
NAD+ Metabolism and Diseases with Motor Dysfunction

{kind=link}
Abstract
:1. Introduction
2. Cellular NAD+ Homeostasis
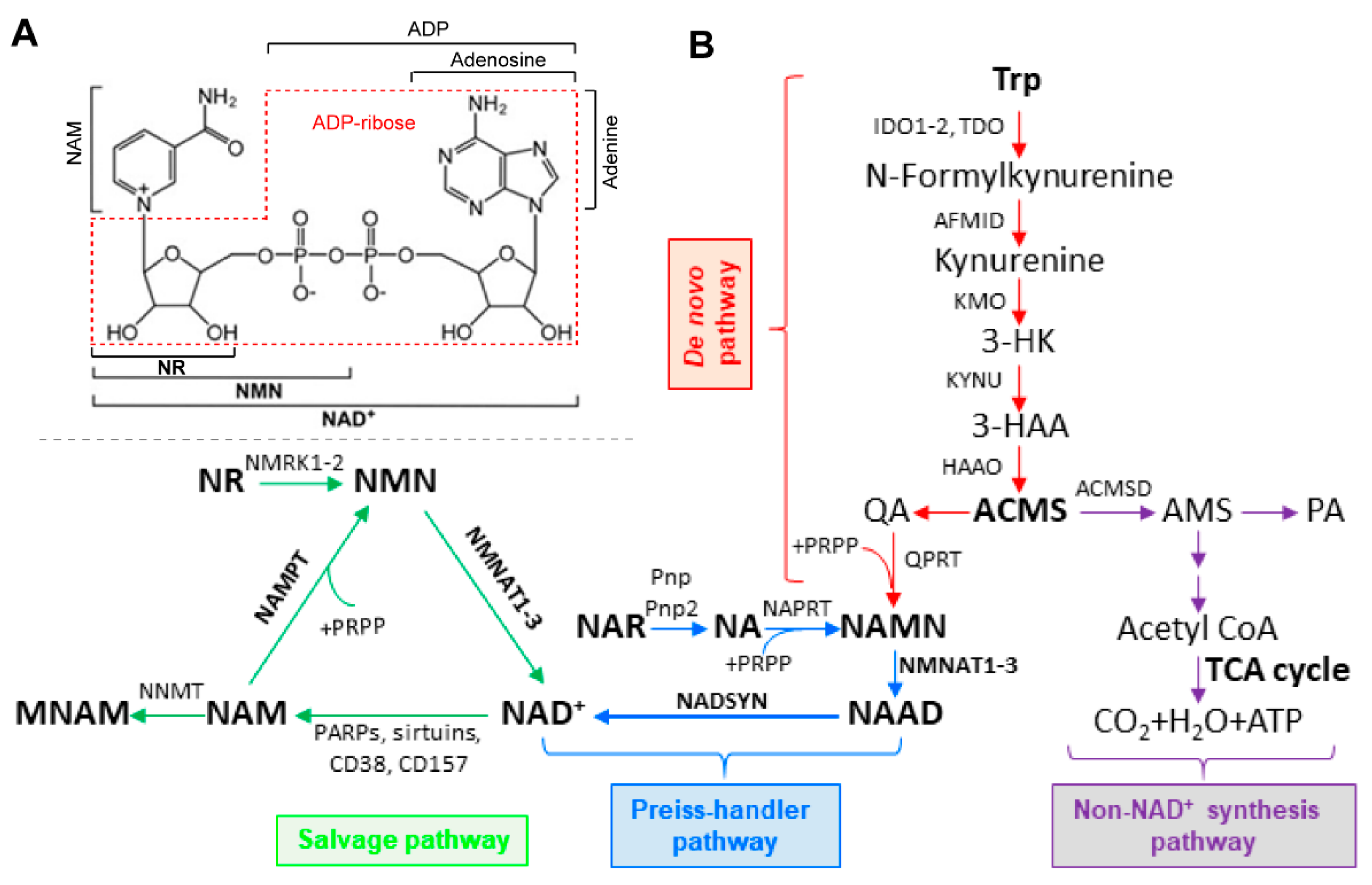
2.1. Intracellular NAD+ Biosynthesis
2.2. Nampt Expression in the CNS
2.3. Cellular Consumption of NAD+
3. NAD+ and Diseases with Motor Dysfunction
3.1. Amyotrophic Lateral Sclerosis
3.2. Charcot–Marie–Tooth Disease
3.3. Parkinson’s Disease
3.4. Huntington’s Disease
3.5. Other Motor Neuron Diseases
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dugger, B.N.; Dickson, D.W. Pathology of Neurodegenerative Diseases. Cold Spring Harb. Perspect. Biol. 2017, 9, a028035. [Google Scholar] [CrossRef]
- Blaszczyk, J.W. Energy Metabolism Decline in the Aging Brain-Pathogenesis of Neurodegenerative Disorders. Metabolites 2020, 10, 450. [Google Scholar] [CrossRef] [PubMed]
- Goutman, S.A. Diagnosis and Clinical Management of Amyotrophic Lateral Sclerosis and Other Motor Neuron Disorders. Continuum 2017, 23, 1332–1359. [Google Scholar] [CrossRef]
- Ragagnin, A.M.G.; Shadfar, S.; Vidal, M.; Jamali, M.S.; Atkin, J.D. Motor Neuron Susceptibility in ALS/FTD. Front. Neurosci. 2019, 13, 532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murthy, M.; Cheng, Y.Y.; Holton, J.L.; Bettencourt, C. Neurodegenerative movement disorders: An epigenetics perspective and promise for the future. Neuropathol. Appl. Neurobiol. 2021, 1–13. [Google Scholar] [CrossRef]
- Lautrup, S.; Sinclair, D.A.; Mattson, M.P.; Fang, E.F. NAD+ in Brain Aging and Neurodegenerative Disorders. Cell Metab. 2019, 30, 630–655. [Google Scholar] [CrossRef]
- Verdin, E. NAD+ in aging, metabolism, and neurodegeneration. Science 2015, 350, 1208–1213. [Google Scholar] [CrossRef]
- Pieper, A.A.; McKnight, S.L. Benefits of Enhancing Nicotinamide Adenine Dinucleotide Levels in Damaged or Diseased Nerve Cells. Cold Spring Harb. Symp. Quant. Biol. 2018, 83, 207–217. [Google Scholar] [CrossRef]
- Rajman, L.; Chwalek, K.; Sinclair, D.A. Therapeutic Potential of NAD-Boosting Molecules: The In Vivo Evidence. Cell Metab. 2018, 27, 529–547. [Google Scholar] [CrossRef] [Green Version]
- Canto, C.; Menzies, K.J.; Auwerx, J. NAD+ Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell Metab. 2015, 22, 31–53. [Google Scholar] [CrossRef] [Green Version]
- Covarrubias, A.J.; Perrone, R.; Grozio, A.; Verdin, E. NAD+ metabolism and its roles in cellular processes during ageing. Nat. Rev. Mol. Cell Biol. 2021, 22, 119–141. [Google Scholar] [CrossRef]
- Gasperi, V.; Sibilano, M.; Savini, I.; Catani, M.V. Niacin in the Central Nervous System: An Update of Biological Aspects and Clinical Applications. Int. J. Mol. Sci. 2019, 20, 974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rongvaux, A.; Andris, F.; Van Gool, F.; Leo, O. Reconstructing eukaryotic NAD metabolism. Bioessays 2003, 25, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Revollo, J.R.; Grimm, A.A.; Imai, S. The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J. Biol. Chem. 2004, 279, 50754–50763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Xie, Y.; Wang, T.; Bi, J.; Li, H.; Zhang, L.Q.; Ye, S.Q.; Ding, S. Neuronal protective role of PBEF in a mouse model of cerebral ischemia. J. Cereb. Blood Flow Metab. 2010, 30, 1962–1971. [Google Scholar] [CrossRef] [Green Version]
- Katsyuba, E.; Auwerx, J. Modulating NAD+ metabolism, from bench to bedside. EMBO J. 2017, 36, 2670–2683. [Google Scholar] [CrossRef]
- Berger, F.; Lau, C.; Dahlmann, M.; Ziegler, M. Subcellular compartmentation and differential catalytic properties of the three human nicotinamide mononucleotide adenylyltransferase isoforms. J. Biol. Chem. 2005, 280, 36334–36341. [Google Scholar] [CrossRef] [Green Version]
- Gilley, J.; Adalbert, R.; Yu, G.; Coleman, M.P. Rescue of peripheral and CNS axon defects in mice lacking NMNAT2. J. Neurosci. 2013, 33, 13410–13424. [Google Scholar] [CrossRef] [Green Version]
- Gilley, J.; Mayer, P.R.; Yu, G.; Coleman, M.P. Low levels of NMNAT2 compromise axon development and survival. Hum. Mol. Genet. 2019, 28, 448–458. [Google Scholar] [CrossRef]
- Russo, A.; Goel, P.; Brace, E.J.; Buser, C.; Dickman, D.; DiAntonio, A. The E3 ligase Highwire promotes synaptic transmission by targeting the NAD-synthesizing enzyme dNmnat. EMBO Rep. 2019, 20, e46975. [Google Scholar] [CrossRef]
- Bieganowski, P.; Brenner, C. Discoveries of nicotinamide riboside as a nutrient and conserved NRK genes establish a Preiss-Handler independent route to NAD+ in fungi and humans. Cell 2004, 117, 495–502. [Google Scholar] [CrossRef] [Green Version]
- Fletcher, R.S.; Ratajczak, J.; Doig, C.L.; Oakey, L.A.; Callingham, R.; Da Silva Xavier, G.; Garten, A.; Elhassan, Y.S.; Redpath, P.; Migaud, M.E.; et al. Nicotinamide riboside kinases display redundancy in mediating nicotinamide mononucleotide and nicotinamide riboside metabolism in skeletal muscle cells. Mol. Metab. 2017, 6, 819–832. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Lautrup, S.; Hou, Y.; Demarest, T.G.; Croteau, D.L.; Mattson, M.P.; Bohr, V.A. NAD+ in Aging: Molecular Mechanisms and Translational Implications. Trends Mol. Med. 2017, 23, 899–916. [Google Scholar] [CrossRef] [PubMed]
- Cambronne, X.A.; Stewart, M.L.; Kim, D.; Jones-Brunette, A.M.; Morgan, R.K.; Farrens, D.L.; Cohen, M.S.; Goodman, R.H. Biosensor reveals multiple sources for mitochondrial NAD+. Science 2016, 352, 1474–1477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garten, A.; Schuster, S.; Penke, M.; Gorski, T.; de Giorgis, T.; Kiess, W. Physiological and pathophysiological roles of NAMPT and NAD metabolism. Nat. Rev. Endocrinol. 2015, 11, 535–546. [Google Scholar] [CrossRef]
