
Hemoglobin A2 and Heterogeneous Diagnostic Relevance Observed in Eight New Variants of the Delta Globin Gene

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
3. Results
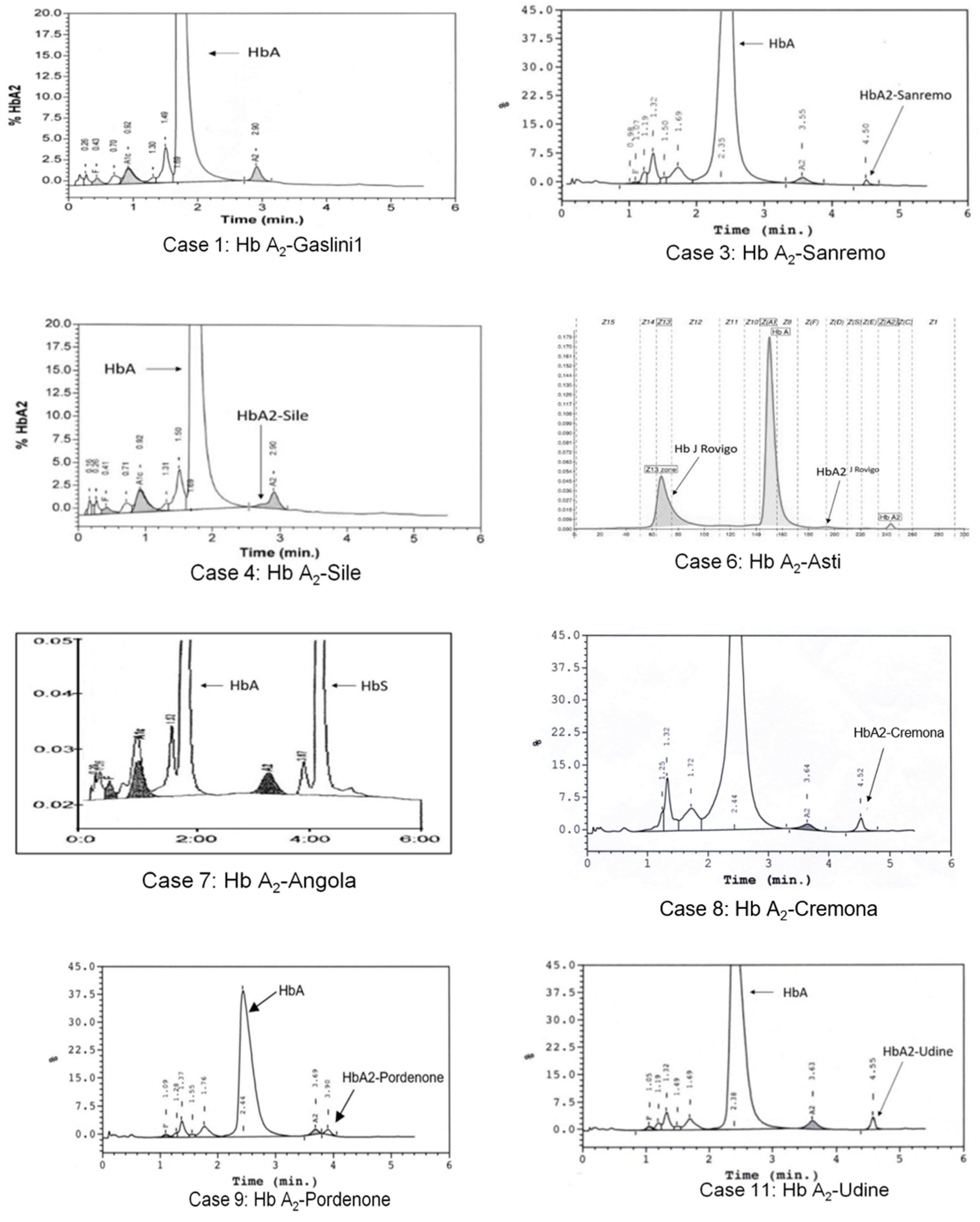
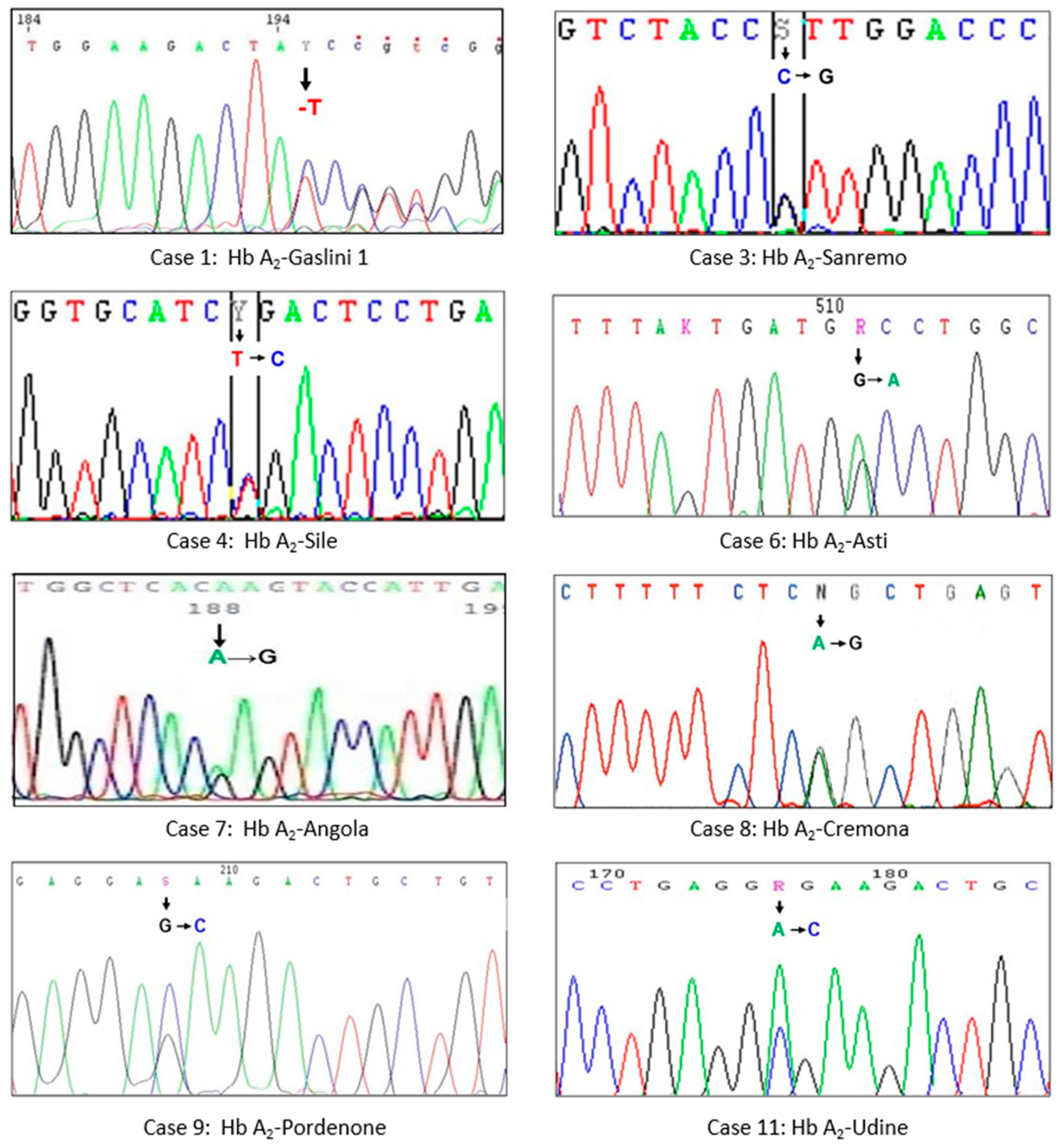
3.1. Hb A2-Gaslini 1
3.2. Hb A2-Sanremo
3.3. Hb A2-Sile
3.4. Hb A2-Asti
3.5. Hb A2-Angola
3.6. Hb A2-Cremona
3.7. Hb A2-Pordenone
3.8. Hb A2-Udine
4. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bain, B.J. Haemoglobin and the Genetics of Haemoglobin Synthesis. In Haemoglobinopathy Diagnosis, 2nd ed.; Blackwell Science: Oxford, UK, 2006; pp. 1–26. [Google Scholar]
- Traeger-Synodinos, J.; Harteveld, C.L.; Old, J.M.; Petrou, M.; Galanello, R.; Giordano, P.; Angastioniotis, M.; De la Salle, B.; Henderson, S.; May, A. EMQN Best Practice Guidelines for molecular and haematology methods for carrier identification and prenatal diagnosis of the haemoglobinopathies. Eur. J. Hum. Genet. 2015, 23, 426–437. [Google Scholar] [CrossRef] [Green Version]
- Giambona, A.; Passarello, C.; Vinciguerra, M.; Li Muli, R.; Teresi, P.; Anzà, M.; Ruggeri, G.; Renda, D.; Maggio, A. Significance of borderline hemoglobin A2 values in an Italian population with a high prevalence of β-thalassemia. Haematologica 2008, 93, 1380–1384. [Google Scholar] [CrossRef] [Green Version]
