.; Hoskins, B.E.; Scambler, P.J.; Davidson, W.S.; Beales, P.L.; Lupski, J.R. Triallelic inheritance in Bardet-Biedl syndrome, a Mendelian recessive disorder. Science 2001, 293, 2256–2259. [Google Scholar] [CrossRef] [Green Version]
- Antignac, C.; Arduy, C.H.; Beckmann, J.S.; Benessy, F.; Gros, F.; Medhioub, M.; Hildebrandt, F.; Dufier, J.L.; Kleinknecht, C.; Broyer, M.; et al. A gene for familial juvenile nephronophthisis (recessive medullary cystic kidney disease) maps to chromosome 2p. Nat. Genet. 1993, 3, 342–345. [Google Scholar] [CrossRef]
- Saunier, S.; Calado, J.; Benessy, F.; Silbermann, F.; Heilig, R.; Weissenbach, J.; Antignac, C. Characterization of the NPHP1 locus: Mutational mechanism involved in deletions in familial juvenile nephronophthisis. Am. J. Hum. Genet. 2000, 66, 778–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otto, E.; Betz, R.; Rensing, C.; Schatzle, S.; Kuntzen, T.; Vetsi, T.; Imm, A.; Hildebrandt, F. A deletion distinct from the classical homologous recombination of juvenile nephronophthisis type 1 (NPH1) allows exact molecular definition of deletion breakpoints. Hum. Mutat. 2000, 16, 211–223. [Google Scholar] [CrossRef]
- Caridi, G.; Murer, L.; Bellantuono, R.; Sorino, P.; Caringella, D.A.; Gusmano, R.; Ghiggeri, G.M. Renal-retinal syndromes: Association of retinal anomalies and recessive nephronophthisis in patients with homozygous deletion of the NPH1 locus. Am. J. Kidney Dis. 1998, 32, 1059–1062. [Google Scholar] [CrossRef]
- Benzing, T.; Gerke, P.; Hopker, K.; Hildebrandt, F.; Kim, E.; Walz, G. Nephrocystin interacts with Pyk2, p130(Cas), and tensin and triggers phosphorylation of Pyk2. Proc. Natl. Acad. Sci. USA 2001, 98, 9784–9789. [Google Scholar] [CrossRef] [Green Version]
- Donaldson, J.C.; Dempsey, P.J.; Reddy, S.; Bouton, A.H.; Coffey, R.J.; Hanks, S.K. Crk-associated substrate p130(Cas) interacts with nephrocystin and both proteins localize to cell-cell contacts of polarized epithelial cells. Exp. Cell Res. 2000, 256, 168–178. [Google Scholar] [CrossRef]
- Donaldson, J.C.; Dise, R.S.; Ritchie, M.D.; Hanks, S.K. Nephrocystin-conserved domains involved in targeting to epithelial cell-cell junctions, interaction with filamins, and establishing cell polarity. J. Biol. Chem. 2002, 277, 29028–29035. [Google Scholar] [CrossRef] [Green Version]
- Schermer, B.; Hopker, K.; Omran, H.; Ghenoiu, C.; Fliegauf, M.; Fekete, A.; Horvath, J.; Kottgen, M.; Hackl, M.; Zschiedrich, S.; et al. Phosphorylation by casein kinase 2 induces PACS-1 binding of nephrocystin and targeting to cilia. EMBO J. 2005, 24, 4415–4424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fliegauf, M.; Horvath, J.; von Schnakenburg, C.; Olbrich, H.; Muller, D.; Thumfart, J.; Schermer, B.; Pazour, G.J.; Neumann, H.P.; Zentgraf, H.; et al. Nephrocystin specifically localizes to the transition zone of renal and respiratory cilia and photoreceptor connecting cilia. J. Am. Soc. Nephrol. 2006, 17, 2424–2433. [Google Scholar] [CrossRef] [Green Version]
- Tory, K.; Rousset-Rouviere, C.; Gubler, M.C.; Moriniere, V.; Pawtowski, A.; Becker, C.; Guyot, C.; Gie, S.; Frishberg, Y.; Nivet, H.; et al. Mutations of NPHP2 and NPHP3 in infantile nephronophthisis. Kidney Int. 2009, 75, 839–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hildebrandt, F. Nephronophthisis and Medullary Cystic Kidney Disease. In Pediatric Nephrology: Sixth Completely Revised, Updated and Enlarged Edition; Avner, E., Harmon, W., Niaudet, P., Yoshikawa, N., Eds.; Springer: Berlin/Heidelberg, Germany, 2009; pp. 831–848. [Google Scholar]
- Bergmann, C.; Fliegauf, M.; Bruchle, N.O.; Frank, V.; Olbrich, H.; Kirschner, J.; Schermer, B.; Schmedding, I.; Kispert, A.; Kranzlin, B.; et al. Loss of nephrocystin-3 function can cause embryonic lethality, Meckel-Gruber-like syndrome, situs inversus, and renal-hepatic-pancreatic dysplasia. Am. J. Hum. Genet. 2008, 82, 959–970. [Google Scholar] [CrossRef] [Green Version]
- Nurnberger, J.; Kribben, A.; Opazo Saez, A.; Heusch, G.; Philipp, T.; Phillips, C.L. The Invs gene encodes a microtubule-associated protein. J. Am. Soc. Nephrol. 2004, 15, 1700–1710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nurnberger, J.; Bacallao, R.L.; Phillips, C.L. Inversin forms a complex with catenins and N-cadherin in polarized epithelial cells. Mol. Biol. Cell 2002, 13, 3096–3106. [Google Scholar] [CrossRef]
- Morgan, D.; Eley, L.; Sayer, J.; Strachan, T.; Yates, L.M.; Craighead, A.S.; Goodship, J.A. Expression analyses and interaction with the anaphase promoting complex protein Apc2 suggest a role for inversin in primary cilia and involvement in the cell cycle. Hum. Mol. Genet. 2002, 11, 3345–3350. [Google Scholar] [CrossRef] [Green Version]
- Simons, M.; Gloy, J.; Ganner, A.; Bullerkotte, A.; Bashkurov, M.; Kronig, C.; Schermer, B.; Benzing, T.; Cabello, O.A.; Jenny, A.; et al. Inversin, the gene product mutated in nephronophthisis type II, functions as a molecular switch between Wnt signaling pathways. Nat. Genet. 2005, 37, 537–543. [Google Scholar] [CrossRef] [Green Version]
- Omran, H.; Fernandez, C.; Jung, M.; Haffner, K.; Fargier, B.; Villaquiran, A.; Waldherr, R.; Gretz, N.; Brandis, M.; Ruschendorf, F.; et al. Identification of a new gene locus for adolescent nephronophthisis, on chromosome 3q22 in a large Venezuelan pedigree. Am. J. Hum. Genet. 2000, 66, 118–127. [Google Scholar] [CrossRef] [Green Version]
- Gattone, V.H., 2nd; Wang, X.; Harris, P.C.; Torres, V.E. Inhibition of renal cystic disease development and progression by a vasopressin V2 receptor antagonist. Nat. Med. 2003, 9, 1323–1326. [Google Scholar] [CrossRef]
- NIH. Tolvaptan Phase 3 Efficacy and Safety Study in Autosomal Dominant Polycystic Kidney Disease (ADPKD) (TEMPO3:4). Available online: https://clinicaltrials.gov/ct2/show/NCT00428948 (accessed on 20 September 2021).
- Schuermann, M.J.; Otto, E.; Becker, A.; Saar, K.; Ruschendorf, F.; Polak, B.C.; Ala-Mello, S.; Hoefele, J.; Wiedensohler, A.; Haller, M.; et al. Mapping of gene loci for nephronophthisis type 4 and Senior-Loken syndrome, to chromosome 1p36. Am. J. Hum. Genet. 2002, 70, 1240–1246. [Google Scholar] [CrossRef] [Green Version]
- Mollet, G.; Salomon, R.; Gribouval, O.; Silbermann, F.; Bacq, D.; Landthaler, G.; Milford, D.; Nayir, A.; Rizzoni, G.; Antignac, C.; et al. The gene mutated in juvenile nephronophthisis type 4 encodes a novel protein that interacts with nephrocystin. Nat. Genet. 2002, 32, 300–305. [Google Scholar] [CrossRef] [PubMed]
- den Hollander, A.I.; Koenekoop, R.K.; Yzer, S.; Lopez, I.; Arends, M.L.; Voesenek, K.E.; Zonneveld, M.N.; Strom, T.M.; Meitinger, T.; Brunner, H.G.; et al. Mutations in the CEP290 (NPHP6) gene are a frequent cause of Leber congenital amaurosis. Am. J. Hum. Genet. 2006, 79, 556–561. [Google Scholar] [CrossRef] [Green Version]
- Leitch, C.C.; Zaghloul, N.A.; Davis, E.E.; Stoetzel, C.; Diaz-Font, A.; Rix, S.; Alfadhel, M.; Lewis, R.A.; Eyaid, W.; Banin, E.; et al. Hypomorphic mutations in syndromic encephalocele genes are associated with Bardet-Biedl syndrome. Nat. Genet. 2008, 40, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Slaats, G.G.; Saldivar, J.C.; Bacal, J.; Zeman, M.K.; Kile, A.C.; Hynes, A.M.; Srivastava, S.; Nazmutdinova, J.; den Ouden, K.; Zagers, M.S.; et al. DNA replication stress underlies renal phenotypes in CEP290-associated Joubert syndrome. J. Clin. Investig. 2015, 125, 3657–3666. [Google Scholar] [CrossRef] [Green Version]
- Lu, D.; Rauhauser, A.; Li, B.; Ren, C.; McEnery, K.; Zhu, J.; Chaki, M.; Vadnagara, K.; Elhadi, S.; Jetten, A.M.; et al. Loss of Glis2/NPHP7 causes kidney epithelial cell senescence and suppresses cyst growth in the Kif3a mouse model of cystic kidney disease. Kidney Int. 2016, 89, 1307–1323. [Google Scholar] [CrossRef] [Green Version]
- Song, L.; Li, Z.Y.; Liu, W.P.; Zhao, M.R. Crosstalk between Wnt/β-catenin and Hedgehog/Gli signaling pathways in colon cancer and implications for therapy. Cancer Biol. Ther. 2015, 16, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Arts, H.H.; Doherty, D.; van Beersum, S.E.; Parisi, M.A.; Letteboer, S.J.; Gorden, N.T.; Peters, T.A.; Marker, T.; Voesenek, K.; Kartono, A.; et al. Mutations in the gene encoding the basal body protein RPGRIP1L, a nephrocystin-4 interactor, cause Joubert syndrome. Nat. Genet. 2007, 39, 882–888. [Google Scholar] [CrossRef] [PubMed]
- Roepman, R.; Letteboer, S.J.; Arts, H.H.; van Beersum, S.E.; Lu, X.; Krieger, E.; Ferreira, P.A.; Cremers, F.P. Interaction of nephrocystin-4 and RPGRIP1 is disrupted by nephronophthisis or Leber congenital amaurosis-associated mutations. Proc. Natl. Acad. Sci. USA 2005, 102, 18520–18525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brancati, F.; Travaglini, L.; Zablocka, D.; Boltshauser, E.; Accorsi, P.; Montagna, G.; Silhavy, J.L.; Barrano, G.; Bertini, E.; Emma, F.; et al. RPGRIP1L mutations are mainly associated with the cerebello-renal phenotype of Joubert syndrome-related disorders. Clin. Genet. 2008, 74, 164–170. [Google Scholar] [CrossRef]
- Liu, S.; Lu, W.; Obara, T.; Kuida, S.; Lehoczky, J.; Dewar, K.; Drummond, I.A.; Beier, D.R. A defect in a novel Nek-family kinase causes cystic kidney disease in the mouse and in zebrafish. Development 2002, 129, 5839–5846. [Google Scholar] [CrossRef] [Green Version]
- Bowers, A.J.; Boylan, J.F. Nek8, a NIMA family kinase member, is overexpressed in primary human breast tumors. Gene 2004, 328, 135–142. [Google Scholar] [CrossRef] [PubMed]
- McCooke, J.K.; Appels, R.; Barrero, R.A.; Ding, A.; Ozimek-Kulik, J.E.; Bellgard, M.I.; Morahan, G.; Phillips, J.K. A novel mutation causing nephronophthisis in the Lewis polycystic kidney rat localises to a conserved RCC1 domain in Nek8. BMC Genom. 2012, 13, 393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, J.K.; Hopwood, D.; Loxley, R.A.; Ghatora, K.; Coombes, J.D.; Tan, Y.S.; Harrison, J.L.; McKitrick, D.J.; Holobotvskyy, V.; Arnolda, L.F.; et al. Temporal relationship between renal cyst development, hypertension and cardiac hypertrophy in a new rat model of autosomal recessive polycystic kidney disease. Kidney Blood Press Res. 2007, 30, 129–144. [Google Scholar] [CrossRef] [Green Version]
- Shiba, D.; Manning, D.K.; Koga, H.; Beier, D.R.; Yokoyama, T. Inv acts as a molecular anchor for Nphp3 and Nek8 in the proximal segment of primary cilia. Cytoskeleton (Hoboken) 2010, 67, 112–119. [Google Scholar] [CrossRef]
- Zalli, D.; Bayliss, R.; Fry, A.M. The Nek8 protein kinase, mutated in the human cystic kidney disease nephronophthisis, is both activated and degraded during ciliogenesis. Hum. Mol. Genet. 2012, 21, 1155–1171. [Google Scholar] [CrossRef] [Green Version]
- Valkova, N.; Yunis, R.; Mak, S.K.; Kang, K.; Kultz, D. Nek8 mutation causes overexpression of galectin-1, sorcin, and vimentin and accumulation of the major urinary protein in renal cysts of jck mice. Mol. Cell Proteom. 2005, 4, 1009–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abeyta, A.; Castella, M.; Jacquemont, C.; Taniguchi, T. NEK8 regulates DNA damage-induced RAD51 foci formation and replication fork protection. Cell Cycle 2017, 16, 335–347. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.J.; Lin, J.R.; Vannier, J.B.; Slaats, G.G.; Kile, A.C.; Paulsen, R.D.; Manning, D.K.; Beier, D.R.; Giles, R.H.; Boulton, S.J.; et al. NEK8 links the ATR-regulated replication stress response and S phase CDK activity to renal ciliopathies. Mol. Cell 2013, 51, 423–439. [Google Scholar] [CrossRef] [Green Version]
- Otto, E.A.; Hurd, T.W.; Airik, R.; Chaki, M.; Zhou, W.; Stoetzel, C.; Patil, S.B.; Levy, S.; Ghosh, A.K.; Murga-Zamalloa, C.A.; et al. Candidate exome capture identifies mutation of SDCCAG8 as the cause of a retinal-renal ciliopathy. Nat. Genet. 2010, 42, 840–850. [Google Scholar] [CrossRef] [Green Version]
- Brancati, F.; Iannicelli, M.; Travaglini, L.; Mazzotta, A.; Bertini, E.; Boltshauser, E.; D’Arrigo, S.; Emma, F.; Fazzi, E.; Gallizzi, R.; et al. MKS3/TMEM67 mutations are a major cause of COACH Syndrome, a Joubert Syndrome related disorder with liver involvement. Hum. Mutat. 2009, 30, E432–E442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otto, E.A.; Tory, K.; Attanasio, M.; Zhou, W.; Chaki, M.; Paruchuri, Y.; Wise, E.L.; Wolf, M.T.; Utsch, B.; Becker, C.; et al. Hypomorphic mutations in meckelin (MKS3/TMEM67) cause nephronophthisis with liver fibrosis (NPHP11). J. Med. Genet. 2009, 46, 663–670. [Google Scholar] [CrossRef]
- Davis, E.E.; Zhang, Q.; Liu, Q.; Diplas, B.H.; Davey, L.M.; Hartley, J.; Stoetzel, C.; Szymanska, K.; Ramaswami, G.; Logan, C.V.; et al. TTC21B contributes both causal and modifying alleles across the ciliopathy spectrum. Nat. Genet. 2011, 43, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Chaki, M.; Airik, R.; Ghosh, A.K.; Giles, R.H.; Chen, R.; Slaats, G.G.; Wang, H.; Hurd, T.W.; Zhou, W.; Cluckey, A.; et al. Exome capture reveals ZNF423 and CEP164 mutations, linking renal ciliopathies to DNA damage response signaling. Cell 2012, 150, 533–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taskiran, E.Z.; Korkmaz, E.; Gucer, S.; Kosukcu, C.; Kaymaz, F.; Koyunlar, C.; Bryda, E.C.; Chaki, M.; Lu, D.; Vadnagara, K.; et al. Mutations in ANKS6 cause a nephronophthisis-like phenotype with ESRD. J. Am. Soc. Nephrol. 2014, 25, 1653–1661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halbritter, J.; Bizet, A.A.; Schmidts, M.; Porath, J.D.; Braun, D.A.; Gee, H.Y.; McInerney-Leo, A.M.; Krug, P.; Filhol, E.; Davis, E.E.; et al. Defects in the IFT-B component IFT172 cause Jeune and Mainzer-Saldino syndromes in humans. Am. J. Hum. Genet. 2013, 93, 915–925. [Google Scholar] [CrossRef] [Green Version]
- Failler, M.; Gee, H.Y.; Krug, P.; Joo, K.; Halbritter, J.; Belkacem, L.; Filhol, E.; Porath, J.D.; Braun, D.A.; Schueler, M.; et al. Mutations of CEP83 cause infantile nephronophthisis and intellectual disability. Am. J. Hum. Genet. 2014, 94, 905–914. [Google Scholar] [CrossRef] [Green Version]
- Schueler, M.; Braun, D.A.; Chandrasekar, G.; Gee, H.Y.; Klasson, T.D.; Halbritter, J.; Bieder, A.; Porath, J.D.; Airik, R.; Zhou, W.; et al. DCDC2 mutations cause a renal-hepatic ciliopathy by disrupting Wnt signaling. Am. J. Hum. Genet. 2015, 96, 81–92. [Google Scholar] [CrossRef] [Green Version]
- Macia, M.S.; Halbritter, J.; Delous, M.; Bredrup, C.; Gutter, A.; Filhol, E.; Mellgren, A.E.C.; Leh, S.; Bizet, A.; Braun, D.A.; et al. Mutations in MAPKBP1 Cause Juvenile or Late-Onset Cilia-Independent Nephronophthisis. Am. J. Hum. Genet. 2017, 100, 323–333. [Google Scholar] [CrossRef] [Green Version]