- Yoshino, J.; Baur, J.A.; Imai, S.I. NAD+ Intermediates: The Biology and Therapeutic Potential of NMN and NR. Cell Metab. 2018, 27, 513–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitani, T.; Okuno, S.; Fujisawa, H. Growth phase-dependent changes in the subcellular localization of pre-B-cell colony-enhancing factor. FEBS Lett. 2003, 544, 74–78. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Yang, T.; Baur, J.A.; Perez, E.; Matsui, T.; Carmona, J.J.; Lamming, D.W.; Souza-Pinto, N.C.; Bohr, V.A.; Rosenzweig, A.; et al. Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell 2007, 130, 1095–1107. [Google Scholar] [CrossRef] [Green Version]
- Pittelli, M.; Formentini, L.; Faraco, G.; Lapucci, A.; Rapizzi, E.; Cialdai, F.; Romano, G.; Moneti, G.; Moroni, F.; Chiarugi, A. Inhibition of nicotinamide phosphoribosyltransferase: Cellular bioenergetics reveals a mitochondrial insensitive NAD pool. J. Biol. Chem. 2010, 285, 34106–34114. [Google Scholar] [CrossRef] [Green Version]
- Morris-Blanco, K.C.; Cohan, C.H.; Neumann, J.T.; Sick, T.J.; Perez-Pinzon, M.A. Protein kinase C epsilon regulates mitochondrial pools of Nampt and NAD following resveratrol and ischemic preconditioning in the rat cortex. J. Cereb. Blood Flow Metab. 2014, 34, 1024–1032. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Zhang, Z.; Zhang, N.; Li, H.; Zhang, L.; Baines, C.P.; Ding, S. Subcellular NAMPT-mediated NAD+ salvage pathways and their roles in bioenergetics and neuronal protection after ischemic injury. J. Neurochem. 2019, 151, 732–748. [Google Scholar] [CrossRef] [PubMed]
- Bogan, K.L.; Brenner, C. Nicotinic acid, nicotinamide, and nicotinamide riboside: A molecular evaluation of NAD+ precursor vitamins in human nutrition. Annu. Rev. Nutr. 2008, 28, 115–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, V.; Amici, A.; Mazzola, F.; Di Stefano, M.; Conforti, L.; Magni, G.; Ruggieri, S.; Raffaelli, N.; Orsomando, G. Metabolic profiling of alternative NAD biosynthetic routes in mouse tissues. PLoS ONE 2014, 9, e113939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujigaki, H.; Yamamoto, Y.; Saito, K. L-Tryptophan-kynurenine pathway enzymes are therapeutic target for neuropsychiatric diseases: Focus on cell type differences. Neuropharmacology 2017, 112, 264–274. [Google Scholar] [CrossRef]
- Revollo, J.R.; Korner, A.; Mills, K.F.; Satoh, A.; Wang, T.; Garten, A.; Dasgupta, B.; Sasaki, Y.; Wolberger, C.; Townsend, R.R.; et al. Nampt/PBEF/Visfatin regulates insulin secretion in β cells as a systemic NAD biosynthetic enzyme. Cell Metab. 2007, 6, 363–375. [Google Scholar] [CrossRef] [Green Version]
- Jing, Z.; Xing, J.; Chen, X.; Stetler, R.A.; Weng, Z.; Gan, Y.; Zhang, F.; Gao, Y.; Chen, J.; Leak, R.K.; et al. Neuronal NAMPT is released after cerebral ischemia and protects against white matter injury. J. Cereb. Blood Flow Metab. 2014, 34, 1613–1621. [Google Scholar] [CrossRef]
- Wang, P.; Xu, T.Y.; Guan, Y.F.; Tian, W.W.; Viollet, B.; Rui, Y.C.; Zhai, Q.W.; Su, D.F.; Miao, C.Y. Nicotinamide phosphoribosyltransferase protects against ischemic stroke through SIRT1-dependent adenosine monophosphate-activated kinase pathway. Ann. Neurol. 2011, 69, 360–374. [Google Scholar] [CrossRef] [PubMed]
- Stein, L.R.; Imai, S. Specific ablation of Nampt in adult neural stem cells recapitulates their functional defects during aging. EMBO J. 2014, 33, 1321–1340. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.Y.; Wang, F.; Zhang, X.Y.; Huang, P.; Lu, Y.B.; Wei, E.Q.; Zhang, W.P. Nicotinamide phosphoribosyltransferase may be involved in age-related brain diseases. PLoS ONE 2012, 7, e44933. [Google Scholar] [CrossRef] [Green Version]
- Caton, P.W.; Kieswich, J.; Yaqoob, M.M.; Holness, M.J.; Sugden, M.C. Nicotinamide mononucleotide protects against pro-inflammatory cytokine-mediated impairment of mouse islet function. Diabetologia 2011, 54, 3083–3092. [Google Scholar] [CrossRef] [Green Version]
- Garten, A.; Petzold, S.; Barnikol-Oettler, A.; Korner, A.; Thasler, W.E.; Kratzsch, J.; Kiess, W.; Gebhardt, R. Nicotinamide phosphoribosyltransferase (NAMPT/PBEF/visfatin) is constitutively released from human hepatocytes. Biochem. Biophys. Res. Commun. 2010, 391, 376–381. [Google Scholar] [CrossRef]
- Friebe, D.; Neef, M.; Kratzsch, J.; Erbs, S.; Dittrich, K.; Garten, A.; Petzold-Quinque, S.; Bluher, S.; Reinehr, T.; Stumvoll, M.; et al. Leucocytes are a major source of circulating nicotinamide phosphoribosyltransferase (NAMPT)/pre-B cell colony (PBEF)/visfatin linking obesity and inflammation in humans. Diabetologia 2011, 54, 1200–1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curat, C.A.; Wegner, V.; Sengenes, C.; Miranville, A.; Tonus, C.; Busse, R.; Bouloumie, A. Macrophages in human visceral adipose tissue: Increased accumulation in obesity and a source of resistin and visfatin. Diabetologia 2006, 49, 744–747. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Satoh, A.; Lin, J.B.; Mills, K.F.; Sasaki, Y.; Rensing, N.; Wong, M.; Apte, R.S.; Imai, S.I. Extracellular Vesicle-Contained eNAMPT Delays Aging and Extends Lifespan in Mice. Cell Metab. 2019, 30, 329–342. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, Q.; Bao, R.; Zhang, N.; Wang, Y.; Polo-Parada, L.; Tarim, A.; Alemifar, A.; Han, X.; Wilkins, H.M.; et al. Deletion of Nampt in Projection Neurons of Adult Mice Leads to Motor Dysfunction, Neurodegeneration, and Death. Cell Rep. 2017, 20, 2184–2200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imai, S.; Guarente, L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014, 24, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Agerholm, M.; Dall, M.; Jensen, B.A.H.; Prats, C.; Madsen, S.; Basse, A.L.; Graae, A.S.; Risis, S.; Goldenbaum, J.; Quistorff, B.; et al. Perturbations of NAD+ salvage systems impact mitochondrial function and energy homeostasis in mouse myoblasts and intact skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2018, 314, E377–E395. [Google Scholar] [CrossRef] [Green Version]