- Van Delft, P.; Lenters, E.; Bakker-Verweij, M.; De Korte, M.; Baylan, U.; Harteveld, C.R.; Giordano, P.C. Evaluating five dedicated automatic devices for hemoglobinopathy diagnostics in multiethnic populations. Int. J. Lab. Hematol. 2009, 31, 484–495. [Google Scholar] [CrossRef]
- Wajcman, H.; Azimi, M.; Cui, J.; Hoppe, C.; Flamini, M.; Ho, C.; Reddy, S. Hemoglobinopathy testing: The significance of accuracy and pitfalls in HbA2 determination. Int. J. Lab. Hematol. 2016, 39, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Paleari, R.; Ceriotti, F.; Harteveld, C.L.; Strollo, M.; Bakker-Verweij, G.; ter Huurne, J.; Bisoen, S.; Mosca, A. Calibration by commutable control materials is able to reduce inter-method differences of current high-performance methods for HbA2. Clin. Chim. Acta 2018, 477, 60–65. [Google Scholar] [CrossRef]
- Stephens, A.D.; Angastiniotis, M.; Baysal, E.; Chan, V.; Fucharoen, S.; Giordano, P.S.; Hoyer, J.D.; Mosca, A.; Wild, B.; International Council for the Standardisation of Haematology (ICSH). ICSH recommendations for the measurement of haemoglobin A2. Int. J. Lab. Hematol. 2012, 34, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Paleari, R.; Caruso, D.; Kaiser, P.; Arsene, C.G.; Schaeffer-Reiss, C.; Van Dorsselaer, A.; Bissé, E.; Ospina, M.; De Jesús, V.R.; Wild, B.; et al. Developing a reference system for the IFCC standardization of HbA2. Clin. Chim. Acta 2017, 467, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Bain, B.J. The Alpha, Beta, Delta and Gamma Thalassemias and Related Conditions. In Haemoglobinopathy Diagnosis, 2nd ed.; Blackwell Science: Oxford, UK, 2006; p. 98. [Google Scholar]