- Dixon-Salazar, T.; Silhavy, J.L.; Marsh, S.E.; Louie, C.M.; Scott, L.C.; Gururaj, A.; Al-Gazali, L.; Al-Tawari, A.A.; Kayserili, H.; Sztriha, L.; et al. Mutations in the AHI1 gene, encoding jouberin, cause Joubert syndrome with cortical polymicrogyria. Am. J. Hum. Genet. 2004, 75, 979–987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferland, R.J.; Eyaid, W.; Collura, R.V.; Tully, L.D.; Hill, R.S.; Al-Nouri, D.; Al-Rumayyan, A.; Topcu, M.; Gascon, G.; Bodell, A.; et al. Abnormal cerebellar development and axonal decussation due to mutations in AHI1 in Joubert syndrome. Nat. Genet. 2004, 36, 1008–1013. [Google Scholar] [CrossRef] [Green Version]
- Tuz, K.; Hsiao, Y.C.; Juarez, O.; Shi, B.; Harmon, E.Y.; Phelps, I.G.; Lennartz, M.R.; Glass, I.A.; Doherty, D.; Ferland, R.J. The Joubert syndrome-associated missense mutation (V443D) in the Abelson-helper integration site 1 (AHI1) protein alters its localization and protein-protein interactions. J. Biol. Chem. 2013, 288, 13676–13694. [Google Scholar] [CrossRef] [Green Version]
- Utsch, B.; Sayer, J.A.; Attanasio, M.; Pereira, R.R.; Eccles, M.; Hennies, H.C.; Otto, E.A.; Hildebrandt, F. Identification of the first AHI1 gene mutations in nephronophthisis-associated Joubert syndrome. Pediatr. Nephrol. 2006, 21, 32–35. [Google Scholar] [CrossRef]
- Gorden, N.T.; Arts, H.H.; Parisi, M.A.; Coene, K.L.; Letteboer, S.J.; van Beersum, S.E.; Mans, D.A.; Hikida, A.; Eckert, M.; Knutzen, D.; et al. CC2D2A is mutated in Joubert syndrome and interacts with the ciliopathy-associated basal body protein CEP290. Am. J. Hum. Genet. 2008, 83, 559–571. [Google Scholar] [CrossRef] [Green Version]
- Otto, E.A.; Ramaswami, G.; Janssen, S.; Chaki, M.; Allen, S.J.; Zhou, W.; Airik, R.; Hurd, T.W.; Ghosh, A.K.; Wolf, M.T.; et al. Mutation analysis of 18 nephronophthisis associated ciliopathy disease genes using a DNA pooling and next generation sequencing strategy. J. Med. Genet. 2011, 48, 105–116. [Google Scholar] [CrossRef] [PubMed]
- O’Toole, J.F.; Liu, Y.; Davis, E.E.; Westlake, C.J.; Attanasio, M.; Otto, E.A.; Seelow, D.; Nurnberg, G.; Becker, C.; Nuutinen, M.; et al. Individuals with mutations in XPNPEP3, which encodes a mitochondrial protein, develop a nephronophthisis-like nephropathy. J. Clin. Investig. 2010, 120, 791–802. [Google Scholar] [CrossRef]
- Grewal, R.P.; Tayag, E.; Figueroa, K.P.; Zu, L.; Durazo, A.; Nunez, C.; Pulst, S.M. Clinical and genetic analysis of a distinct autosomal dominant spinocerebellar ataxia. Neurology 1998, 51, 1423–1426. [Google Scholar] [CrossRef] [PubMed]
- Hurd, T.W.; Otto, E.A.; Mishima, E.; Gee, H.Y.; Inoue, H.; Inazu, M.; Yamada, H.; Halbritter, J.; Seki, G.; Konishi, M.; et al. Mutation of the Mg2+ transporter SLC41A1 results in a nephronophthisis-like phenotype. J. Am. Soc. Nephrol. 2013, 24, 967–977. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.J.; Halbritter, J.; Braun, D.A.; Schueler, M.; Schapiro, D.; Rim, J.H.; Nandadasa, S.; Choi, W.I.; Widmeier, E.; Shril, S.; et al. Mutations of ADAMTS9 Cause Nephronophthisis-Related Ciliopathy. Am. J. Hum. Genet. 2019, 104, 45–54. [Google Scholar] [CrossRef] [Green Version]
- McKusick, N. Online Mendelian Inheritance in Man, OMIM (TM). Available online: https://www.ncbi.nlm.nih.gov/omim (accessed on 20 September 2021).
- UniProt, C. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- The European Polycystic Kidney Disease Consortium. The polycystic kidney disease 1 gene encodes a 14 kb transcript and lies within a duplicated region on chromosome 16. Cell 1994, 77, 881–894. [Google Scholar] [CrossRef] [Green Version]
- Mochizuki, T.; Wu, G.; Hayashi, T.; Xenophontos, S.L.; Veldhuisen, B.; Saris, J.J.; Reynolds, D.M.; Cai, Y.; Gabow, P.A.; Pierides, A.; et al. PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science 1996, 272, 1339–1342. [Google Scholar] [CrossRef] [PubMed]
- Pazour, G.J.; San Agustin, J.T.; Follit, J.A.; Rosenbaum, J.L.; Witman, G.B. Polycystin-2 localizes to kidney cilia and the ciliary level is elevated in orpk mice with polycystic kidney disease. Curr. Biol. 2002, 12, R378–R380. [Google Scholar] [CrossRef] [Green Version]
- Taulman, P.D.; Haycraft, C.J.; Balkovetz, D.F.; Yoder, B.K. Polaris, a protein involved in left-right axis patterning, localizes to basal bodies and cilia. Mol. Biol. Cell 2001, 12, 589–599. [Google Scholar] [CrossRef] [Green Version]
- Pazour, G.J.; Dickert, B.L.; Vucica, Y.; Seeley, E.S.; Rosenbaum, J.L.; Witman, G.B.; Cole, D.G. Chlamydomonas IFT88 and its mouse homologue, polycystic kidney disease gene tg737, are required for assembly of cilia and flagella. J. Cell Biol. 2000, 151, 709–718. [Google Scholar] [CrossRef] [PubMed]
- Moyer, J.H.; Lee-Tischler, M.J.; Kwon, H.Y.; Schrick, J.J.; Avner, E.D.; Sweeney, W.E.; Godfrey, V.L.; Cacheiro, N.L.; Wilkinson, J.E.; Woychik, R.P. Candidate gene associated with a mutation causing recessive polycystic kidney disease in mice. Science 1994, 264, 1329–1333. [Google Scholar] [CrossRef] [Green Version]
- Haycraft, C.J.; Zhang, Q.; Song, B.; Jackson, W.S.; Detloff, P.J.; Serra, R.; Yoder, B.K. Intraflagellar transport is essential for endochondral bone formation. Development 2007, 134, 307–316. [Google Scholar] [CrossRef] [Green Version]
- Chizhikov, V.V.; Davenport, J.; Zhang, Q.; Shih, E.K.; Cabello, O.A.; Fuchs, J.L.; Yoder, B.K.; Millen, K.J. Cilia proteins control cerebellar morphogenesis by promoting expansion of the granule progenitor pool. J. Neurosci. 2007, 27, 9780–9789. [Google Scholar] [CrossRef]
- Zhang, Q.; Davenport, J.R.; Croyle, M.J.; Haycraft, C.J.; Yoder, B.K. Disruption of IFT results in both exocrine and endocrine abnormalities in the pancreas of Tg737(orpk) mutant mice. Lab. Investig. 2005, 85, 45–64. [Google Scholar] [CrossRef] [Green Version]
- Banizs, B.; Pike, M.M.; Millican, C.L.; Ferguson, W.B.; Komlosi, P.; Sheetz, J.; Bell, P.D.; Schwiebert, E.M.; Yoder, B.K. Dysfunctional cilia lead to altered ependyma and choroid plexus function, and result in the formation of hydrocephalus. Development 2005, 132, 5329–5339. [Google Scholar] [CrossRef] [Green Version]
- Cano, D.A.; Murcia, N.S.; Pazour, G.J.; Hebrok, M. Orpk mouse model of polycystic kidney disease reveals essential role of primary cilia in pancreatic tissue organization. Development 2004, 131, 3457–3467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pazour, G.J.; Baker, S.A.; Deane, J.A.; Cole, D.G.; Dickert, B.L.; Rosenbaum, J.L.; Witman, G.B.; Besharse, J.C. The intraflagellar transport protein, IFT88, is essential for vertebrate photoreceptor assembly and maintenance. J. Cell Biol. 2002, 157, 103–113. [Google Scholar] [CrossRef] [Green Version]
- Yoder, B.K.; Hou, X.; Guay-Woodford, L.M. The polycystic kidney disease proteins, polycystin-1, polycystin-2, polaris, and cystin, are co-localized in renal cilia. J. Am. Soc. Nephrol. 2002, 13, 2508–2516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watnick, T.; Germino, G. From cilia to cyst. Nat. Genet. 2003, 34, 355–356. [Google Scholar] [CrossRef]
- Hildebrandt, F.; Otto, E. Cilia and centrosomes: A unifying pathogenic concept for cystic kidney disease? Nat. Rev. Genet. 2005, 6, 928–940. [Google Scholar] [CrossRef]
- Zimmermann, K.W. Beiträge zur Kenntnis einiger Drusen und Epithelien. Arch. Mikrosk Anat. 1898, 52, 552–706. [Google Scholar] [CrossRef]
- Praetorius, H.A.; Frokiaer, J.; Nielsen, S.; Spring, K.R. Bending the primary cilium opens Ca2+-sensitive intermediate-conductance K+ channels in MDCK cells. J. Membr. Biol. 2003, 191, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Praetorius, H.A.; Spring, K.R. Removal of the MDCK cell primary cilium abolishes flow sensing. J. Membr. Biol. 2003, 191, 69–76. [Google Scholar] [CrossRef]
- Praetorius, H.A.; Spring, K.R. The renal cell primary cilium functions as a flow sensor. Curr. Opin. Nephrol. Hypertens 2003, 12, 517–520. [Google Scholar] [CrossRef]
- Liu, W.; Murcia, N.S.; Duan, Y.; Weinbaum, S.; Yoder, B.K.; Schwiebert, E.; Satlin, L.M. Mechanoregulation of intracellular Ca2+ concentration is attenuated in collecting duct of monocilium-impaired orpk mice. Am. J. Physiol. Renal. Physiol. 2005, 289, F978–F988. [Google Scholar] [CrossRef] [Green Version]
- Nauli, S.M.; Alenghat, F.J.; Luo, Y.; Williams, E.; Vassilev, P.; Li, X.; Elia, A.E.; Lu, W.; Brown, E.M.; Quinn, S.J.; et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat. Genet. 2003, 33, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Manning, D.K.; Sergeev, M.; van Heesbeen, R.G.; Wong, M.D.; Oh, J.H.; Liu, Y.; Henkelman, R.M.; Drummond, I.; Shah, J.V.; Beier, D.R. Loss of the ciliary kinase Nek8 causes left-right asymmetry defects. J. Am. Soc. Nephrol. 2013, 24, 100–112. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Zhang, J.; Nauli, S.M.; Li, X.; Starremans, P.G.; Luo, Y.; Roberts, K.A.; Zhou, J. Fibrocystin/polyductin, found in the same protein complex with polycystin-2, regulates calcium responses in kidney epithelia. Mol. Cell Biol. 2007, 27, 3241–3252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kottgen, M.; Buchholz, B.; Garcia-Gonzalez, M.A.; Kotsis, F.; Fu, X.; Doerken, M.; Boehlke, C.; Steffl, D.; Tauber, R.; Wegierski, T.; et al. TRPP2 and TRPV4 form a polymodal sensory channel complex. J. Cell Biol. 2008, 182, 437–447. [Google Scholar] [CrossRef]
- Pieczynski, J.N.; Yoder, B.K. Chapter 11—Renal Cilia Structure, Function, and Physiology, 5th ed.; Elsevier: Amsterdam, The Netherlands, 2013. [Google Scholar]
- Ferreira, R.R.; Fukui, H.; Chow, R.; Vilfan, A.; Vermot, J. The cilium as a force sensor-myth versus reality. J. Cell Sci. 2019, 132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davenport, J.R.; Watts, A.J.; Roper, V.C.; Croyle, M.J.; van Groen, T.; Wyss, J.M.; Nagy, T.R.; Kesterson, R.A.; Yoder, B.K. Disruption of intraflagellar transport in adult mice leads to obesity and slow-onset cystic kidney disease. Curr. Biol. 2007, 17, 1586–1594. [Google Scholar] [CrossRef] [Green Version]
- Lantinga-van Leeuwen, I.S.; Leonhard, W.N.; van der Wal, A.; Breuning, M.H.; de Heer, E.; Peters, D.J. Kidney-specific inactivation of the Pkd1 gene induces rapid cyst formation in developing kidneys and a slow onset of disease in adult mice. Hum. Mol. Genet. 2007, 16, 3188–3196. [Google Scholar] [CrossRef] [Green Version]
- Piontek, K.; Menezes, L.F.; Garcia-Gonzalez, M.A.; Huso, D.L.; Germino, G.G. A critical developmental switch defines the kinetics of kidney cyst formation after loss of Pkd1. Nat. Med. 2007, 13, 1490–1495. [Google Scholar] [CrossRef]
- Smith, L.A.; Bukanov, N.O.; Husson, H.; Russo, R.J.; Barry, T.C.; Taylor, A.L.; Beier, D.R.; Ibraghimov-Beskrovnaya, O. Development of polycystic kidney disease in juvenile cystic kidney mice: Insights into pathogenesis, ciliary abnormalities, and common features with human disease. J. Am. Soc. Nephrol. 2006, 17, 2821–2831. [Google Scholar] [CrossRef] [Green Version]
- Sohara, E.; Luo, Y.; Zhang, J.; Manning, D.K.; Beier, D.R.; Zhou, J. Nek8 regulates the expression and localization of polycystin-1 and polycystin-2. J. Am. Soc. Nephrol. 2008, 19, 469–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trapp, M.L.; Galtseva, A.; Manning, D.K.; Beier, D.R.; Rosenblum, N.D.; Quarmby, L.M. Defects in ciliary localization of Nek8 is associated with cystogenesis. Pediatr. Nephrol. 2008, 23, 377–387. [Google Scholar] [CrossRef]
- Vivante, A.; Hildebrandt, F. Exploring the genetic basis of early-onset chronic kidney disease. Nat. Rev. Nephrol. 2016, 12, 133–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaika, O.; Mamenko, M.; Berrout, J.; Boukelmoune, N.; O’Neil, R.G.; Pochynyuk, O. TRPV4 dysfunction promotes renal cystogenesis in autosomal recessive polycystic kidney disease. J. Am. Soc. Nephrol. 2013, 24, 604–616. [Google Scholar] [CrossRef] [Green Version]
- Delling, M.; DeCaen, P.G.; Doerner, J.F.; Febvay, S.; Clapham, D.E. Primary cilia are specialized calcium signalling organelles. Nature 2013, 504, 311–314. [Google Scholar] [CrossRef] [Green Version]
- Su, S.; Phua, S.C.; DeRose, R.; Chiba, S.; Narita, K.; Kalugin, P.N.; Katada, T.; Kontani, K.; Takeda, S.; Inoue, T. Genetically encoded calcium indicator illuminates calcium dynamics in primary cilia. Nat. Methods 2013, 10, 1105–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, X.; Mohieldin, A.M.; Muntean, B.S.; Green, J.A.; Shah, J.V.; Mykytyn, K.; Nauli, S.M. Cilioplasm is a cellular compartment for calcium signaling in response to mechanical and chemical stimuli. Cell Mol. Life Sci. 2014, 71, 2165–2178. [Google Scholar] [CrossRef] [Green Version]
- Yuan, S.; Zhao, L.; Brueckner, M.; Sun, Z. Intraciliary calcium oscillations initiate vertebrate left-right asymmetry. Curr. Biol. 2015, 25, 556–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeCaen, P.G.; Delling, M.; Vien, T.N.; Clapham, D.E. Direct recording and molecular identification of the calcium channel of primary cilia. Nature 2013, 504, 315–318. [Google Scholar] [CrossRef] [Green Version]
- Delling, M.; Indzhykulian, A.A.; Liu, X.; Li, Y.; Xie, T.; Corey, D.P.; Clapham, D.E. Primary cilia are not calcium-responsive mechanosensors. Nature 2016, 531, 656–660. [Google Scholar] [CrossRef] [Green Version]
- Sigg, M.A.; Menchen, T.; Lee, C.; Johnson, J.; Jungnickel, M.K.; Choksi, S.P.; Garcia, G., 3rd; Busengdal, H.; Dougherty, G.W.; Pennekamp, P.; et al. Evolutionary Proteomics Uncovers Ancient Associations of Cilia with Signaling Pathways. Dev. Cell 2017, 43, 744–762.e711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Mathur, J.; Vessieres, E.; Hammack, S.; Nonomura, K.; Favre, J.; Grimaud, L.; Petrus, M.; Francisco, A.; Li, J.; et al. GPR68 Senses Flow and Is Essential for Vascular Physiology. Cell 2018, 173, 762–775.e716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ong, A.C.M. Making sense of polycystic kidney disease. Lancet 2017, 389, 1780–1782. [Google Scholar] [CrossRef]
- Kim, S.; Nie, H.; Nesin, V.; Tran, U.; Outeda, P.; Bai, C.X.; Keeling, J.; Maskey, D.; Watnick, T.; Wessely, O.; et al. The polycystin complex mediates Wnt/Ca(2+) signalling. Nat. Cell Biol. 2016, 18, 752–764. [Google Scholar] [CrossRef] [PubMed]
- Robertson, G.L. Antidiuretic hormone. Normal and disordered function. Endocrinol. Metab. Clin. N. Am. 2001, 30, 671–694. [Google Scholar] [CrossRef]
- Cuzzo, B.; Lappin, S.L. Vasopressin (Antidiuretic Hormone, ADH). Available online: https://www.ncbi.nlm.nih.gov/books/NBK526069/ (accessed on 20 September 2021).