- Costford, S.R.; Brouwers, B.; Hopf, M.E.; Sparks, L.M.; Dispagna, M.; Gomes, A.P.; Cornnell, H.H.; Petucci, C.; Phelan, P.; Xie, H.; et al. Skeletal muscle overexpression of nicotinamide phosphoribosyl transferase in mice coupled with voluntary exercise augments exercise endurance. Mol. Metab. 2018, 7, 1–11. [Google Scholar] [CrossRef]
- Frederick, D.W.; Davis, J.G.; Davila, A., Jr.; Agarwal, B.; Michan, S.; Puchowicz, M.A.; Nakamaru-Ogiso, E.; Baur, J.A. Increasing NAD synthesis in muscle via nicotinamide phosphoribosyltransferase is not sufficient to promote oxidative metabolism. J. Biol. Chem. 2015, 290, 1546–1558. [Google Scholar] [CrossRef] [Green Version]
- Frederick, D.W.; Loro, E.; Liu, L.; Davila, A., Jr.; Chellappa, K.; Silverman, I.M.; Quinn, W.J., 3rd; Gosai, S.J.; Tichy, E.D.; Davis, J.G.; et al. Loss of NAD Homeostasis Leads to Progressive and Reversible Degeneration of Skeletal Muscle. Cell Metab. 2016, 24, 269–282. [Google Scholar] [CrossRef] [Green Version]
- Yoshino, J.; Mills, K.F.; Yoon, M.J.; Imai, S. Nicotinamide mononucleotide, a key NAD+ intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011, 14, 528–536. [Google Scholar] [CrossRef] [Green Version]
- Araki, T.; Sasaki, Y.; Milbrandt, J. Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science 2004, 305, 1010–1013. [Google Scholar] [CrossRef] [Green Version]
- Corpas, R.; Grinan-Ferre, C.; Rodriguez-Farre, E.; Pallas, M.; Sanfeliu, C. Resveratrol Induces Brain Resilience against Alzheimer Neurodegeneration through Proteostasis Enhancement. Mol. Neurobiol. 2019, 56, 1502–1516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, W.; Qian, L.; Zhang, J.; Zhang, W.; Morrison, A.; Hayes, P.; Wilson, S.; Chen, T.; Zhao, J. Sirt1 overexpression in neurons promotes neurite outgrowth and cell survival through inhibition of the mTOR signaling. J. Neurosci. Res. 2011, 89, 1723–1736. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.D.; Guo, S.Q.; Hu, Z.W.; Li, W.L. NAMPT protects against 6-hydroxydopamine-induced neurotoxicity in PC12 cells through modulating SIRT1 activity. Mol. Med. Rep. 2016, 13, 4058–4064. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Ageta-Ishihara, N.; Nagatsu, S.; Takao, K.; Komine, O.; Endo, F.; Miyakawa, T.; Misawa, H.; Takahashi, R.; Kinoshita, M.; et al. SIRT1 overexpression ameliorates a mouse model of SOD1-linked amyotrophic lateral sclerosis via HSF1/HSP70i chaperone system. Mol. Brain 2014, 7, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.Q.; Van Haandel, L.; Xiong, M.; Huang, P.; Heruth, D.P.; Bi, C.; Gaedigk, R.; Jiang, X.; Li, D.Y.; Wyckoff, G.; et al. Metabolic and molecular insights into an essential role of nicotinamide phosphoribosyltransferase. Cell Death Dis. 2017, 8, e2705. [Google Scholar] [CrossRef] [PubMed]
- Mouchiroud, L.; Houtkooper, R.H.; Moullan, N.; Katsyuba, E.; Ryu, D.; Canto, C.; Mottis, A.; Jo, Y.S.; Viswanathan, M.; Schoonjans, K.; et al. The NAD+/Sirtuin Pathway Modulates Longevity through Activation of Mitochondrial UPR and FOXO Signaling. Cell 2013, 154, 430–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roichman, A.; Elhanati, S.; Aon, M.A.; Abramovich, I.; Di Francesco, A.; Shahar, Y.; Avivi, M.Y.; Shurgi, M.; Rubinstein, A.; Wiesner, Y.; et al. Restoration of energy homeostasis by SIRT6 extends healthy lifespan. Nat. Commun. 2021, 12, 3208. [Google Scholar] [CrossRef]
- Bai, P.; Canto, C. The role of PARP-1 and PARP-2 enzymes in metabolic regulation and disease. Cell Metab. 2012, 16, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Krishnakumar, R.; Kraus, W.L. The PARP side of the nucleus: Molecular actions, physiological outcomes, and clinical targets. Mol. Cell 2010, 39, 8–24. [Google Scholar] [CrossRef] [Green Version]
- Rongvaux, A.; Galli, M.; Denanglaire, S.; Van Gool, F.; Dreze, P.L.; Szpirer, C.; Bureau, F.; Andris, F.; Leo, O. Nicotinamide phosphoribosyl transferase/pre-B cell colony-enhancing factor/visfatin is required for lymphocyte development and cellular resistance to genotoxic stress. J. Immunol. 2008, 181, 4685–4695. [Google Scholar] [CrossRef] [Green Version]
- Alano, C.C.; Garnier, P.; Ying, W.; Higashi, Y.; Kauppinen, T.M.; Swanson, R.A. NAD+ depletion is necessary and sufficient for poly(ADP-ribose) polymerase-1-mediated neuronal death. J. Neurosci. 2010, 30, 2967–2978. [Google Scholar] [CrossRef] [PubMed]
- Rappou, E.; Jukarainen, S.; Rinnankoski-Tuikka, R.; Kaye, S.; Heinonen, S.; Hakkarainen, A.; Lundbom, J.; Lundbom, N.; Saunavaara, V.; Rissanen, A.; et al. Weight Loss Is Associated With Increased NAD+/SIRT1 Expression But Reduced PARP Activity in White Adipose Tissue. J. Clin. Endocrinol. Metab. 2016, 101, 1263–1273. [Google Scholar] [CrossRef]
- Ryu, D.; Zhang, H.; Ropelle, E.R.; Sorrentino, V.; Mazala, D.A.; Mouchiroud, L.; Marshall, P.L.; Campbell, M.D.; Ali, A.S.; Knowels, G.M.; et al. NAD+ repletion improves muscle function in muscular dystrophy and counters global PARylation. Sci. Transl. Med. 2016, 8, 361ra139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.H.; Long, A.; Owens, K.; Kristian, T. Nicotinamide mononucleotide inhibits post-ischemic NAD+ degradation and dramatically ameliorates brain damage following global cerebral ischemia. Neurobiol. Dis. 2016, 95, 102–110. [Google Scholar] [CrossRef] [Green Version]
- Cameron, A.M.; Castoldi, A.; Sanin, D.E.; Flachsmann, L.J.; Field, C.S.; Puleston, D.J.; Kyle, R.L.; Patterson, A.E.; Hassler, F.; Buescher, J.M.; et al. Inflammatory macrophage dependence on NAD+ salvage is a consequence of reactive oxygen species-mediated DNA damage. Nat. Immunol. 2019, 20, 420–432. [Google Scholar] [CrossRef] [PubMed]
- Wilk, A.; Hayat, F.; Cunningham, R.; Li, J.; Garavaglia, S.; Zamani, L.; Ferraris, D.M.; Sykora, P.; Andrews, J.; Clark, J.; et al. Extracellular NAD+ enhances PARP-dependent DNA repair capacity independently of CD73 activity. Sci. Rep. 2020, 10, 651. [Google Scholar] [CrossRef] [Green Version]
- Aksoy, P.; White, T.A.; Thompson, M.; Chini, E.N. Regulation of intracellular levels of NAD: A novel role for CD38. Biochem. Biophys. Res. Commun. 2006, 345, 1386–1392. [Google Scholar] [CrossRef]
- Sasaki, Y.; Vohra, B.P.S.; Lund, F.E.; Milbrandt, J. Nicotinamide Mononucleotide Adenylyl Transferase-Mediated Axonal Protection Requires Enzymatic Activity But Not Increased Levels of Neuronal Nicotinamide Adenine Dinucleotide. J. Neurosci. 2009, 29, 5525–5535. [Google Scholar] [CrossRef]
- Camacho-Pereira, J.; Tarrago, M.G.; Chini, C.C.S.; Nin, V.; Escande, C.; Warner, G.M.; Puranik, A.S.; Schoon, R.A.; Reid, J.M.; Galina, A.; et al. CD38 Dictates Age-Related NAD Decline and Mitochondrial Dysfunction through an SIRT3-Dependent Mechanism. Cell Metab. 2016, 23, 1127–1139. [Google Scholar] [CrossRef] [Green Version]
- Levy, A.; Bercovich-Kinori, A.; Alexandrovich, A.G.; Tsenter, J.; Trembovler, V.; Lund, F.E.; Shohami, E.; Stein, R.; Mayo, L. CD38 facilitates recovery from traumatic brain injury. J. Neurotrauma 2009, 26, 1521–1533. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, K.; Esposito, E.; Wang, X.; Terasaki, Y.; Liu, Y.; Xing, C.; Ji, X.; Lo, E.H. Transfer of mitochondria from astrocytes to neurons after stroke. Nature 2016, 535, 551–555. [Google Scholar] [CrossRef] [Green Version]
- Long, A.; Park, J.H.; Klimova, N.; Fowler, C.; Loane, D.J.; Kristian, T. CD38 Knockout Mice Show Significant Protection against Ischemic Brain Damage Despite High Level Poly-ADP-Ribosylation. Neurochem. Res. 2017, 42, 283–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Essuman, K.; Summers, D.W.; Sasaki, Y.; Mao, X.; DiAntonio, A.; Milbrandt, J. The SARM1 Toll/Interleukin-1 Receptor Domain Possesses Intrinsic NAD+ Cleavage Activity that Promotes Pathological Axonal Degeneration. Neuron 2017, 93, 1334–1343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, L.; Essuman, K.; Anderson, R.G.; Sasaki, Y.; Monteiro, F.; Chung, E.H.; Osborne Nishimura, E.; DiAntonio, A.; Milbrandt, J.; Dangl, J.L.; et al. TIR domains of plant immune receptors are NAD+-cleaving enzymes that promote cell death. Science 2019, 365, 799–803. [Google Scholar] [CrossRef]
- Horsefield, S.; Burdett, H.; Zhang, X.; Manik, M.K.; Shi, Y.; Chen, J.; Qi, T.; Gilley, J.; Lai, J.S.; Rank, M.X.; et al. NAD+ cleavage activity by animal and plant TIR domains in cell death pathways. Science 2019, 365, 793–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, Y.; Nakagawa, T.; Mao, X.; DiAntonio, A.; Milbrandt, J. NMNAT1 inhibits axon degeneration via blockade of SARM1-mediated NAD+ depletion. Elife 2016, 5, e19749. [Google Scholar] [CrossRef]
- Gilley, J.; Ribchester, R.R.; Coleman, M.P. Sarm1 Deletion, but Not WldS, Confers Lifelong Rescue in a Mouse Model of Severe Axonopathy. Cell Rep. 2017, 21, 10–16. [Google Scholar] [CrossRef] [Green Version]
- Loreto, A.; Hill, C.S.; Hewitt, V.L.; Orsomando, G.; Angeletti, C.; Gilley, J.; Lucci, C.; Sanchez-Martinez, A.; Whitworth, A.J.; Conforti, L.; et al. Mitochondrial impairment activates the Wallerian pathway through depletion of NMNAT2 leading to SARM1-dependent axon degeneration. Neurobiol. Dis. 2020, 134, 104678. [Google Scholar] [CrossRef]
- Figley, M.D.; Gu, W.; Nanson, J.D.; Shi, Y.; Sasaki, Y.; Cunnea, K.; Malde, A.K.; Jia, X.; Luo, Z.; Saikot, F.K.; et al. SARM1 is a metabolic sensor activated by an increased NMN/NAD+ ratio to trigger axon degeneration. Neuron 2021, 109, 1118–1136. [Google Scholar] [CrossRef]
- Sasaki, Y.; Zhu, J.; Shi, Y.; Gu, W.; Kobe, B.; Ve, T.; DiAntonio, A.; Milbrandt, J. Nicotinic acid mononucleotide is an allosteric SARM1 inhibitor promoting axonal protection. Exp. Neurol. 2021, 345, 113842. [Google Scholar] [CrossRef]
- Bloom, A.J.; Mao, X.; Strickland, A.; Sasaki, Y.; Milbrandt, J.; DiAntonio, A. Constitutively active SARM1 variants found in ALS patients induce neuropathy. bioRxiv 2021. [Google Scholar] [CrossRef]
- Gilley, J.; Jackson, O.; Pipis, M.; Estiar, M.A.; Gan-Or, Z.; Goutman, S.A.; Harms, M.B.; Kaye, J.; Lima, L.; Genomics, Q.S.; et al. Enrichment of SARM1 alleles encoding variants with constitutively hyperactive NADase in patients with ALS and other motor nerve disorders. medRxiv 2021. [Google Scholar] [CrossRef]
- Hopkins, E.L.; Gu, W.; Kobe, B.; Coleman, M.P. A Novel NAD Signaling Mechanism in Axon Degeneration and its Relationship to Innate Immunity. Front. Mol. Biosci. 2021, 8, 703532. [Google Scholar] [CrossRef] [PubMed]
- Pandya, V.A.; Patani, R. Decoding the relationship between ageing and amyotrophic lateral sclerosis: A cellular perspective. Brain 2020, 143, 1057–1072. [Google Scholar] [CrossRef]
- Cappello, V.; Francolini, M. Neuromuscular Junction Dismantling in Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2017, 18, 2092. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.P.; Brown, R.H., Jr.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Guillemin, G.J. Kynurenine pathway metabolites in humans: Disease and healthy States. Int. J. Tryptophan Res. 2009, 2, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Blacher, E.; Bashiardes, S.; Shapiro, H.; Rothschild, D.; Mor, U.; Dori-Bachash, M.; Kleimeyer, C.; Moresi, C.; Harnik, Y.; Zur, M.; et al. Potential roles of gut microbiome and metabolites in modulating ALS in mice. Nature 2019, 572, 474–480. [Google Scholar] [CrossRef] [PubMed]