- Davies, A.G. Antidiuretic and growth hormones. Br. Med. J. 1972, 2, 282–284. [Google Scholar] [CrossRef] [Green Version]
- Boone, M.; Deen, P.M. Physiology and pathophysiology of the vasopressin-regulated renal water reabsorption. Pflugers. Arch. 2008, 456, 1005–1024. [Google Scholar] [CrossRef] [Green Version]
- Fushimi, K.; Sasaki, S.; Marumo, F. Phosphorylation of serine 256 is required for cAMP-dependent regulatory exocytosis of the aquaporin-2 water channel. J. Biol. Chem. 1997, 272, 14800–14804. [Google Scholar] [CrossRef] [Green Version]
- Chou, C.L.; Yip, K.P.; Michea, L.; Kador, K.; Ferraris, J.D.; Wade, J.B.; Knepper, M.A. Regulation of aquaporin-2 trafficking by vasopressin in the renal collecting duct. Roles of ryanodine-sensitive Ca2+ stores and calmodulin. J. Biol. Chem. 2000, 275, 36839–36846. [Google Scholar] [CrossRef] [Green Version]
- Deen, P.M.; Knoers, N.V. Physiology and pathophysiology of the aquaporin-2 water channel. Curr. Opin. Nephrol. Hypertens. 1998, 7, 37–42. [Google Scholar] [CrossRef]
- Nielsen, S.; Fror, J.; Knepper, M.A. Renal aquaporins: Key roles in water balance and water balance disorders. Curr. Opin. Nephrol. Hypertens. 1998, 7, 509–516. [Google Scholar] [CrossRef]
- Yamamoto, T.; Sasaki, S.; Fushimi, K.; Ishibashi, K.; Yaoita, E.; Kawasaki, K.; Marumo, F.; Kihara, I. Vasopressin increases AQP-CD water channel in apical membrane of collecting duct cells in Brattleboro rats. Am. J. Physiol. 1995, 268, C1546–C1551. [Google Scholar] [CrossRef] [PubMed]
- Finley, J.J.T.; Konstam, M.A.; Udelson, J.E. Arginine vasopressin antagonists for the treatment of heart failure and hyponatremia. Circulation 2008, 118, 410–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y.H.; Suzuki, A.; Hajarnis, S.; Ma, Z.; Chapin, H.C.; Caplan, M.J.; Pontoglio, M.; Somlo, S.; Igarashi, P. Polycystin-2 and phosphodiesterase 4C are components of a ciliary A-kinase anchoring protein complex that is disrupted in cystic kidney diseases. Proc. Natl. Acad. Sci. USA 2011, 108, 10679–10684. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Cobo-Stark, P.; Patel, V.; Somlo, S.; Han, P.L.; Igarashi, P. Adenylyl cyclase 5 deficiency reduces renal cyclic AMP and cyst growth in an orthologous mouse model of polycystic kidney disease. Kidney Int. 2018, 93, 403–415. [Google Scholar] [CrossRef] [PubMed]
- Cantero Mdel, R.; Velazquez, I.F.; Streets, A.J.; Ong, A.C.; Cantiello, H.F. The cAMP Signaling Pathway and Direct Protein Kinase A Phosphorylation Regulate Polycystin-2 (TRPP2) Channel Function. J. Biol. Chem. 2015, 290, 23888–23896. [Google Scholar] [CrossRef] [Green Version]
- Jin, X.; Muntean, B.S.; Aal-Aaboda, M.S.; Duan, Q.; Zhou, J.; Nauli, S.M. L-type calcium channel modulates cystic kidney phenotype. Biochim. Biophys. Acta 2014, 1842, 1518–1526. [Google Scholar] [CrossRef] [Green Version]
- Tamma, G.; Di Mise, A.; Ranieri, M.; Geller, A.; Tamma, R.; Zallone, A.; Valenti, G. The V2 receptor antagonist tolvaptan raises cytosolic calcium and prevents AQP2 trafficking and function: An in vitro and in vivo assessment. J. Cell Mol. Med. 2017, 21, 1767–1780. [Google Scholar] [CrossRef] [Green Version]
- FDA. FDA Drug Safety Communication: FDA Limits Duration and Usage of Samsca (Tolvaptan) Due to Possible Liver Injury Leading to Organ Transplant or Death. Available online: https://www.fda.gov/drugs/drug-safety-and-availability/fda-drug-safety-communication-fda-limits-duration-and-usage-samsca-tolvaptan-due-possible-liver (accessed on 20 September 2021).
- Meijer, E.; Gansevoort, R.T. Vasopressin V2 receptor antagonists in autosomal dominant polycystic kidney disease: Efficacy, safety, and tolerability. Kidney Int. 2020, 98, 289–293. [Google Scholar] [CrossRef]
- Hopp, K.; Hommerding, C.J.; Wang, X.; Ye, H.; Harris, P.C.; Torres, V.E. Tolvaptan plus pasireotide shows enhanced efficacy in a PKD1 model. J. Am. Soc. Nephrol. 2015, 26, 39–47. [Google Scholar] [CrossRef] [Green Version]
- Ye, H.; Wang, X.; Sussman, C.R.; Hopp, K.; Irazabal, M.V.; Bakeberg, J.L.; LaRiviere, W.B.; Manganiello, V.C.; Vorhees, C.V.; Zhao, H.; et al. Modulation of Polycystic Kidney Disease Severity by Phosphodiesterase 1 and 3 Subfamilies. J. Am. Soc. Nephrol. 2016, 27, 1312–1320. [Google Scholar] [CrossRef] [Green Version]
- Margaria, J.P.; Campa, C.C.; De Santis, M.C.; Hirsch, E.; Franco, I. The PI3K/Akt/mTOR pathway in polycystic kidney disease: A complex interaction with polycystins and primary cilium. Cell Signal. 2020, 66, 109468. [Google Scholar] [CrossRef] [PubMed]
- Ibraghimov-Beskrovnaya, O.; Natoli, T.A. mTOR signaling in polycystic kidney disease. Trends Mol. Med. 2011, 17, 625–633. [Google Scholar] [CrossRef]
- Torres, V.E.; Boletta, A.; Chapman, A.; Gattone, V.; Pei, Y.; Qian, Q.; Wallace, D.P.; Weimbs, T.; Wuthrich, R.P. Prospects for mTOR inhibitor use in patients with polycystic kidney disease and hamartomatous diseases. Clin. J. Am. Soc. Nephrol. 2010, 5, 1312–1329. [Google Scholar] [CrossRef]
- Shillingford, J.M.; Murcia, N.S.; Larson, C.H.; Low, S.H.; Hedgepeth, R.; Brown, N.; Flask, C.A.; Novick, A.C.; Goldfarb, D.A.; Kramer-Zucker, A.; et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc. Natl. Acad. Sci. USA 2006, 103, 5466–5471. [Google Scholar] [CrossRef] [Green Version]
- Guertin, D.A.; Sabatini, D.M. Defining the role of mTOR in cancer. Cancer Cell 2007, 12, 9–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sengupta, S.; Peterson, T.R.; Sabatini, D.M. Regulation of the mTOR complex 1 pathway by nutrients, growth factors, and stress. Mol. Cell 2010, 40, 310–322. [Google Scholar] [CrossRef] [Green Version]
- Boehlke, C.; Kotsis, F.; Patel, V.; Braeg, S.; Voelker, H.; Bredt, S.; Beyer, T.; Janusch, H.; Hamann, C.; Godel, M.; et al. Primary cilia regulate mTORC1 activity and cell size through Lkb1. Nat. Cell Biol. 2010, 12, 1115–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zullo, A.; Iaconis, D.; Barra, A.; Cantone, A.; Messaddeq, N.; Capasso, G.; Dolle, P.; Igarashi, P.; Franco, B. Kidney-specific inactivation of Ofd1 leads to renal cystic disease associated with upregulation of the mTOR pathway. Hum. Mol. Genet. 2010, 19, 2792–2803. [Google Scholar] [CrossRef] [Green Version]
- Bell, P.D.; Fitzgibbon, W.; Sas, K.; Stenbit, A.E.; Amria, M.; Houston, A.; Reichert, R.; Gilley, S.; Siegal, G.P.; Bissler, J.; et al. Loss of primary cilia upregulates renal hypertrophic signaling and promotes cystogenesis. J. Am. Soc. Nephrol. 2011, 22, 839–848. [Google Scholar] [CrossRef] [Green Version]
- Dere, R.; Wilson, P.D.; Sandford, R.N.; Walker, C.L. Carboxy terminal tail of polycystin-1 regulates localization of TSC2 to repress mTOR. PLoS ONE 2010, 5, e9239. [Google Scholar] [CrossRef] [PubMed]
- Chauvet, V.; Tian, X.; Husson, H.; Grimm, D.H.; Wang, T.; Hiesberger, T.; Igarashi, P.; Bennett, A.M.; Ibraghimov-Beskrovnaya, O.; Somlo, S.; et al. Mechanical stimuli induce cleavage and nuclear translocation of the polycystin-1 C terminus. J. Clin. Investig. 2004, 114, 1433–1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mekahli, D.; Decuypere, J.P.; Sammels, E.; Welkenhuyzen, K.; Schoeber, J.; Audrezet, M.P.; Corvelyn, A.; Dechenes, G.; Ong, A.C.; Wilmer, M.J.; et al. Polycystin-1 but not polycystin-2 deficiency causes upregulation of the mTOR pathway and can be synergistically targeted with rapamycin and metformin. Pflugers Arch. 2014, 466, 1591–1604. [Google Scholar] [CrossRef]
- Qin, S.; Taglienti, M.; Nauli, S.M.; Contrino, L.; Takakura, A.; Zhou, J.; Kreidberg, J.A. Failure to ubiquitinate c-Met leads to hyperactivation of mTOR signaling in a mouse model of autosomal dominant polycystic kidney disease. J. Clin. Investig. 2010, 120, 3617–3628. [Google Scholar] [CrossRef] [Green Version]
- Takiar, V.; Nishio, S.; Seo-Mayer, P.; King, J.D., Jr.; Li, H.; Zhang, L.; Karihaloo, A.; Hallows, K.R.; Somlo, S.; Caplan, M.J. Activating AMP-activated protein kinase (AMPK) slows renal cystogenesis. Proc. Natl. Acad. Sci. USA 2011, 108, 2462–2467. [Google Scholar] [CrossRef] [Green Version]
- Frew, I.J.; Thoma, C.R.; Georgiev, S.; Minola, A.; Hitz, M.; Montani, M.; Moch, H.; Krek, W. pVHL and PTEN tumour suppressor proteins cooperatively suppress kidney cyst formation. EMBO J. 2008, 27, 1747–1757. [Google Scholar] [CrossRef] [Green Version]