- Roderer, P.; Klatt, L.; John, F.; Theis, V.; Winklhofer, K.F.; Theiss, C.; Matschke, V. Increased ROS Level in Spinal Cord of Wobbler Mice due to Nmnat2 Downregulation. Mol. Neurobiol. 2018, 55, 8414–8424. [Google Scholar] [CrossRef]
- Obrador, E.; Salvador, R.; Marchio, P.; Lopez-Blanch, R.; Jihad-Jebbar, A.; Rivera, P.; Valles, S.L.; Banacloche, S.; Alcacer, J.; Colomer, N.; et al. Nicotinamide Riboside and Pterostilbene Cooperatively Delay Motor Neuron Failure in ALS SOD1G93A Mice. Mol. Neurobiol. 2021, 58, 1345–1371. [Google Scholar] [CrossRef] [PubMed]
- Lundt, S.; Zhang, N.; Wang, X.; Polo-Parada, L.; Ding, S. The effect of NAMPT deletion in projection neurons on the function and structure of neuromuscular junction (NMJ) in mice. Sci. Rep. 2020, 10, 99. [Google Scholar] [CrossRef] [PubMed]
- Tesla, R.; Wolf, H.P.; Xu, P.; Drawbridge, J.; Estill, S.J.; Huntington, P.; McDaniel, L.; Knobbe, W.; Burket, A.; Tran, S.; et al. Neuroprotective efficacy of aminopropyl carbazoles in a mouse model of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2012, 109, 17016–17021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harlan, B.A.; Killoy, K.M.; Pehar, M.; Liu, L.; Auwerx, J.; Vargas, M.R. Evaluation of the NAD+ biosynthetic pathway in ALS patients and effect of modulating NAD+ levels in hSOD1-linked ALS mouse models. Exp. Neurol. 2020, 327, 113219. [Google Scholar] [CrossRef]
- Zwilling, M.; Theiss, C.; Matschke, V. Caffeine and NAD+ Improve Motor Neural Integrity of Dissociated Wobbler Cells in vitro. Antioxidants 2020, 9, 460. [Google Scholar] [CrossRef]
- Hor, J.H.; Santosa, M.M.; Lim, V.J.W.; Ho, B.X.; Taylor, A.; Khong, Z.J.; Ravits, J.; Fan, Y.; Liou, Y.C.; Soh, B.S.; et al. ALS motor neurons exhibit hallmark metabolic defects that are rescued by SIRT3 activation. Cell Death Differ. 2021, 28, 1379–1397. [Google Scholar] [CrossRef]
- Zhou, Q.; Zhu, L.; Qiu, W.; Liu, Y.; Yang, F.; Chen, W.; Xu, R. Nicotinamide Riboside Enhances Mitochondrial Proteostasis and Adult Neurogenesis through Activation of Mitochondrial Unfolded Protein Response Signaling in the Brain of ALS SOD1G93A Mice. Int. J. Biol. Sci. 2020, 16, 284–297. [Google Scholar] [CrossRef] [Green Version]
- Harlan, B.A.; Pehar, M.; Killoy, K.M.; Vargas, M.R. Enhanced SIRT6 activity abrogates the neurotoxic phenotype of astrocytes expressing ALS-linked mutant SOD1. FASEB J. 2019, 33, 7084–7091. [Google Scholar] [CrossRef]
- Harlan, B.A.; Pehar, M.; Sharma, D.R.; Beeson, G.; Beeson, C.C.; Vargas, M.R. Enhancing NAD+ Salvage Pathway Reverts the Toxicity of Primary Astrocytes Expressing Amyotrophic Lateral Sclerosis-linked Mutant Superoxide Dismutase 1 (SOD1). J. Biol. Chem. 2016, 291, 10836–10846. [Google Scholar] [CrossRef] [Green Version]
- Mancuso, R.; del Valle, J.; Modol, L.; Martinez, A.; Granado-Serrano, A.B.; Ramirez-Nunez, O.; Pallas, M.; Portero-Otin, M.; Osta, R.; Navarro, X. Resveratrol improves motoneuron function and extends survival in SOD1G93A ALS mice. Neurotherapeutics 2014, 11, 419–432. [Google Scholar] [CrossRef] [Green Version]
- Herskovits, A.Z.; Hunter, T.A.; Maxwell, N.; Pereira, K.; Whittaker, C.A.; Valdez, G.; Guarente, L.P. SIRT1 deacetylase in aging-induced neuromuscular degeneration and amyotrophic lateral sclerosis. Aging Cell 2018, 17, e12839. [Google Scholar] [CrossRef] [PubMed]
- Buck, E.; Bayer, H.; Lindenberg, K.S.; Hanselmann, J.; Pasquarelli, N.; Ludolph, A.C.; Weydt, P.; Witting, A. Comparison of Sirtuin 3 Levels in ALS and Huntington’s Disease-Differential Effects in Human Tissue Samples vs. Transgenic Mouse Models. Front. Mol. Neurosci. 2017, 10, 156. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.G.; Shorter, J.; Wobst, H.J. Emerging small-molecule therapeutic approaches for amyotrophic lateral sclerosis and frontotemporal dementia. Bioorg. Med. Chem. Lett. 2020, 30, 126942. [Google Scholar] [CrossRef]
- McGurk, L.; Mojsilovic-Petrovic, J.; Van Deerlin, V.M.; Shorter, J.; Kalb, R.G.; Lee, V.M.; Trojanowski, J.Q.; Lee, E.B.; Bonini, N.M. Nuclear poly(ADP-ribose) activity is a therapeutic target in amyotrophic lateral sclerosis. Acta Neuropathol. Commun. 2018, 6, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veriepe, J.; Fossouo, L.; Parker, J.A. Neurodegeneration in C. elegans models of ALS requires TIR-1/Sarm1 immune pathway activation in neurons. Nat. Commun. 2015, 6, 7319. [Google Scholar] [CrossRef] [Green Version]
- White, M.A.; Lin, Z.; Kim, E.; Henstridge, C.M.; Pena Altamira, E.; Hunt, C.K.; Burchill, E.; Callaghan, I.; Loreto, A.; Brown-Wright, H.; et al. Sarm1 deletion suppresses TDP-43-linked motor neuron degeneration and cortical spine loss. Acta Neuropathol. Commun. 2019, 7, 166. [Google Scholar] [CrossRef]
- Peters, O.M.; Lewis, E.A.; Osterloh, J.M.; Weiss, A.; Salameh, J.S.; Metterville, J.; Brown, R.H.; Freeman, M.R. Loss of Sarm1 does not suppress motor neuron degeneration in the SOD1G93A mouse model of amyotrophic lateral sclerosis. Hum. Mol. Genet. 2018, 27, 3761–3771. [Google Scholar] [CrossRef]
- de la Rubia, J.E.; Drehmer, E.; Platero, J.L.; Benlloch, M.; Caplliure-Llopis, J.; Villaron-Casales, C.; de Bernardo, N.; AlarcOn, J.; Fuente, C.; Carrera, S.; et al. Efficacy and tolerability of EH301 for amyotrophic lateral sclerosis: A randomized, double-blind, placebo-controlled human pilot study. Amyotroph. Lateral Scler. Front. Degener. 2019, 20, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Stavrou, M.; Sargiannidou, I.; Georgiou, E.; Kagiava, A.; Kleopa, K.A. Emerging Therapies for Charcot-Marie-Tooth Inherited Neuropathies. Int. J. Mol. Sci. 2021, 22, 6048. [Google Scholar] [CrossRef] [PubMed]
- Moss, K.R.; Hoke, A. Targeting the programmed axon degeneration pathway as a potential therapeutic for Charcot-Marie-Tooth disease. Brain Res. 2020, 1727, 146539. [Google Scholar] [CrossRef]
- Freeman, M.R. Signaling mechanisms regulating Wallerian degeneration. Curr. Opin. Neurobiol. 2014, 27, 224–231. [Google Scholar] [CrossRef] [Green Version]
- Meyer zu Horste, G.; Miesbach, T.A.; Muller, J.I.; Fledrich, R.; Stassart, R.M.; Kieseier, B.C.; Coleman, M.P.; Sereda, M.W. The Wlds transgene reduces axon loss in a Charcot-Marie-Tooth disease 1A rat model and nicotinamide delays post-traumatic axonal degeneration. Neurobiol. Dis. 2011, 42, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.Y.; Gu, M.M.; Sun, L.H.; Guo, W.T.; Zhu, H.B.; Ma, J.F.; Yuan, W.T.; Kuang, Y.; Ji, B.J.; Wu, X.L.; et al. A nonsense mutation in DHTKD1 causes Charcot-Marie-Tooth disease type 2 in a large Chinese pedigree. Am. J. Hum. Genet. 2012, 91, 1088–1094. [Google Scholar] [CrossRef] [Green Version]
- Bolino, A.; Piguet, F.; Alberizzi, V.; Pellegatta, M.; Rivellini, C.; Guerrero-Valero, M.; Noseda, R.; Brombin, C.; Nonis, A.; D’Adamo, P.; et al. Niacin-mediated Tace activation ameliorates CMT neuropathies with focal hypermyelination. EMBO Mol. Med. 2016, 8, 1438–1454. [Google Scholar] [CrossRef] [PubMed]
- Cassereau, J.; Chevrollier, A.; Codron, P.; Goizet, C.; Gueguen, N.; Verny, C.; Reynier, P.; Bonneau, D.; Lenaers, G.; Procaccio, V. Oxidative stress contributes differentially to the pathophysiology of Charcot-Marie-Tooth disease type 2K. Exp. Neurol. 2020, 323, 113069. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Xie, L.; Shen, C.; Qi, Q.; Qin, Y.; Xing, J.; Zhou, D.; Qi, Y.; Yan, Z.; Lin, X.; et al. SIRT2-knockdown rescues GARS-induced Charcot-Marie-Tooth neuropathy. Aging Cell 2021, 20, e13391. [Google Scholar] [CrossRef]
- Chini, C.C.S.; Zeidler, J.D.; Kashyap, S.; Warner, G.; Chini, E.N. Evolving concepts in NAD+ metabolism. Cell Metab. 2021, 33, 1076–1087. [Google Scholar] [CrossRef]
- Hayes, M.T. Parkinson’s Disease and Parkinsonism. Am. J. Med. 2019, 132, 802–807. [Google Scholar] [CrossRef]
- Thirtamara-Rajamani, K.; Li, P.; Escobar Galvis, M.L.; Labrie, V.; Brundin, P.; Brundin, L. Is the Enzyme ACMSD a Novel Therapeutic Target in Parkinson’s Disease? J. Parkinsons Dis. 2017, 7, 577–587. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, P.J.; Cryan, J.F.; Dinan, T.G.; Clarke, G. Kynurenine pathway metabolism and the microbiota-gut-brain axis. Neuropharmacology 2017, 112, 399–412. [Google Scholar] [CrossRef] [PubMed]
- O’Mahony, S.M.; Clarke, G.; Borre, Y.E.; Dinan, T.G.; Cryan, J.F. Serotonin, tryptophan metabolism and the brain-gut-microbiome axis. Behav. Brain Res. 2015, 277, 32–48. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, D.; Iyer, M.; Narayanasamy, A.; Siva, K.; Vellingiri, B. Kynurenine pathway in Parkinson’s disease-An update. Eneurologicalsci 2020, 21, 100270. [Google Scholar] [CrossRef]
- Schwab, A.J.; Sison, S.L.; Meade, M.R.; Broniowska, K.A.; Corbett, J.A.; Ebert, A.D. Decreased Sirtuin Deacetylase Activity in LRRK2 G2019S iPSC-Derived Dopaminergic Neurons. Stem Cell Rep. 2017, 9, 1839–1852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, L.; Tang, L.; Wei, W.; Hong, Y.; Chen, H.; Ying, W.; Chen, S. Nicotinamide mononucleotide improves energy activity and survival rate in an in vitro model of Parkinson’s disease. Exp. Ther. Med. 2014, 8, 943–950. [Google Scholar] [CrossRef] [Green Version]
- Sheline, C.T.; Zhu, J.; Zhang, W.; Shi, C.; Cai, A.L. Mitochondrial inhibitor models of Huntington’s disease and Parkinson’s disease induce zinc accumulation and are attenuated by inhibition of zinc neurotoxicity in vitro or in vivo. Neurodegener. Dis. 2013, 11, 49–58. [Google Scholar] [CrossRef]
- Jia, H.; Li, X.; Gao, H.; Feng, Z.; Li, X.; Zhao, L.; Jia, X.; Zhang, H.; Liu, J. High doses of nicotinamide prevent oxidative mitochondrial dysfunction in a cellular model and improve motor deficit in a Drosophila model of Parkinson’s disease. J. Neurosci. Res. 2008, 86, 2083–2090. [Google Scholar] [CrossRef]
- Lehmann, S.; Costa, A.C.; Celardo, I.; Loh, S.H.; Martins, L.M. Parp mutations protect against mitochondrial dysfunction and neurodegeneration in a PARKIN model of Parkinson’s disease. Cell Death Dis. 2016, 7, e2166. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, S.; Loh, S.H.; Martins, L.M. Enhancing NAD+ salvage metabolism is neuroprotective in a PINK1 model of Parkinson’s disease. Biol. Open 2017, 6, 141–147. [Google Scholar] [CrossRef] [Green Version]
- Schondorf, D.C.; Ivanyuk, D.; Baden, P.; Sanchez-Martinez, A.; De Cicco, S.; Yu, C.; Giunta, I.; Schwarz, L.K.; Di Napoli, G.; Panagiotakopoulou, V.; et al. The NAD+ Precursor Nicotinamide Riboside Rescues Mitochondrial Defects and Neuronal Loss in iPSC and Fly Models of Parkinson’s Disease. Cell Rep. 2018, 23, 2976–2988. [Google Scholar] [CrossRef]
- Shan, C.; Gong, Y.L.; Zhuang, Q.Q.; Hou, Y.F.; Wang, S.M.; Zhu, Q.; Huang, G.R.; Tao, B.; Sun, L.H.; Zhao, H.Y.; et al. Protective effects of β- nicotinamide adenine dinucleotide against motor deficits and dopaminergic neuronal damage in a mouse model of Parkinson’s disease. Prog. Neuropsychopharmacol. Biol. Psychiatry 2019, 94, 109670. [Google Scholar] [CrossRef] [PubMed]
- De Jesus-Cortes, H.; Xu, P.; Drawbridge, J.; Estill, S.J.; Huntington, P.; Tran, S.; Britt, J.; Tesla, R.; Morlock, L.; Naidoo, J.; et al. Neuroprotective efficacy of aminopropyl carbazoles in a mouse model of Parkinson disease. Proc. Natl. Acad. Sci. USA 2012, 109, 17010–17015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naidoo, J.; De Jesus-Cortes, H.; Huntington, P.; Estill, S.; Morlock, L.K.; Starwalt, R.; Mangano, T.J.; Williams, N.S.; Pieper, A.A.; Ready, J.M. Discovery of a neuroprotective chemical, (S)-N-(3-(3,6-dibromo-9H-carbazol-9-yl)-2-fluoropropyl)-6-methoxypyridin-2-amine [(-)-P7C3-S243], with improved druglike properties. J. Med. Chem. 2014, 57, 3746–3754. [Google Scholar] [CrossRef]