- Hasumi, Y.; Baba, M.; Ajima, R.; Hasumi, H.; Valera, V.A.; Klein, M.E.; Haines, D.C.; Merino, M.J.; Hong, S.B.; Yamaguchi, T.P.; et al. Homozygous loss of BHD causes early embryonic lethality and kidney tumor development with activation of mTORC1 and mTORC2. Proc. Natl. Acad. Sci. USA 2009, 106, 18722–18727. [Google Scholar] [CrossRef] [Green Version]
- Hartman, T.R.; Liu, D.; Zilfou, J.T.; Robb, V.; Morrison, T.; Watnick, T.; Henske, E.P. The tuberous sclerosis proteins regulate formation of the primary cilium via a rapamycin-insensitive and polycystin 1-independent pathway. Hum. Mol. Genet. 2009, 18, 151–163. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Zhang, T.; Wang, G.; Wang, G.; Chi, W.; Jiang, Q.; Zhang, C. GSK3beta-Dzip1-Rab8 cascade regulates ciliogenesis after mitosis. PLoS Biol. 2015, 13, e1002129. [Google Scholar] [CrossRef]
- Thoma, C.R.; Frew, I.J.; Hoerner, C.R.; Montani, M.; Moch, H.; Krek, W. pVHL and GSK3beta are components of a primary cilium-maintenance signalling network. Nat. Cell Biol. 2007, 9, 588–595. [Google Scholar] [CrossRef] [PubMed]
- Hakim, S.; Dyson, J.M.; Feeney, S.J.; Davies, E.M.; Sriratana, A.; Koenig, M.N.; Plotnikova, O.V.; Smyth, I.M.; Ricardo, S.D.; Hobbs, R.M.; et al. Inpp5e suppresses polycystic kidney disease via inhibition of PI3K/Akt-dependent mTORC1 signaling. Hum. Mol. Genet. 2016, 25, 2295–2313. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Wen, X.; Chih, B.; Nelson, C.D.; Lane, W.S.; Scales, S.J.; Jackson, P.K. TULP3 bridges the IFT-A complex and membrane phosphoinositides to promote trafficking of G protein-coupled receptors into primary cilia. Genes Dev. 2010, 24, 2180–2193. [Google Scholar] [CrossRef] [Green Version]
- Margaria, J.P.; Ratto, E.; Gozzelino, L.; Li, H.; Hirsch, E. Class II PI3Ks at the Intersection between Signal Transduction and Membrane Trafficking. Biomolecules 2019, 9, 104. [Google Scholar] [CrossRef] [Green Version]
- Canaud, G.; Knebelmann, B.; Harris, P.C.; Vrtovsnik, F.; Correas, J.M.; Pallet, N.; Heyer, C.M.; Letavernier, E.; Bienaime, F.; Thervet, E.; et al. Therapeutic mTOR inhibition in autosomal dominant polycystic kidney disease: What is the appropriate serum level? Am. J. Transplant. 2010, 10, 1701–1706. [Google Scholar] [CrossRef]
- Zafar, I.; Ravichandran, K.; Belibi, F.A.; Doctor, R.B.; Edelstein, C.L. Sirolimus attenuates disease progression in an orthologous mouse model of human autosomal dominant polycystic kidney disease. Kidney Int. 2010, 78, 754–761. [Google Scholar] [CrossRef] [Green Version]
- Natoli, T.A.; Smith, L.A.; Rogers, K.A.; Wang, B.; Komarnitsky, S.; Budman, Y.; Belenky, A.; Bukanov, N.O.; Dackowski, W.R.; Husson, H.; et al. Inhibition of glucosylceramide accumulation results in effective blockade of polycystic kidney disease in mouse models. Nat. Med. 2010, 16, 788–792. [Google Scholar] [CrossRef] [Green Version]
- Shillingford, J.M.; Piontek, K.B.; Germino, G.G.; Weimbs, T. Rapamycin ameliorates PKD resulting from conditional inactivation of Pkd1. J. Am. Soc. Nephrol. 2010, 21, 489–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gattone, V.H., 2nd; Sinders, R.M.; Hornberger, T.A.; Robling, A.G. Late progression of renal pathology and cyst enlargement is reduced by rapamycin in a mouse model of nephronophthisis. Kidney Int. 2009, 76, 178–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruggenenti, P.; Gentile, G.; Perico, N.; Perna, A.; Barcella, L.; Trillini, M.; Cortinovis, M.; Ferrer Siles, C.P.; Reyes Loaeza, J.A.; Aparicio, M.C.; et al. Effect of Sirolimus on Disease Progression in Patients with Autosomal Dominant Polycystic Kidney Disease and CKD Stages 3b-4. Clin. J. Am. Soc. Nephrol. 2016, 11, 785–794. [Google Scholar] [CrossRef] [Green Version]
- Serra, A.L.; Poster, D.; Kistler, A.D.; Krauer, F.; Raina, S.; Young, J.; Rentsch, K.M.; Spanaus, K.S.; Senn, O.; Kristanto, P.; et al. Sirolimus and kidney growth in autosomal dominant polycystic kidney disease. N. Engl. J. Med. 2010, 363, 820–829. [Google Scholar] [CrossRef] [Green Version]
- Walz, G.; Budde, K.; Mannaa, M.; Nurnberger, J.; Wanner, C.; Sommerer, C.; Kunzendorf, U.; Banas, B.; Horl, W.H.; Obermuller, N.; et al. Everolimus in patients with autosomal dominant polycystic kidney disease. N. Engl. J. Med. 2010, 363, 830–840. [Google Scholar] [CrossRef] [Green Version]
- Shillingford, J.M.; Leamon, C.P.; Vlahov, I.R.; Weimbs, T. Folate-conjugated rapamycin slows progression of polycystic kidney disease. J. Am. Soc. Nephrol. 2012, 23, 1674–1681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravichandran, K.; Zafar, I.; Ozkok, A.; Edelstein, C.L. An mTOR kinase inhibitor slows disease progression in a rat model of polycystic kidney disease. Nephrol. Dial. Transplant. 2015, 30, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S.; Avizonis, D.; Reczek, C.R.; Weinberg, S.E.; Menz, S.; Neuhaus, R.; Christian, S.; Haegebarth, A.; Algire, C.; Pollak, M. Are Metformin Doses Used in Murine Cancer Models Clinically Relevant? Cell Metab. 2016, 23, 569–570. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Wondisford, F.E. Metformin action: Concentrations matter. Cell Metab. 2015, 21, 159–162. [Google Scholar] [CrossRef] [Green Version]
- Leonhard, W.N.; Song, X.; Kanhai, A.A.; Iliuta, I.A.; Bozovic, A.; Steinberg, G.R.; Peters, D.J.M.; Pei, Y. Salsalate, but not metformin or canagliflozin, slows kidney cyst growth in an adult-onset mouse model of polycystic kidney disease. EBioMedicine 2019, 47, 436–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, D.A. The Jak/STAT pathway. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Shea, J.J.; Schwartz, D.M.; Villarino, A.V.; Gadina, M.; McInnes, I.B.; Laurence, A. The JAK-STAT pathway: Impact on human disease and therapeutic intervention. Annu. Rev. Med. 2015, 66, 311–328. [Google Scholar] [CrossRef] [Green Version]
- Chuang, P.Y.; He, J.C. JAK/STAT signaling in renal diseases. Kidney Int. 2010, 78, 231–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patera, F.; Cudzich-Madry, A.; Huang, Z.; Fragiadaki, M. Renal expression of JAK2 is high in polycystic kidney disease and its inhibition reduces cystogenesis. Sci. Rep. 2019, 9, 4491. [Google Scholar] [CrossRef]
- Gardner, K.D., Jr.; Burnside, J.S.; Elzinga, L.W.; Locksley, R.M. Cytokines in fluids from polycystic kidneys. Kidney Int. 1991, 39, 718–724. [Google Scholar] [CrossRef] [Green Version]
- Mrug, M.; Zhou, J.; Woo, Y.; Cui, X.; Szalai, A.J.; Novak, J.; Churchill, G.A.; Guay-Woodford, L.M. Overexpression of innate immune response genes in a model of recessive polycystic kidney disease. Kidney Int. 2008, 73, 63–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karihaloo, A.; Koraishy, F.; Huen, S.C.; Lee, Y.; Merrick, D.; Caplan, M.J.; Somlo, S.; Cantley, L.G. Macrophages promote cyst growth in polycystic kidney disease. J. Am. Soc. Nephrol. 2011, 22, 1809–1814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Huen, S.; Nishio, H.; Nishio, S.; Lee, H.K.; Choi, B.S.; Ruhrberg, C.; Cantley, L.G. Distinct macrophage phenotypes contribute to kidney injury and repair. J. Am. Soc. Nephrol. 2011, 22, 317–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talbot, J.J.; Shillingford, J.M.; Vasanth, S.; Doerr, N.; Mukherjee, S.; Kinter, M.T.; Watnick, T.; Weimbs, T. Polycystin-1 regulates STAT activity by a dual mechanism. Proc. Natl. Acad. Sci. USA 2011, 108, 7985–7990. [Google Scholar] [CrossRef] [Green Version]
- Low, S.H.; Vasanth, S.; Larson, C.H.; Mukherjee, S.; Sharma, N.; Kinter, M.T.; Kane, M.E.; Obara, T.; Weimbs, T. Polycystin-1, STAT6, and P100 function in a pathway that transduces ciliary mechanosensation and is activated in polycystic kidney disease. Dev. Cell 2006, 10, 57–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhunia, A.K.; Piontek, K.; Boletta, A.; Liu, L.; Qian, F.; Xu, P.N.; Germino, F.J.; Germino, G.G. PKD1 induces p21(waf1) and regulation of the cell cycle via direct activation of the JAK-STAT signaling pathway in a process requiring PKD2. Cell 2002, 109, 157–168. [Google Scholar] [CrossRef] [Green Version]
- Fragiadaki, M.; Lannoy, M.; Themanns, M.; Maurer, B.; Leonhard, W.N.; Peters, D.J.; Moriggl, R.; Ong, A.C. STAT5 drives abnormal proliferation in autosomal dominant polycystic kidney disease. Kidney Int. 2017, 91, 575–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.H.; Weiss, R.H. GHetting to know ADPKD proliferative signaling, STAT. Kidney Int. 2017, 91, 524–526. [Google Scholar] [CrossRef] [Green Version]