- De Jesus-Cortes, H.; Miller, A.D.; Britt, J.K.; DeMarco, A.J.; De Jesus-Cortes, M.; Stuebing, E.; Naidoo, J.; Vazquez-Rosa, E.; Morlock, L.; Williams, N.S.; et al. Protective efficacy of P7C3-S243 in the 6-hydroxydopamine model of Parkinson’s disease. NPJ Parkinsons Dis. 2015, 1, 15010. [Google Scholar] [CrossRef] [Green Version]
- Santiago, J.A.; Littlefield, A.M.; Potashkin, J.A. Integrative transcriptomic meta-analysis of Parkinson’s disease and depression identifies NAMPT as a potential blood biomarker for de novo Parkinson’s disease. Sci. Rep. 2016, 6, 34579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.; Wu, Q.; Shi, J.; Zhou, S. Regulation of SIRT3 on mitochondrial functions and oxidative stress in Parkinson’s disease. Biomed. Pharmacother. 2020, 132, 110928. [Google Scholar] [CrossRef] [PubMed]
- Maszlag-Torok, R.; Boros, F.A.; Vecsei, L.; Klivenyi, P. Gene variants and expression changes of SIRT1 and SIRT6 in peripheral blood are associated with Parkinson’s disease. Sci. Rep. 2021, 11, 10677. [Google Scholar] [CrossRef]
- Li, X.; Liu, T.; Wu, T.T.; Feng, Y.; Peng, S.J.; Yin, H.; Wu, Y.C. SIRT1 Deacetylates TET2 and Promotes Its Ubiquitination Degradation to Achieve Neuroprotection Against Parkinson’s Disease. Front. Neurol. 2021, 12, 652882. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.D.; Tan, E.K. Oxidized nicotinamide adenine dinucleotide-dependent mitochondrial deacetylase sirtuin-3 as a potential therapeutic target of Parkinson’s disease. Ageing Res. Rev. 2020, 62, 101107. [Google Scholar] [CrossRef]
- Thapa, K.; Khan, H.; Sharma, U.; Grewal, A.K.; Singh, T.G. Poly (ADP-ribose) polymerase-1 as a promising drug target for neurodegenerative diseases. Life Sci. 2021, 267, 118975. [Google Scholar] [CrossRef]
- Salemi, M.; Mazzetti, S.; De Leonardis, M.; Giampietro, F.; Medici, V.; Poloni, T.E.; Cannarella, R.; Giaccone, G.; Pezzoli, G.; Cappelletti, G.; et al. Poly (ADP-ribose) polymerase 1 and Parkinson’s disease: A study in post-mortem human brain. Neurochem. Int. 2021, 144, 104978. [Google Scholar] [CrossRef] [PubMed]
- Wakade, C.; Chong, R.; Bradley, E.; Morgan, J.C. Low-dose niacin supplementation modulates GPR109A, niacin index and ameliorates Parkinson’s disease symptoms without side effects. Clin. Case Rep. 2015, 3, 635–637. [Google Scholar] [CrossRef]
- Chong, R.; Wakade, C.; Seamon, M.; Giri, B.; Morgan, J.; Purohit, S. Niacin Enhancement for Parkinson’s Disease: An Effectiveness Trial. Front. Aging Neurosci. 2021, 13, 667032. [Google Scholar] [CrossRef] [PubMed]
- Stoker, T.B.; Mason, S.L.; Greenland, J.C.; Holden, S.T.; Santini, H.; Barker, R.A. Huntington’s disease: Diagnosis and management. Pract. Neurol. 2021. [Google Scholar] [CrossRef]
- Kim, A.; Lalonde, K.; Truesdell, A.; Gomes Welter, P.; Brocardo, P.S.; Rosenstock, T.R.; Gil-Mohapel, J. New Avenues for the Treatment of Huntington’s Disease. Int. J. Mol. Sci. 2021, 22, 8363. [Google Scholar] [CrossRef] [PubMed]
- McGarry, A.; Gaughan, J.; Hackmyer, C.; Lovett, J.; Khadeer, M.; Shaikh, H.; Pradhan, B.; Ferraro, T.N.; Wainer, I.W.; Moaddel, R. Cross-sectional analysis of plasma and CSF metabolomic markers in Huntington’s disease for participants of varying functional disability: A pilot study. Sci. Rep. 2020, 10, 20490. [Google Scholar] [CrossRef]
- Singh, V.; Sharma, R.K.; Athilingam, T.; Sinha, P.; Sinha, N.; Thakur, A.K. NMR Spectroscopy-based Metabolomics of Drosophila Model of Huntington’s Disease Suggests Altered Cell Energetics. J. Proteome Res. 2017, 16, 3863–3872. [Google Scholar] [CrossRef]
- Bertrand, M.; Decoville, M.; Meudal, H.; Birman, S.; Landon, C. Metabolomic Nuclear Magnetic Resonance Studies at Presymptomatic and Symptomatic Stages of Huntington’s Disease on a Drosophila Model. J. Proteome Res. 2020, 19, 4034–4045. [Google Scholar] [CrossRef]
- Pallos, J.; Bodai, L.; Lukacsovich, T.; Purcell, J.M.; Steffan, J.S.; Thompson, L.M.; Marsh, J.L. Inhibition of specific HDACs and sirtuins suppresses pathogenesis in a Drosophila model of Huntington’s disease. Hum. Mol. Genet. 2008, 17, 3767–3775. [Google Scholar] [CrossRef] [Green Version]
- Hathorn, T.; Snyder-Keller, A.; Messer, A. Nicotinamide improves motor deficits and upregulates PGC-1α and BDNF gene expression in a mouse model of Huntington’s disease. Neurobiol. Dis. 2011, 41, 43–50. [Google Scholar] [CrossRef] [Green Version]
- Naia, L.; Rosenstock, T.R.; Oliveira, A.M.; Oliveira-Sousa, S.I.; Caldeira, G.L.; Carmo, C.; Laco, M.N.; Hayden, M.R.; Oliveira, C.R.; Rego, A.C. Comparative Mitochondrial-Based Protective Effects of Resveratrol and Nicotinamide in Huntington’s Disease Models. Mol. Neurobiol. 2017, 54, 5385–5399. [Google Scholar] [CrossRef]
- Caldwell, C.C.; Petzinger, G.M.; Jakowec, M.W.; Cadenas, E. Treadmill exercise rescues mitochondrial function and motor behavior in the CAG140 knock-in mouse model of Huntington’s disease. Chem. Biol. Interact. 2020, 315, 108907. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Li, C.; Tao, X.; Brazill, J.M.; Park, J.; Diaz-Perez, Z.; Zhai, R.G. Nmnat restores neuronal integrity by neutralizing mutant Huntingtin aggregate-induced progressive toxicity. Proc. Natl. Acad. Sci. USA 2019, 116, 19165–19175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lloret, A.; Beal, M.F. PGC-1α, Sirtuins and PARPs in Huntington’s Disease and Other Neurodegenerative Conditions: NAD+ to Rule Them All. Neurochem. Res. 2019, 44, 2423–2434. [Google Scholar] [CrossRef]
- Chopra, V.; Quinti, L.; Kim, J.; Vollor, L.; Narayanan, K.L.; Edgerly, C.; Cipicchio, P.M.; Lauver, M.A.; Choi, S.H.; Silverman, R.B.; et al. The sirtuin 2 inhibitor AK-7 is neuroprotective in Huntington’s disease mouse models. Cell Rep. 2012, 2, 1492–1497. [Google Scholar] [CrossRef] [Green Version]
- Bobrowska, A.; Donmez, G.; Weiss, A.; Guarente, L.; Bates, G. SIRT2 ablation has no effect on tubulin acetylation in brain, cholesterol biosynthesis or the progression of Huntington’s disease phenotypes in vivo. PLoS ONE 2012, 7, e34805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, D.J.; Calingasan, N.Y.; Wille, E.; Dumont, M.; Beal, M.F. Resveratrol protects against peripheral deficits in a mouse model of Huntington’s disease. Exp. Neurol. 2010, 225, 74–84. [Google Scholar] [CrossRef]
- Jeong, H.; Cohen, D.E.; Cui, L.; Supinski, A.; Savas, J.N.; Mazzulli, J.R.; Yates, J.R., 3rd; Bordone, L.; Guarente, L.; Krainc, D. Sirt1 mediates neuroprotection from mutant huntingtin by activation of the TORC1 and CREB transcriptional pathway. Nat. Med. 2011, 18, 159–165. [Google Scholar] [CrossRef]
- Tulino, R.; Benjamin, A.C.; Jolinon, N.; Smith, D.L.; Chini, E.N.; Carnemolla, A.; Bates, G.P. SIRT1 Activity Is Linked to Its Brain Region-Specific Phosphorylation and Is Impaired in Huntington’s Disease Mice. PLoS ONE 2016, 11, e0145425. [Google Scholar]
- Fu, J.; Jin, J.; Cichewicz, R.H.; Hageman, S.A.; Ellis, T.K.; Xiang, L.; Peng, Q.; Jiang, M.; Arbez, N.; Hotaling, K.; et al. Trans-(-)-epsilon-Viniferin increases mitochondrial sirtuin 3 (SIRT3), activates AMP-activated protein kinase (AMPK), and protects cells in models of Huntington Disease. J. Biol. Chem. 2012, 287, 24460–24472. [Google Scholar] [CrossRef] [Green Version]
- Vis, J.C.; Schipper, E.; de Boer-van Huizen, R.T.; Verbeek, M.M.; de Waal, R.M.; Wesseling, P.; ten Donkelaar, H.J.; Kremer, B. Expression pattern of apoptosis-related markers in Huntington’s disease. Acta Neuropathol. 2005, 109, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Paldino, E.; Cardinale, A.; D’Angelo, V.; Sauve, I.; Giampa, C.; Fusco, F.R. Selective Sparing of Striatal Interneurons after Poly (ADP-Ribose) Polymerase 1 Inhibition in the R6/2 Mouse Model of Huntington’s Disease. Front. Neuroanat. 2017, 11, 61. [Google Scholar] [CrossRef] [Green Version]
- Chidambaram, S.B.; Vijayan, R.; Sekar, S.; Mani, S.; Rajamani, B.; Ganapathy, R. Simultaneous blockade of NMDA receptors and PARP-1 activity synergistically alleviate immunoexcitotoxicity and bioenergetics in 3-nitropropionic acid intoxicated mice: Evidences from memantine and 3-aminobenzamide interventions. Eur. J. Pharmacol. 2017, 803, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Ozdinler, P.H.; Gautam, M.; Gozutok, O.; Konrad, C.; Manfredi, G.; Area Gomez, E.; Mitsumoto, H.; Erb, M.L.; Tian, Z.; Haase, G. Better understanding the neurobiology of primary lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener 2020, 21, 35–46. [Google Scholar] [CrossRef]
- Geser, F.; Stein, B.; Partain, M.; Elman, L.B.; McCluskey, L.F.; Xie, S.X.; Van Deerlin, V.M.; Kwong, L.K.; Lee, V.M.; Trojanowski, J.Q. Motor neuron disease clinically limited to the lower motor neuron is a diffuse TDP-43 proteinopathy. Acta Neuropathol. 2011, 121, 509–517. [Google Scholar] [CrossRef] [Green Version]
- Pourshafie, N.; Masati, E.; Bunker, E.; Nickolls, A.R.; Thepmankorn, P.; Johnson, K.; Feng, X.; Ekins, T.; Grunseich, C.; Fischbeck, K.H. Linking epigenetic dysregulation, mitochondrial impairment, and metabolic dysfunction in SBMA motor neurons. JCI Insight 2020, 5, e136539. [Google Scholar] [CrossRef]
- Scatozza, F.; Moschella, F.; D’Arcangelo, D.; Rossi, S.; Tabolacci, C.; Giampietri, C.; Proietti, E.; Facchiano, F.; Facchiano, A. Nicotinamide inhibits melanoma in vitro and in vivo. J. Exp. Clin. Cancer Res. 2020, 39, 211. [Google Scholar] [CrossRef] [PubMed]
- Deguise, M.O.; Chehade, L.; Kothary, R. Metabolic Dysfunction in Spinal Muscular Atrophy. Int. J. Mol. Sci. 2021, 22, 5913. [Google Scholar] [CrossRef]
- Narver, H.L.; Kong, L.; Burnett, B.G.; Choe, D.W.; Bosch-Marce, M.; Taye, A.A.; Eckhaus, M.A.; Sumner, C.J. Sustained improvement of spinal muscular atrophy mice treated with trichostatin A plus nutrition. Ann. Neurol. 2008, 64, 465–470. [Google Scholar] [CrossRef]
- Bowerman, M.; Murray, L.M.; Scamps, F.; Schneider, B.L.; Kothary, R.; Raoul, C. Pathogenic commonalities between spinal muscular atrophy and amyotrophic lateral sclerosis: Converging roads to therapeutic development. Eur. J. Med. Genet. 2018, 61, 685–698. [Google Scholar] [CrossRef]
- Braidy, N.; Poljak, A.; Grant, R.; Jayasena, T.; Mansour, H.; Chan-Ling, T.; Guillemin, G.J.; Smythe, G.; Sachdev, P. Mapping NAD+ metabolism in the brain of ageing Wistar rats: Potential targets for influencing brain senescence. Biogerontology 2014, 15, 177–198. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.; Wozniak, D.F.; Imai, S. CA1 Nampt knockdown recapitulates hippocampal cognitive phenotypes in old mice which nicotinamide mononucleotide improves. NPJ Aging Mech. Dis. 2018, 4, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lundt, S.; Ding, S. NAD+ Metabolism and Diseases with Motor Dysfunction. Genes 2021, 12, 1776. https://doi.org/10.3390/genes12111776
Lundt S, Ding S. NAD+ Metabolism and Diseases with Motor Dysfunction. Genes. 2021; 12(11):1776. https://doi.org/10.3390/genes12111776
Chicago/Turabian StyleLundt, Samuel, and Shinghua Ding. 2021. "NAD+ Metabolism and Diseases with Motor Dysfunction" Genes 12, no. 11: 1776. https://doi.org/10.3390/genes12111776
APA StyleLundt, S., & Ding, S. (2021). NAD+ Metabolism and Diseases with Motor Dysfunction. Genes, 12(11), 1776. https://doi.org/10.3390/genes12111776