- Cassini, M.F.; Kakade, V.R.; Kurtz, E.; Sulkowski, P.; Glazer, P.; Torres, R.; Somlo, S.; Cantley, L.G. Mcp1 Promotes Macrophage-Dependent Cyst Expansion in Autosomal Dominant Polycystic Kidney Disease. J. Am. Soc. Nephrol. 2018, 29, 2471–2481. [Google Scholar] [CrossRef] [Green Version]
- Zheng, D.; Wolfe, M.; Cowley, B.D., Jr.; Wallace, D.P.; Yamaguchi, T.; Grantham, J.J. Urinary excretion of monocyte chemoattractant protein-1 in autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 2003, 14, 2588–2595. [Google Scholar] [CrossRef] [Green Version]
- Anastas, J.N.; Moon, R.T. WNT signalling pathways as therapeutic targets in cancer. Nat. Rev. Cancer 2013, 13, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Benzing, T.; Simons, M.; Walz, G. Wnt signaling in polycystic kidney disease. J. Am. Soc. Nephrol. 2007, 18, 1389–1398. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/β-catenin signaling: Components, mechanisms, and diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, E.C.; Katsanis, N. Context-dependent regulation of Wnt signaling through the primary cilium. J. Am. Soc. Nephrol. 2013, 24, 10–18. [Google Scholar] [CrossRef] [Green Version]
- Behrens, J.; von Kries, J.P.; Kuhl, M.; Bruhn, L.; Wedlich, D.; Grosschedl, R.; Birchmeier, W. Functional interaction of β-catenin with the transcription factor LEF-1. Nature 1996, 382, 638–642. [Google Scholar] [CrossRef]
- Molenaar, M.; van de Wetering, M.; Oosterwegel, M.; Peterson-Maduro, J.; Godsave, S.; Korinek, V.; Roose, J.; Destree, O.; Clevers, H. XTcf-3 transcription factor mediates β-catenin-induced axis formation in Xenopus embryos. Cell 1996, 86, 391–399. [Google Scholar] [CrossRef] [Green Version]
- Stamos, J.L.; Weis, W.I. The β-catenin destruction complex. Cold Spring Harb. Perspect. Biol. 2013, 5, a007898. [Google Scholar] [CrossRef]
- Butler, M.T.; Wallingford, J.B. Planar cell polarity in development and disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 375–388. [Google Scholar] [CrossRef]
- Fischer, E.; Legue, E.; Doyen, A.; Nato, F.; Nicolas, J.F.; Torres, V.; Yaniv, M.; Pontoglio, M. Defective planar cell polarity in polycystic kidney disease. Nat. Genet. 2006, 38, 21–23. [Google Scholar] [CrossRef]
- Matakatsu, H.; Blair, S.S. Interactions between Fat and Dachsous and the regulation of planar cell polarity in the Drosophila wing. Development 2004, 131, 3785–3794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, S.; Kim, S.; Yoon, J.; Adler, P.N.; Yim, J. The balance between the novel protein target of wingless and the Drosophila Rho-associated kinase pathway regulates planar cell polarity in the Drosophila wing. Genetics 2007, 176, 891–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodrich, L.V.; Strutt, D. Principles of planar polarity in animal development. Development 2011, 138, 1877–1892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davey, C.F.; Moens, C.B. Planar cell polarity in moving cells: Think globally, act locally. Development 2017, 144, 187–200. [Google Scholar] [CrossRef] [Green Version]
- Minegishi, K.; Hashimoto, M.; Ajima, R.; Takaoka, K.; Shinohara, K.; Ikawa, Y.; Nishimura, H.; McMahon, A.P.; Willert, K.; Okada, Y.; et al. A Wnt5 Activity Asymmetry and Intercellular Signaling via PCP Proteins Polarize Node Cells for Left-Right Symmetry Breaking. Dev. Cell 2017, 40, 439–452.e434. [Google Scholar] [CrossRef] [Green Version]
- Lancaster, M.A.; Schroth, J.; Gleeson, J.G. Subcellular spatial regulation of canonical Wnt signalling at the primary cilium. Nat Cell Biol. 2011, 13, 700–707. [Google Scholar] [CrossRef] [Green Version]
- Huang, P.; Schier, A.F. Dampened Hedgehog signaling but normal Wnt signaling in zebrafish without cilia. Development 2009, 136, 3089–3098. [Google Scholar] [CrossRef] [Green Version]
- Lancaster, M.A.; Gleeson, J.G. Cystic kidney disease: The role of Wnt signaling. Trends Mol. Med. 2010, 16, 349–360. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhou, C.J.; Liu, Y. Wnt Signaling in Kidney Development and Disease. Prog. Mol. Biol. Transl. Sci. 2018, 153, 181–207. [Google Scholar] [CrossRef]
- Zhou, D.; Tan, R.J.; Fu, H.; Liu, Y. Wnt/β-catenin signaling in kidney injury and repair: A double-edged sword. Lab. Investig. 2016, 96, 156–167. [Google Scholar] [CrossRef] [Green Version]
- Kawakami, T.; Ren, S.; Duffield, J.S. Wnt signalling in kidney diseases: Dual roles in renal injury and repair. J. Pathol. 2013, 229, 221–231. [Google Scholar] [CrossRef]
- Sugiyama, N.; Tsukiyama, T.; Yamaguchi, T.P.; Yokoyama, T. The canonical Wnt signaling pathway is not involved in renal cyst development in the kidneys of inv mutant mice. Kidney Int. 2011, 79, 957–965. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, T.; Saito, D.; Kawasumi, A.; Shinohara, K.; Asai, Y.; Takaoka, K.; Dong, F.; Takamatsu, A.; Belo, J.A.; Mochizuki, A.; et al. Fluid flow and interlinked feedback loops establish left-right asymmetric decay of Cerl2 mRNA. Nat. Commun. 2012, 3, 1322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karner, C.M.; Chirumamilla, R.; Aoki, S.; Igarashi, P.; Wallingford, J.B.; Carroll, T.J. Wnt9b signaling regulates planar cell polarity and kidney tubule morphogenesis. Nat. Genet. 2009, 41, 793–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saburi, S.; Hester, I.; Fischer, E.; Pontoglio, M.; Eremina, V.; Gessler, M.; Quaggin, S.E.; Harrison, R.; Mount, R.; McNeill, H. Loss of Fat4 disrupts PCP signaling and oriented cell division and leads to cystic kidney disease. Nat. Genet. 2008, 40, 1010–1015. [Google Scholar] [CrossRef]
- Mao, Y.; Mulvaney, J.; Zakaria, S.; Yu, T.; Morgan, K.M.; Allen, S.; Basson, M.A.; Francis-West, P.; Irvine, K.D. Characterization of a Dchs1 mutant mouse reveals requirements for Dchs1-Fat4 signaling during mammalian development. Development 2011, 138, 947–957. [Google Scholar] [CrossRef] [Green Version]
- Kunimoto, K.; Bayly, R.D.; Vladar, E.K.; Vonderfecht, T.; Gallagher, A.R.; Axelrod, J.D. Disruption of Core Planar Cell Polarity Signaling Regulates Renal Tubule Morphogenesis but Is Not Cystogenic. Curr. Biol. 2017, 27, 3120–3131.e3124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simons, M.; Walz, G. Polycystic kidney disease: Cell division without a c(l)ue? Kidney Int. 2006, 70, 854–864. [Google Scholar] [CrossRef] [Green Version]
- Hildebrandt, F.; Benzing, T.; Katsanis, N. Ciliopathies. N. Engl. J. Med. 2011, 364, 1533–1543. [Google Scholar] [CrossRef] [Green Version]
- De, A. Wnt/Ca2+ signaling pathway: A brief overview. Acta Biochim. Biophys. Sin. (Shanghai) 2011, 43, 745–756. [Google Scholar] [CrossRef]
- Briscoe, J.; Therond, P.P. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat. Rev. Mol. Cell Biol. 2013, 14, 416–429. [Google Scholar] [CrossRef]
- Rohatgi, R.; Milenkovic, L.; Scott, M.P. Patched1 regulates hedgehog signaling at the primary cilium. Science 2007, 317, 372–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corbit, K.C.; Aanstad, P.; Singla, V.; Norman, A.R.; Stainier, D.Y.; Reiter, J.F. Vertebrate Smoothened functions at the primary cilium. Nature 2005, 437, 1018–1021. [Google Scholar] [CrossRef] [PubMed]
- Niewiadomski, P.; Kong, J.H.; Ahrends, R.; Ma, Y.; Humke, E.W.; Khan, S.; Teruel, M.N.; Novitch, B.G.; Rohatgi, R. Gli protein activity is controlled by multisite phosphorylation in vertebrate Hedgehog signaling. Cell Rep. 2014, 6, 168–181. [Google Scholar] [CrossRef] [Green Version]
- Haycraft, C.J.; Banizs, B.; Aydin-Son, Y.; Zhang, Q.; Michaud, E.J.; Yoder, B.K. Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet. 2005, 1, e53. [Google Scholar] [CrossRef] [PubMed]
- Christensen, S.T.; Ott, C.M. Cell signaling. A ciliary signaling switch. Science 2007, 317, 330–331. [Google Scholar] [CrossRef]
- Eggenschwiler, J.T.; Anderson, K.V. Cilia and developmental signaling. Annu. Rev. Cell Dev. Biol 2007, 23, 345–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eguether, T.; San Agustin, J.T.; Keady, B.T.; Jonassen, J.A.; Liang, Y.; Francis, R.; Tobita, K.; Johnson, C.A.; Abdelhamed, Z.A.; Lo, C.W.; et al. IFT27 links the BBSome to IFT for maintenance of the ciliary signaling compartment. Dev. Cell 2014, 31, 279–290. [Google Scholar] [CrossRef] [Green Version]
- Keady, B.T.; Samtani, R.; Tobita, K.; Tsuchya, M.; San Agustin, J.T.; Follit, J.A.; Jonassen, J.A.; Subramanian, R.; Lo, C.W.; Pazour, G.J. IFT25 links the signal-dependent movement of Hedgehog components to intraflagellar transport. Dev. Cell 2012, 22, 940–951. [Google Scholar] [CrossRef] [Green Version]
- Liew, G.M.; Ye, F.; Nager, A.R.; Murphy, J.P.; Lee, J.S.; Aguiar, M.; Breslow, D.K.; Gygi, S.P.; Nachury, M.V. The intraflagellar transport protein IFT27 promotes BBSome exit from cilia through the GTPase ARL6/BBS3. Dev. Cell 2014, 31, 265–278. [Google Scholar] [CrossRef] [Green Version]
- Hu, M.C.; Mo, R.; Bhella, S.; Wilson, C.W.; Chuang, P.T.; Hui, C.C.; Rosenblum, N.D. GLI3-dependent transcriptional repression of Gli1, Gli2 and kidney patterning genes disrupts renal morphogenesis. Development 2006, 133, 569–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cain, J.E.; Islam, E.; Haxho, F.; Chen, L.; Bridgewater, D.; Nieuwenhuis, E.; Hui, C.C.; Rosenblum, N.D. GLI3 repressor controls nephron number via regulation of Wnt11 and Ret in ureteric tip cells. PLoS ONE 2009, 4, e7313. [Google Scholar] [CrossRef] [Green Version]
- Cain, J.E.; Islam, E.; Haxho, F.; Blake, J.; Rosenblum, N.D. GLI3 repressor controls functional development of the mouse ureter. J. Clin. Investig. 2011, 121, 1199–1206. [Google Scholar] [CrossRef] [Green Version]
- Cain, J.E.; Rosenblum, N.D. Control of mammalian kidney development by the Hedgehog signaling pathway. Pediatr. Nephrol. 2011, 26, 1365–1371. [Google Scholar] [CrossRef]
- Song, X.; Di Giovanni, V.; He, N.; Wang, K.; Ingram, A.; Rosenblum, N.D.; Pei, Y. Systems biology of autosomal dominant polycystic kidney disease (ADPKD): Computational identification of gene expression pathways and integrated regulatory networks. Hum. Mol. Genet. 2009, 18, 2328–2343. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Rauhauser, A.A.; Dai, J.; Sakthivel, R.; Igarashi, P.; Jetten, A.M.; Attanasio, M. Increased hedgehog signaling in postnatal kidney results in aberrant activation of nephron developmental programs. Hum. Mol. Genet. 2011, 20, 4155–4166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, P.V.; Haycraft, C.J.; Besschetnova, T.Y.; Turbe-Doan, A.; Stottmann, R.W.; Herron, B.J.; Chesebro, A.L.; Qiu, H.; Scherz, P.J.; Shah, J.V.; et al. THM1 negatively modulates mouse sonic hedgehog signal transduction and affects retrograde intraflagellar transport in cilia. Nat. Genet. 2008, 40, 403–410. [Google Scholar] [CrossRef] [Green Version]
- Jonassen, J.A.; SanAgustin, J.; Baker, S.P.; Pazour, G.J. Disruption of IFT complex A causes cystic kidneys without mitotic spindle misorientation. J. Am. Soc. Nephrol. 2012, 23, 641–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, P.V.; Talbott, G.C.; Turbe-Doan, A.; Jacobs, D.T.; Schonfeld, M.P.; Silva, L.M.; Chatterjee, A.; Prysak, M.; Allard, B.A.; Beier, D.R. Downregulating hedgehog signaling reduces renal cystogenic potential of mouse models. J. Am. Soc. Nephrol. 2014, 25, 2201–2212. [Google Scholar] [CrossRef]
- Chan, S.K.; Riley, P.R.; Price, K.L.; McElduff, F.; Winyard, P.J.; Welham, S.J.; Woolf, A.S.; Long, D.A. Corticosteroid-induced kidney dysmorphogenesis is associated with deregulated expression of known cystogenic molecules, as well as Indian hedgehog. Am. J. Physiol. Renal. Physiol. 2010, 298, F346–F356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, M.; Legue, E.; Tian, X.; Somlo, S.; Liem, K.F., Jr. Cell-Autonomous Hedgehog Signaling Is Not Required for Cyst Formation in Autosomal Dominant Polycystic Kidney Disease. J. Am. Soc. Nephrol. 2019, 30, 2103–2111. [Google Scholar] [CrossRef]
- Yu, F.X.; Guan, K.L. The Hippo pathway: Regulators and regulations. Genes Dev. 2013, 27, 355–371. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.L.; Choi, S.H.; Mo, J.S. Role of the Hippo Pathway in Fibrosis and Cancer. Cells 2019, 8, 468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, W.; Jho, E.H. The history and regulatory mechanism of the Hippo pathway. BMB Rep. 2018, 51, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Muller, R.U.; Schermer, B. Hippo signaling-a central player in cystic kidney disease? Pediatr. Nephrol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Guan, K.L. Polycystic kidney disease: A Hippo connection. Genes Dev. 2018, 32, 737–739. [Google Scholar] [CrossRef]
- Habbig, S.; Bartram, M.P.; Muller, R.U.; Schwarz, R.; Andriopoulos, N.; Chen, S.; Sagmuller, J.G.; Hoehne, M.; Burst, V.; Liebau, M.C.; et al. NPHP4, a cilia-associated protein, negatively regulates the Hippo pathway. J. Cell Biol. 2011, 193, 633–642. [Google Scholar] [CrossRef] [Green Version]
- Habbig, S.; Bartram, M.P.; Sagmuller, J.G.; Griessmann, A.; Franke, M.; Muller, R.U.; Schwarz, R.; Hoehne, M.; Bergmann, C.; Tessmer, C.; et al. The ciliopathy disease protein NPHP9 promotes nuclear delivery and activation of the oncogenic transcriptional regulator TAZ. Hum. Mol. Genet. 2012, 21, 5528–5538. [Google Scholar] [CrossRef] [Green Version]
- Cai, J.; Song, X.; Wang, W.; Watnick, T.; Pei, Y.; Qian, F.; Pan, D. A RhoA-YAP-c-Myc signaling axis promotes the development of polycystic kidney disease. Genes Dev. 2018, 32, 781–793. [Google Scholar] [CrossRef] [Green Version]
- Reginensi, A.; Scott, R.P.; Gregorieff, A.; Bagherie-Lachidan, M.; Chung, C.; Lim, D.S.; Pawson, T.; Wrana, J.; McNeill, H. Yap- and Cdc42-dependent nephrogenesis and morphogenesis during mouse kidney development. PLoS Genet. 2013, 9, e1003380. [Google Scholar] [CrossRef] [Green Version]
- Kai, T.; Tsukamoto, Y.; Hijiya, N.; Tokunaga, A.; Nakada, C.; Uchida, T.; Daa, T.; Iha, H.; Takahashi, M.; Nomura, T.; et al. Kidney-specific knockout of Sav1 in the mouse promotes hyperproliferation of renal tubular epithelium through suppression of the Hippo pathway. J. Pathol. 2016, 239, 97–108. [Google Scholar] [CrossRef]
- Tian, Y.; Kolb, R.; Hong, J.H.; Carroll, J.; Li, D.; You, J.; Bronson, R.; Yaffe, M.B.; Zhou, J.; Benjamin, T. TAZ promotes PC2 degradation through a SCFbeta-Trcp E3 ligase complex. Mol. Cell Biol. 2007, 27, 6383–6395. [Google Scholar] [CrossRef] [Green Version]
- Park, H.W.; Guan, K.L. Regulation of the Hippo pathway and implications for anticancer drug development. Trends Pharmacol. Sci. 2013, 34, 581–589. [Google Scholar] [CrossRef] [Green Version]
- Johnson, C.A.; Collis, S.J. Ciliogenesis and the DNA damage response: A stressful relationship. Cilia 2016, 5, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, P.K. Nek8 couples renal ciliopathies to DNA damage and checkpoint control. Mol. Cell 2013, 51, 407–408. [Google Scholar] [CrossRef] [Green Version]
- Awasthi, P.; Foiani, M.; Kumar, A. ATM and ATR signaling at a glance. J. Cell Sci. 2015, 128, 4255–4262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef]
- Mullee, L.I.; Morrison, C.G. Centrosomes in the DNA damage response--the hub outside the centre. Chromosome Res. 2016, 24, 35–51. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, D.; Feijoo, P.; Bernal, A.; Ercilla, A.; Agell, N.; Genesca, A.; Tusell, L. Centrosome aberrations in human mammary epithelial cells driven by cooperative interactions between p16INK4a deficiency and telomere-dependent genotoxic stress. Oncotarget 2015, 6, 28238–28256. [Google Scholar] [CrossRef] [Green Version]
- Katsura, M.; Tsuruga, T.; Date, O.; Yoshihara, T.; Ishida, M.; Tomoda, Y.; Okajima, M.; Takaku, M.; Kurumizaka, H.; Kinomura, A.; et al. The ATR-Chk1 pathway plays a role in the generation of centrosome aberrations induced by Rad51C dysfunction. Nucleic Acids Res. 2009, 37, 3959–3968. [Google Scholar] [CrossRef] [Green Version]
- Mahjoub, M.R.; Stearns, T. Supernumerary centrosomes nucleate extra cilia and compromise primary cilium signaling. Curr. Biol. 2012, 22, 1628–1634. [Google Scholar] [CrossRef] [Green Version]
- Kodani, A.; Yu, T.W.; Johnson, J.R.; Jayaraman, D.; Johnson, T.L.; Al-Gazali, L.; Sztriha, L.; Partlow, J.N.; Kim, H.; Krup, A.L.; et al. Centriolar satellites assemble centrosomal microcephaly proteins to recruit CDK2 and promote centriole duplication. Elife 2015, 4. [Google Scholar] [CrossRef] [Green Version]
- Airik, R.; Slaats, G.G.; Guo, Z.; Weiss, A.C.; Khan, N.; Ghosh, A.; Hurd, T.W.; Bekker-Jensen, S.; Schroder, J.M.; Elledge, S.J.; et al. Renal-retinal ciliopathy gene Sdccag8 regulates DNA damage response signaling. J. Am. Soc. Nephrol. 2014, 25, 2573–2583. [Google Scholar] [CrossRef] [Green Version]
- Grampa, V.; Delous, M.; Zaidan, M.; Odye, G.; Thomas, S.; Elkhartoufi, N.; Filhol, E.; Niel, O.; Silbermann, F.; Lebreton, C.; et al. Novel NEK8 Mutations Cause Severe Syndromic Renal Cystic Dysplasia through YAP Dysregulation. PLoS Genet. 2016, 12, e1005894. [Google Scholar] [CrossRef]
- Stiff, T.; Casar Tena, T.; O’Driscoll, M.; Jeggo, P.A.; Philipp, M. ATR promotes cilia signalling: Links to developmental impacts. Hum. Mol. Genet. 2016, 25, 1574–1587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Mazzanti, M.; Mistrik, M.; Kosar, M.; Beznoussenko, G.V.; Mironov, A.A.; Garre, M.; Parazzoli, D.; Shivashankar, G.V.; Scita, G.; et al. ATR mediates a checkpoint at the nuclear envelope in response to mechanical stress. Cell 2014, 158, 633–646. [Google Scholar] [CrossRef] [Green Version]
- Ma, M.; Gallagher, A.R.; Somlo, S. Ciliary Mechanisms of Cyst Formation in Polycystic Kidney Disease. Cold Spring Harb. Perspect. Biol. 2017, 9. [Google Scholar] [CrossRef] [Green Version]
- Ma, M.; Tian, X.; Igarashi, P.; Pazour, G.J.; Somlo, S. Loss of cilia suppresses cyst growth in genetic models of autosomal dominant polycystic kidney disease. Nat. Genet. 2013, 45, 1004–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hildebrandt, F.; Strahm, B.; Nothwang, H.G.; Gretz, N.; Schnieders, B.; Singh-Sawhney, I.; Kutt, R.; Vollmer, M.; Brandis, M. Molecular genetic identification of families with juvenile nephronophthisis type 1: Rate of progression to renal failure. Kidney Int. 1997, 51, 261–269. [Google Scholar] [CrossRef] [Green Version]
- Caliskan, Y.; Gharavi, A.G. Working Out Nephronophthisis Genetics One Family at a Time. J. Am. Soc. Nephrol. 2013, 24, 865–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- König, J.; Kranz, B.; König, S.; Schlingmann, K.P.; Titieni, A.; Tönshoff, B.; Habbig, S.; Pape, L.; Häffner, K.; Hansen, M.; et al. Phenotypic Spectrum of Children with Nephronophthisis and Related Ciliopathies. CJASN 2017, 12, 1974–1983. [Google Scholar] [CrossRef]
- Mallawaarachchi, A.; Mallett, A.; Sawyer, A.; McCarthy, H.; Fletcher, J.; Chapman, J.; Bennetts, B.; Ho, G.; Jueppner, H.; Hahn, D.; et al. Utilising exome sequencing to identify nephronophthisis mutations within an australian clinical cohort. Nephrology 2014, 19, 71. [Google Scholar]
- Jayasinghe, K.; Quinlan, C.; Stark, Z.; Patel, C.; Mallawaarachchi, A.; Wardrop, L.; Kerr, P.G.; Trnka, P.; Mallett, A.J.; Collaborative, K. Renal genetics in Australia: Kidney medicine in the genomic age. Nephrology (Carlton) 2019, 24, 279–286. [Google Scholar] [CrossRef] [Green Version]
- Mallett, A.J.; McCarthy, H.J.; Ho, G.; Holman, K.; Farnsworth, E.; Patel, C.; Fletcher, J.T.; Mallawaarachchi, A.; Quinlan, C.; Bennetts, B.; et al. Massively parallel sequencing and targeted exomes in familial kidney disease can diagnose underlying genetic disorders. Kidney Int. 2017, 92, 1493–1506. [Google Scholar] [CrossRef] [PubMed]
- Braun, D.A.; Schueler, M.; Halbritter, J.; Gee, H.Y.; Porath, J.D.; Lawson, J.A.; Airik, R.; Shril, S.; Allen, S.J.; Stein, D.; et al. Whole exome sequencing identifies causative mutations in the majority of consanguineous or familial cases with childhood-onset increased renal echogenicity. Kidney Int. 2016, 89, 468–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaff, C.L.; Winship, I.M.; Forrest, S.M.; Hansen, D.P.; Clark, J.; Waring, P.M.; South, M.; Sinclair, A.H. Preparing for genomic medicine: A real world demonstration of health system change. NPJ Genom. Med. 2017, 2017, 16. [Google Scholar] [CrossRef]
- Renkema, K.Y.; Giles, R.H.; Lilien, M.R.; Beales, P.L.; Roepman, R.; Oud, M.M.; Arts, H.H.; Knoers, N.V.A.M. The KOUNCIL consortium: From genetic defects to therapeutic development for nephronophthisis. Front. Pediatr. 2018, 6, 131. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Liu, C.; Liu, X.; Chen, J.; Fan, X.; Liu, J.; Ma, D.; Cao, G.; Chen, Z.; Xu, D.; et al. Phenotype and genotype spectra of a Chinese cohort with nephronophthisis-related ciliopathy. Med. Genet. 2020. [Google Scholar] [CrossRef]
- Stokman, M.F.; van der Zwaag, B.; van de Kar, N.C.A.J.; van Haelst, M.M.; van Eerde, A.M.; van der Heijden, J.W.; Kroes, H.Y.; Ippel, E.; Schulp, A.J.A.; van Gassen, K.L.; et al. Clinical and genetic analyses of a Dutch cohort of 40 patients with a nephronophthisis-related ciliopathy. Pediatr. Nephrol. 2018, 33, 1701–1712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapman, A.B.; Devuyst, O.; Eckardt, K.; Gansevoort, R.T.; Harris, T.; Horie, S.; Kasiske, B.L.; Odland, D.; Pei, Y.; Perrone, R.D.; et al. Autosomal-dominant polycystic kidney disease (ADPKD): Executive summary from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2015, 88, 17–27. [Google Scholar] [CrossRef] [Green Version]
- Torres, V.E.; Chapman, A.B.; Devuyst, O.; Gansevoort, R.T.; Grantham, J.J.; Higashihara, E.; Perrone, R.D.; Krasa, H.B.; Ouyang, J.; Czerwiec, F.S.; et al. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N. Engl. J. Med. 2012, 367, 2407–2418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forsythe, E.; Kenny, J.; Bacchelli, C.; Beales, P.L. Managing Bardet-Biedl Syndrome—Now and in the future. Front. Pediatr. 2018, 6, 23. [Google Scholar] [CrossRef] [Green Version]
- Amirou, M.; Bourdat-Michel, G.; Pinel, N.; Huet, G.; Gaultier, J.; Cochat, P. Brief report: Successful renal transplantation in Jeune syndrome type 2. Pediatr. Nephrol 1998, 12, 293–294. [Google Scholar] [CrossRef] [PubMed]
- Ramadani, H.M.; Nasrat, H.A. Prenatal diagnosis of recurrent Meckel syndrome. Int. J. Gynaecol. Obstet. 1992, 39, 327–332. [Google Scholar] [CrossRef]
- Hamiwka, L.A.; Midgley, J.P.; Wade, A.W.; Martz, K.L.; Grisaru, S. Outcomes of kidney transplantation in children with nephronophthisis: An analysis of the North American Pediatric Renal Trials and Collaborative Studies (NAPRTCS) Registry. Pediatr. Transplant. 2008, 12, 878–882. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Mo, H.; Chung, C.T.Y.; Kim, H.K.; Ko, H.; Han, A.; Min, S.; Ha, J. Long-term survival of kidney transplants in pediatric patients with nephronophthisis. Transplantation 2020, 104, S554. [Google Scholar] [CrossRef]
- Collin, R.W.; Den Hollander, A.I.; Der Velde-Visser, S.D.V.; Bennicelli, J.; Bennett, J.; Cremers, F.P. Antisense oligonucleotide (AON)-based therapy for leber congenital amaurosis caused by a frequent mutation in CEP290. Mol. Ther. Nucleic Acids 2012, 1, e14. [Google Scholar] [CrossRef]
- Cideciyan, A.V.; Jacobson, S.G.; Drack, A.V.; Ho, A.C.; Charng, J.; Garafalo, A.V.; Roman, A.J.; Sumaroka, A.; Han, I.C.; Hochstedler, M.D.; et al. Effect of an intravitreal antisense oligonucleotide on vision in Leber congenital amaurosis due to a photoreceptor cilium defect. Nat. Med. 2019, 25, 225–228. [Google Scholar] [CrossRef]
- Vertii, A.; Bright, A.; Delaval, B.; Hehnly, H.; Doxsey, S. New frontiers: Discovering cilia-independent functions of cilia proteins. EMBO Rep. 2015, 16, 1275–1287. [Google Scholar] [CrossRef] [Green Version]
- Hua, K.; Ferland, R.J. Primary cilia proteins: Ciliary and extraciliary sites and functions. Cell Mol. Life Sci. 2018, 75, 1521–1540. [Google Scholar] [CrossRef]












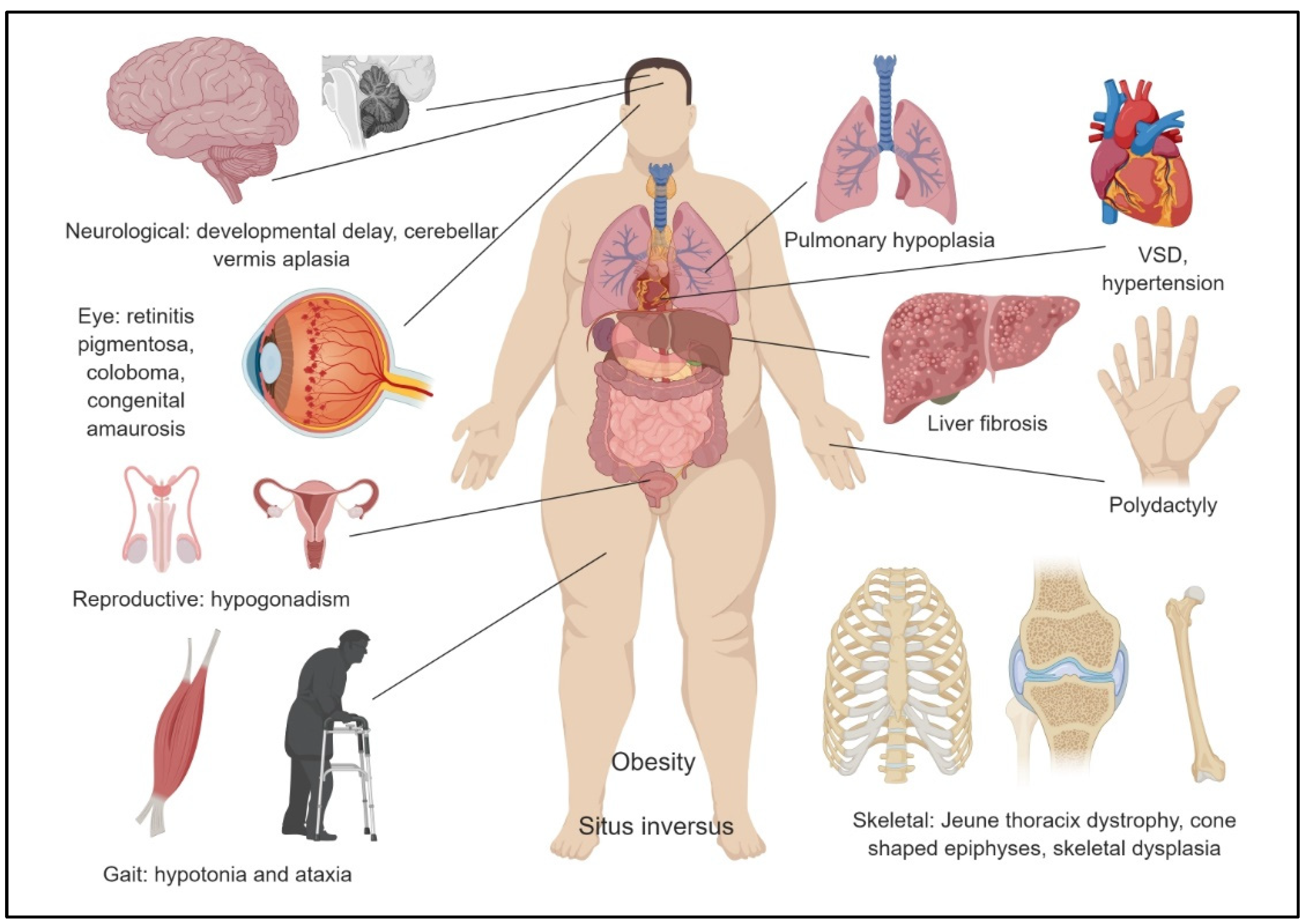{kind=link}
{kind=link}
{kind=link}
{kind=link}
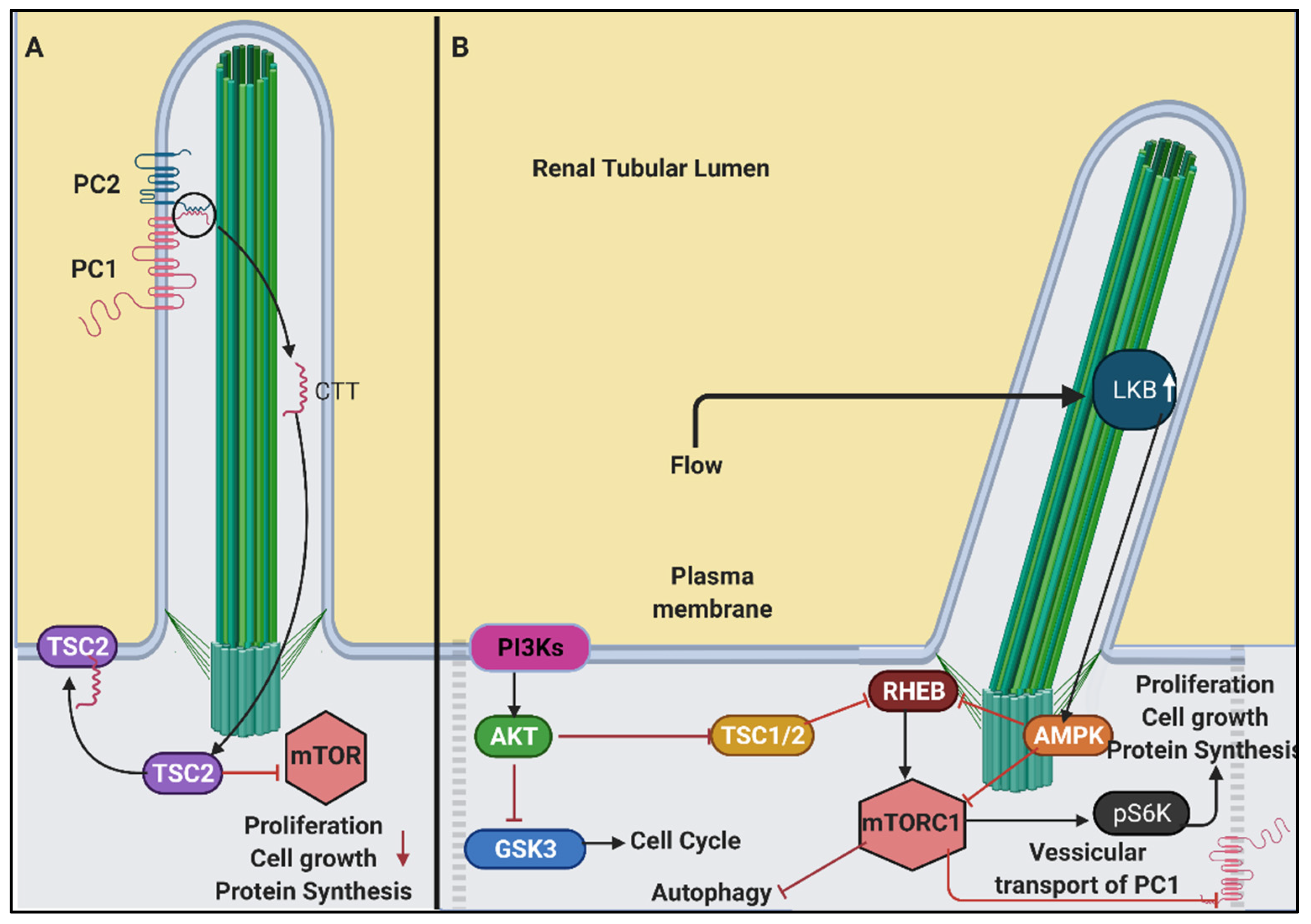{kind=link}
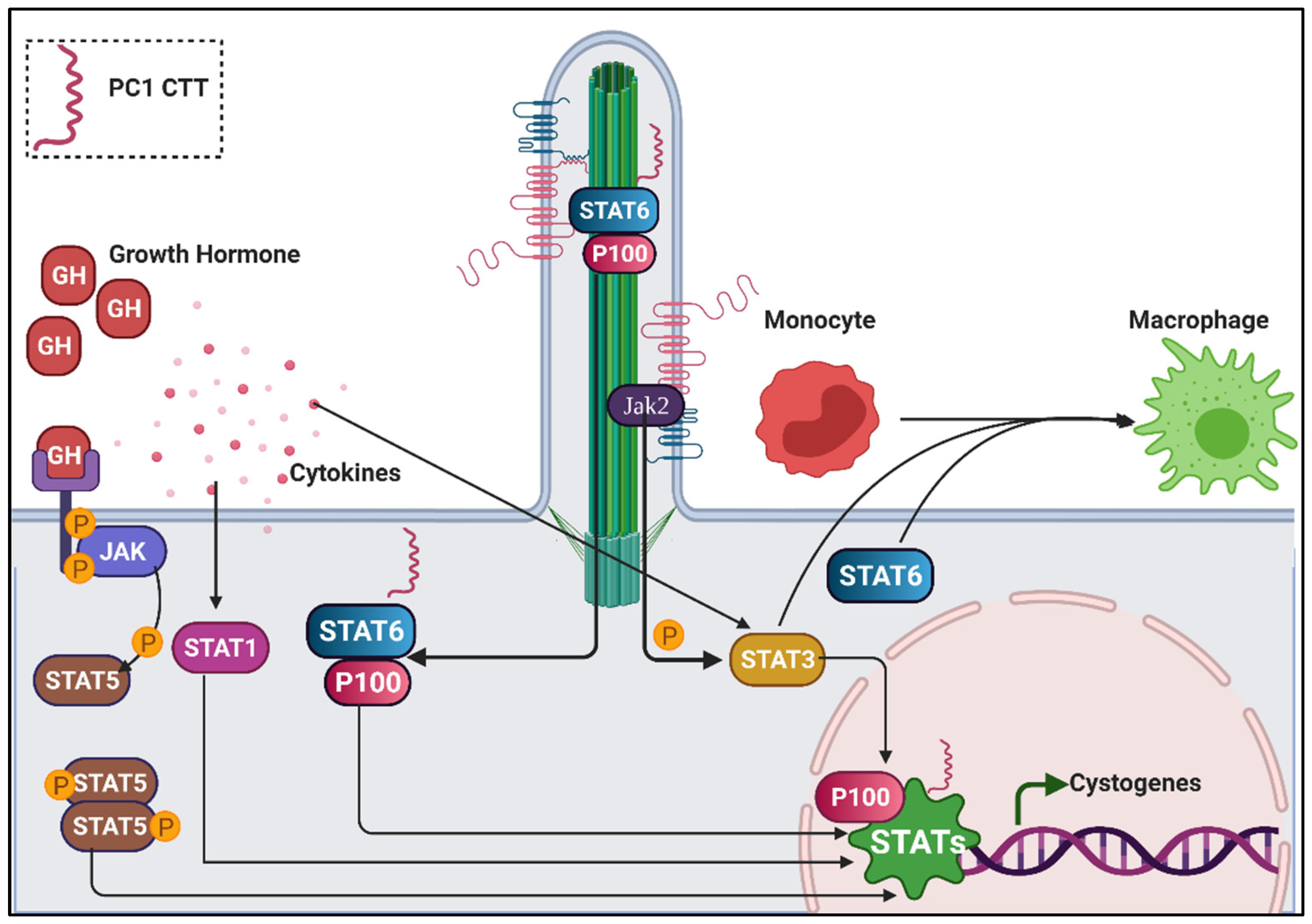{kind=link}
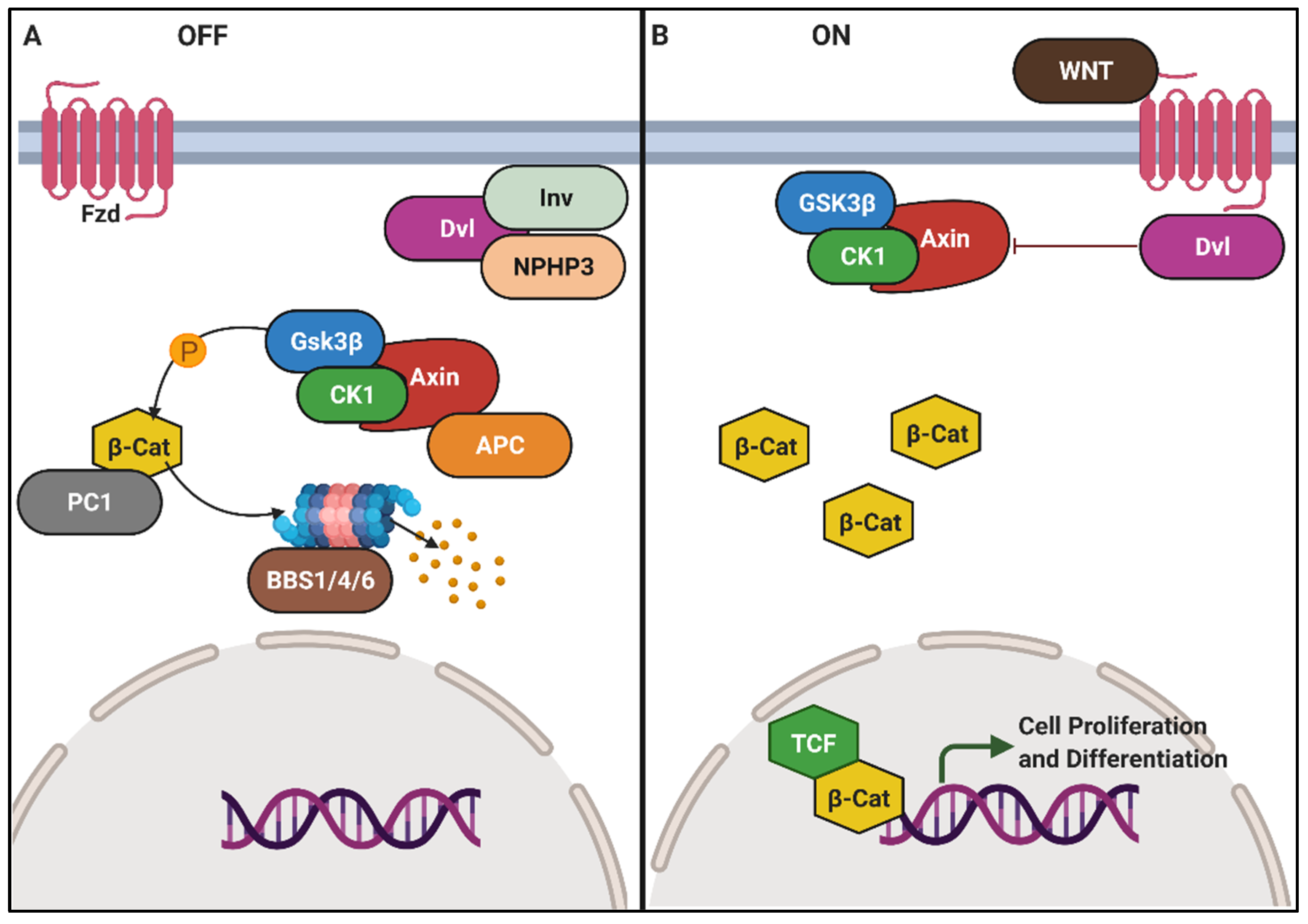{kind=link}
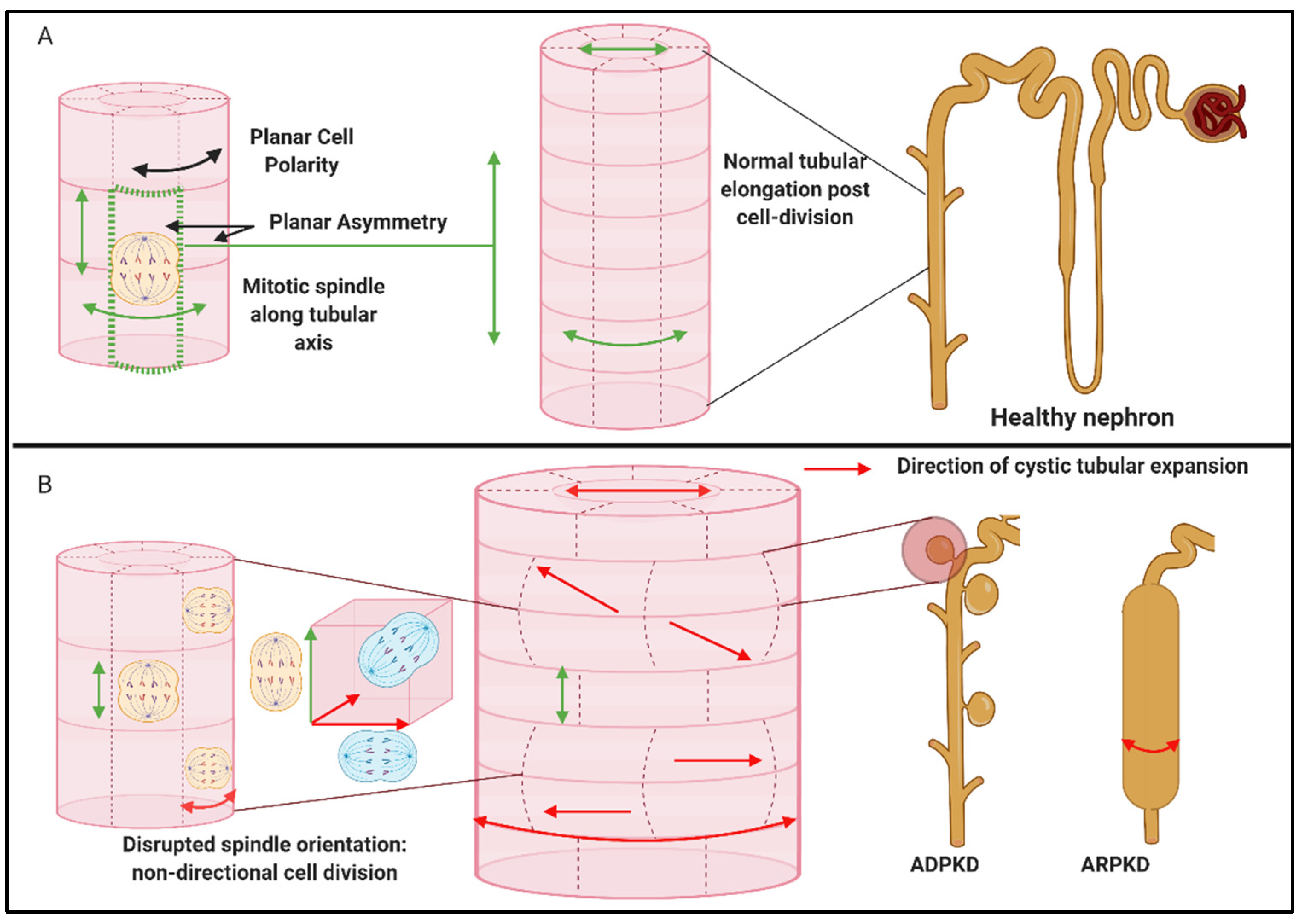{kind=link}
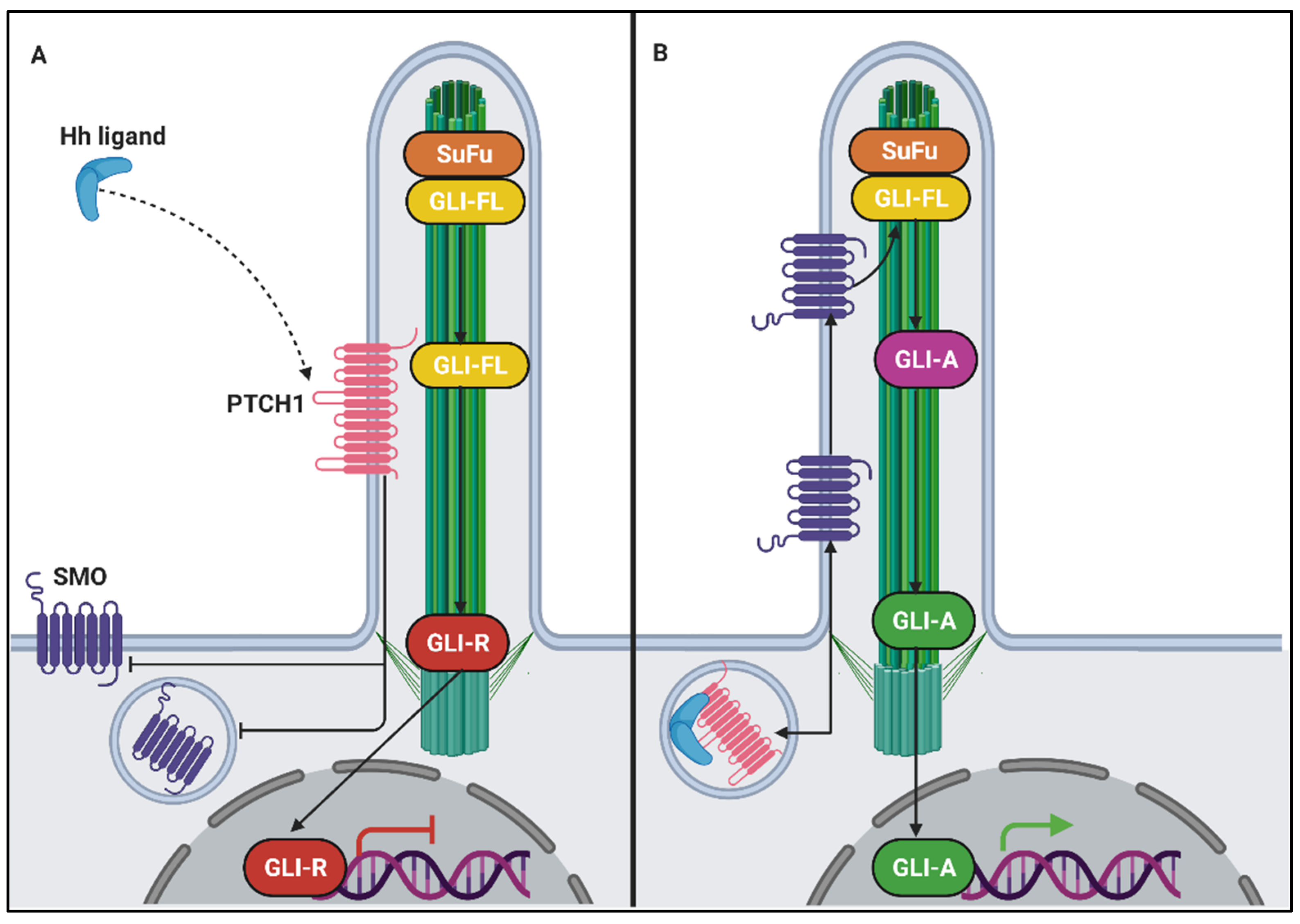{kind=link}
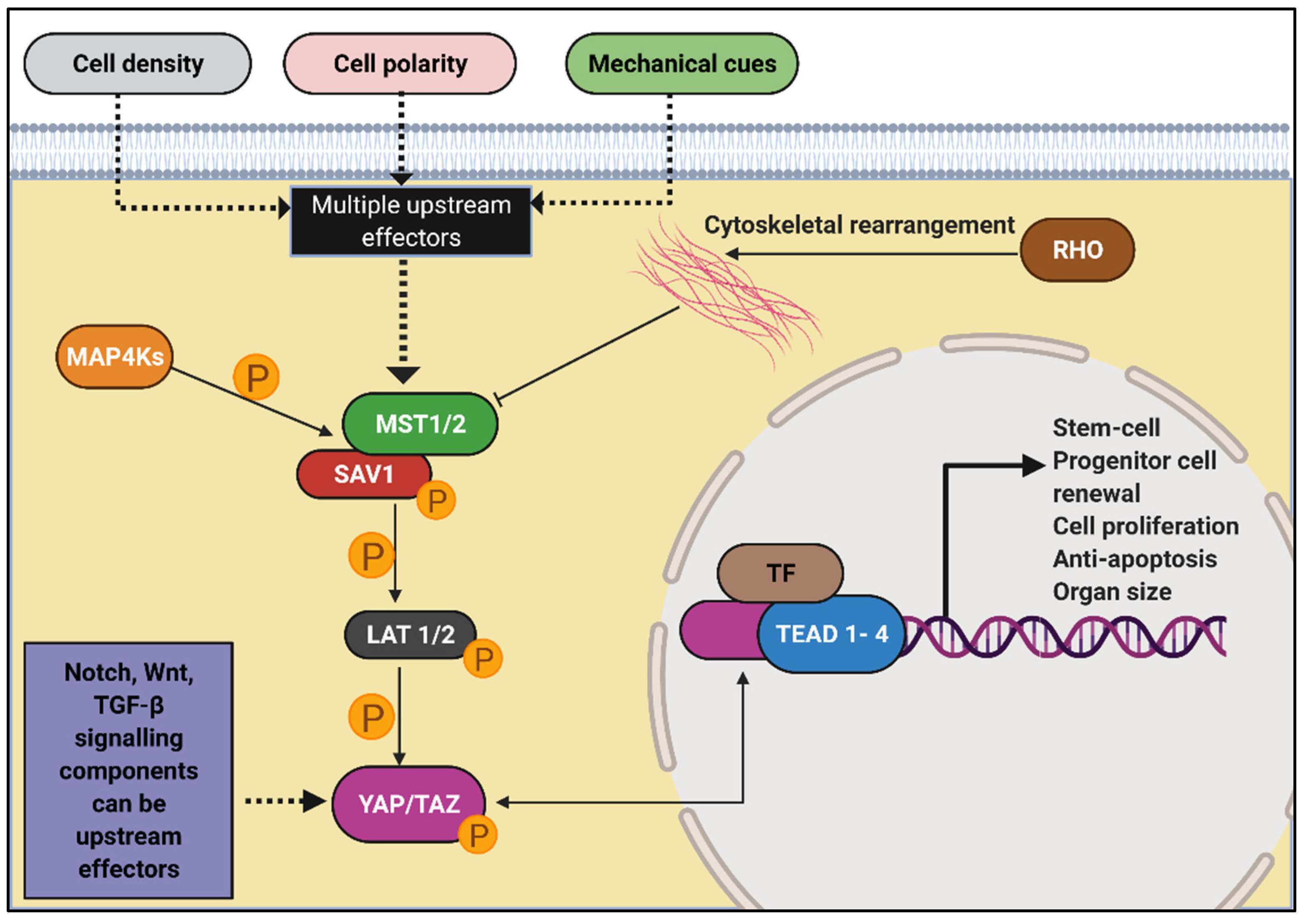{kind=link}
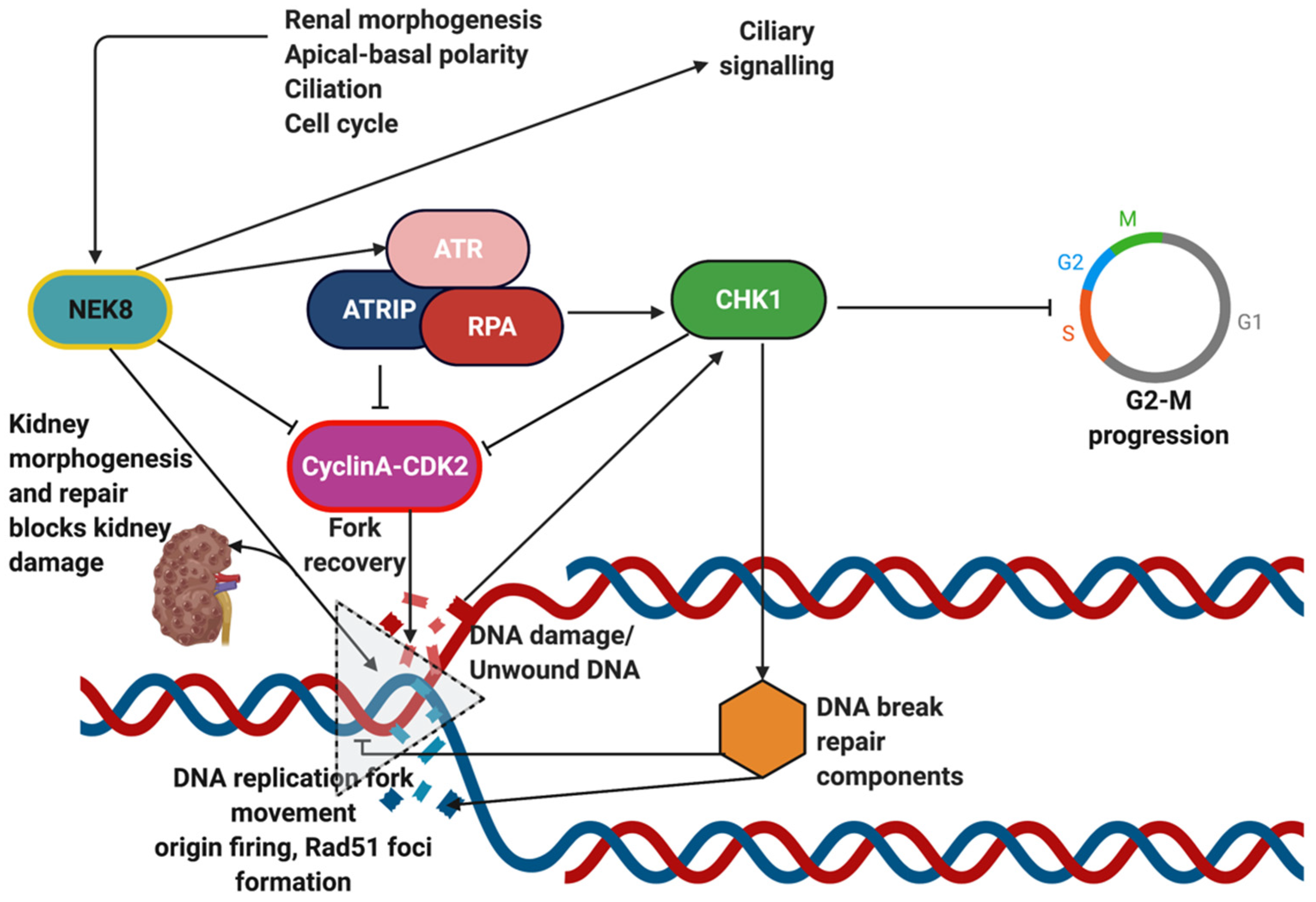{kind=link}
| Gene | Chromosome | Protein | Location | Mode of Inheritance | Animal Model | Protein Families | Human Disease Phenotype * | |
|---|---|---|---|---|---|---|---|---|
| Kidney | Extra Renal Presentations/Syndromes | |||||||
| NPHP1 | 2q12.3 | Nephrocystin-1 | Adherens junctions, focal adhesion, transition zone | AR | Mice: Nphp1 del20/del20; Zebrafish: MO, morpholino antisense oligonucleotides | Nephrocystin-1; Domain: coiled-coil, SH3 domain | NPHP juvenile | Eye (retinitis pigmentosa, oculomotor apraxia), brain (cerebellar vermis hypoplasia) |
| NPHP2/INVS | 9q31.1 | Inversin | Inversin compartment | AR | Mice: Inv -/-, mice, Zebrafish: MO, morpholino antisense oligonucleotides | Domain: ANK repeat, Repeat | NPHP infantile | Eye (retinitis pigmentosa), liver (bile duct proliferation, liver fibrosis), laterality defects (Situs inversus), pulmonary hypoplasia, oligohydramnios |
| NPHP3 | 3q22.1 | Nephrocystin-3 | Inversin compartment, axoneme | AR | Mice: pcy, Nphp-/- Zebrafish: MO, morpholino antisense oligonucleotides | Coiled-coil, Repeat, TPR repeat | NPHP (including infantile) | Eye (retinitis pigmentosa), liver (liver fibrosis, liver cysts), laterality defects (Situs inversus), heart (congenital heart disease), multiple organ polycystic disease, Meckel-–Gruber syndrome |
| NPHP4 | 1p36.31 | Nephrocystin-4 | Transition zone | AR | Mice: Nphp4em1(IMPC)Bay; Zebrafish: MO, morpholino antisense oligonucleotides | NPHP4 Family | NPHP | Eye (retinitis pigmentosa, oculomotor apraxia), liver (bile duct proliferation, liver fibrosis), laterality defects (heterotaxia), heart (congenital heart disease) |
| NPHP5/IQCB1 | 3q13.33 | Nephrocystin-5/IQ motif containing B1 | Transition zone, basal body | AR | Mice: Iqcb1em1(IMPC)Bay; Zebrafish: MO, morpholino antisense oligonucleotides | Coiled-coil, Repeat | NPHP | Eye (retinitis pigmentosa) |
| NPHP6/CEP290 | 12q21.32 | Nephrocystin-6/centrosomal protein 290 | Transition zone, centrosome | AR | Mice: Cep290em1(IMPC) Mbp; rd16; Zebrafish: MO, morpholino antisense oligonucleotides- | Coiled-coil | NPHP | Eye (retinitis pigmentosa), brain (cerebellar vermis hypoplasia, congenital brain defect), liver (bile duct proliferation, liver fibrosis), skeletal defects (polydactyly), heart (ventricular septal defect), Meckel–Gruber syndrome, Bardet–Biedl syndrome |
| NPHP7/GLIS2 | 16p13.3 | Nephrocystin-7/GLI similar 2 | Nucleus | AR | Mice:Glis2tm1Tre/Glis2tm1Tre; Zebrafish: MO, morpholino antisense oligonucleotides | GLI C2H2-type zinc-finger protein family. | NPHP | |
| NPHP8/RPGRIP1L/MKS5 | 16q12.2 | Nephrocystin-8/RPGRIP1-like | Transition zone | AR | Mice: Rpgrip1l -/- embryos; Ftm | Coiled-coil, Signal | NPHP | Eye (retinitis pigmentosa, oculomotor apraxia), brain (cerebellar vermis hypoplasia, congenital brain defect), liver (bile duct proliferation, liver fibrosis), skeletal defects (polydactyly), Meckel–Gruber syndrome, Bardet–Biedl syndrome |
| NPHP9/NEK8 | 17q11.2 | Nephrocystin-9/NIMA-related kinase 8 | Inversin compartment | AR | Mice: Nek8 jck; Nek8-Zebrafish: MO, atg morpholino (5′-TTCTCATACTTCTCCATGTTTTCGC-3′); Rat: LPK | Protein kinase superfamily. NEK Ser/Thr protein kinase family. NIMA subfamily | NPHP (including infantile) | Liver (liver fibrosis), heart (congenital heart disease), Meckel–Gruber syndrome |
| NPHP10/SDCCAG8/SLSN7 | 1q43-q44 | Nephrocystin-10/Serologically defined colon cancer antigen 8 | Basal body | AR | Mice: Sdccag8tm1a(EUCOMM)Wtsi; Zebrafish: MO, morpholino antisense oligonucleotides | Domain: coiled-coil | NPHP | Eye (retinitis pigmentosa), brain (intellectual disability), Bardet–Biedl syndrome |
| NPHP11/TMEM67/MKS3 | 8q22.1 | Nephrocystin-11/Transmembrane protein 67 | Transition zone | AR | Domain: Signal, Transmembrane, Transmembrane helix | NPHP | Eye (retinitis pigmentosa, oculomotor apraxia, coloboma), brain (cerebellar vermis hypoplasia), liver (bile duct proliferation, liver fibrosis), skeletal defects (polydactyly), Meckel–Gruber syndrome | |
| NPHP12/TTC21B/JBTS11 | 2q24.3 | Nephrocystin-12/Intraflagellar transport protein | IFT-A | AR | TTC21 family; Domain: Repeat, TPR repeat | NPHP | Brain (cerebellar vermis hypoplasia), skeletal defects (Jeune asphyxiating thoracic dystrophy), Meckel–Gruber syndrome | |
| NPHP13/WDR19 | 4p14 | Nephrocystin-13/WD repeat domain 19/IFT protein 144 | IFT-A | AR | Domain: Repeat, TPR repeat, WD repeat | NPHP | Eye (retinitis pigmentosa), liver (liver fibrosis), skeletal defects (Jeune asphyxiating thoracic dystrophy, cranioectodermal dysplasia) | |
| NPHP14/ZNF423 | 16q12.1 | Nephrocystin-14/Zinc finger protein 423 | Nucleus | AR/AD | Krueppel C2H2-type zinc-finger protein family; Domain: Repeat, Zinc-finger | NPHP | Eye (retinitis pigmentosa), brain (cerebellar vermis hypoplasia), laterality defects (situs inversus) | |
| NPHP15/CEP164 | 11q23.3 | Nephrocystin-15 centrosomal protein 164 | Basal body | AR | Domain: Coiled-coil | NPHP | Eye (retinitis pigmentosa), brain (cerebellar vermis hypoplasia, intellectual disability), liver (liver fibrosis), skeletal defects (polydactyly), obesity | |
| NPHP16/ANKS6 | 9q22.33 | Nephrocystin-16/ANKS6 | Axoneme, inversion compartment | AR | Mice: Anks6tm1b(KOMP)Wtsi; Zebrafish: MO, antisense morpholinos | Domain: Ankyrin repeat, Repeat | NPHP | Liver (liver fibrosis), laterality defects (situs inversus), heart (congenital heart disease) |
| NPHP17/IFT172 | 2p23.3 | Nephrocystin-17/IFT protein 172 | IFT-B | AR | IFT172 family- Domain: Repeat, TPR repeat, WD repeat | NPHP | Eye (retinitis pigmentosa), brain (cerebellar vermis hypoplasia, intellectual disability), liver (liver fibrosis), skeletal defects (short-rib thoracic dysplasia, polydactyly), ventricular septal defect, obesity | |
| NPHP18/CEP83 | 12q22 | Nephrocystin-18/centrosomal protein 83 | Basal body | AR | CEP83 family; Domain: coiled-coil | NPHP (including infantile) | Eye (retinitis pigmentosa), brain (hydrocephalus, intellectual disability), liver (liver fibrosis) | |
| NPHP19/DCDC2 | 6p22.3 | Doublecortin domain-containing protein 2 | Axoneme | AR | Domain: Repeat | NPHP | Liver (liver fibrosis) | |
| NPHP20/MAPKBP1 | 15q15.1 | Mitogen-activated protein kinase binding protein 1 | Cytoplasm | AR | Domain: Repeat, WD repeat | NPHP | ||
| NPHP1L/XPNPEP3 | 22q13 | X-prolyl aminopeptidase 3 | Mitochondria | AR | Peptidase M24B family; Domain: Transit peptide | NPHP, gout | Neurological disorder (essential tremor), ear (high frequency sensorineural hearing loss), brain (arachnoid cysts) | |
| NPHP2L/SLC41A1 | 1q32.1 | Solute carrier family 41-member 1 | Tubules at the borders of the cortex and medulla | AR | SLC41A transporter family; Domain: Repeat, Transmembrane, Transmembrane helix | NPHP | Skeletal defects (ventral body curvature), brain (hydrocephalus) | |
| TRAF3IP1 | 2q37.3 | TRAF3 interacting protein 1 | Axonemes, basal bodies | AR | Mice: Traf3ip1em1(IMPC)Bay | TRAF3IP1 family; Domain: coiled-coil | NPHP | Eye (retinitis pigmentosa), brain (intellectual disability), skeletal anomaly (brachydactyly) |
| AH11/JBTS3 | 6q23.3 | Jouberin | Basal bodies | AR | Mice:Ahi1tm1Jgg/Ahi1tm1Jgg | Domain: coiled-coil, Repeat, SH3 domain, WD repeat | NPHP, rare | Eye (retinitis pigmentosa, oculomotor apraxia), brain (cerebellar vermis hypoplasia, intellectual disability) |
| JBTS9/CC2D2A/MKS6 | 4p15.32 | Coiled-coil and C2 domain containing 2A | Basal bodies | AR | Mice: Cc2d2atm1a(EUCOMM)Wtsi | Domain: coiled-coil | NPHP | Eye (retinitis pigmentosa, oculomotor apraxia), brain (cerebellar vermis hypoplasia, intellectual disability), liver (liver fibrosis), Meckel–Gruber syndrome |
| FAN1 KIAA1018, MTMR15 | 15q13.3 | Fanconi-associated nuclease 1 | nucleus | AR | Mice: Fan1(nd/nd) Zebrafish: MO, antisense morpholinos | Domain: coiled-coil, D-box, KEN box | NPHP, Interstitial nephritis, karyomegalic | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gupta, S.; Ozimek-Kulik, J.E.; Phillips, J.K. Nephronophthisis-Pathobiology and Molecular Pathogenesis of a Rare Kidney Genetic Disease. Genes 2021, 12, 1762. https://doi.org/10.3390/genes12111762
Gupta S, Ozimek-Kulik JE, Phillips JK. Nephronophthisis-Pathobiology and Molecular Pathogenesis of a Rare Kidney Genetic Disease. Genes. 2021; 12(11):1762. https://doi.org/10.3390/genes12111762
Chicago/Turabian StyleGupta, Shabarni, Justyna E. Ozimek-Kulik, and Jacqueline Kathleen Phillips. 2021. "Nephronophthisis-Pathobiology and Molecular Pathogenesis of a Rare Kidney Genetic Disease" Genes 12, no. 11: 1762. https://doi.org/10.3390/genes12111762
APA StyleGupta, S., Ozimek-Kulik, J. E., & Phillips, J. K. (2021). Nephronophthisis-Pathobiology and Molecular Pathogenesis of a Rare Kidney Genetic Disease. Genes, 12(11), 1762. https://doi.org/10.3390/genes12111762