- Mosca, A.; Paleari, R.; Ivaldi, G.; Galanello, R.; Giordano, P.C. The role of haemoglobin A2 testing in the diagnosis of thalassaemias and related haemoglobinopathies. J. Clin. Pathol. 2009, 62, 13–17. [Google Scholar] [CrossRef]
- Old, J. Prevention and Diagnosis of Haemoglobinopathies: A Short Guide for Health Professionals and Laboratory Scientists; Publishers Thalassaemia International Federation No. 21; Thalassaemia International Federation: Strovolos, Cyprus, 2016. [Google Scholar]
- Giordano, P.C. Strategies for basic laboratory diagnostics of the hemoglobinopathies in multi-ethnic societies: Interpretation of results and pitfalls. Int. J. Lab. Hematol. 2013, 35, 465–479. [Google Scholar] [CrossRef]
- Ivaldi, G.; Barberio, G.; Harteveld, C.; Giordano, P. Hb A2 measurements in beta-thalassemia and in other conditions. Thalassemia Rep. 2014, 4, 45–48. [Google Scholar]
- Paglietti, M.E.; Satta, S.; Sollaino, M.C.; Barella, S.; Ventrella, A.; Desogus, M.F.; Demartis, F.R.; Manunza, L.; Origa, R. The Problem of Borderline Hemoglobin A2 Levels in the Screening for β-Thalassemia Carriers in Sardinia. Acta Haematol. 2016, 135, 193–199. [Google Scholar] [CrossRef]
- Giambona, A.; Passarello, C.; Ruggeri, G.; Renda, D.; Teresi, P.; Anza, M.; Maggio, A. Analysis of δ-globin gene alleles in the Sicilian population: Identification of five new mutations. Hematologica 2006, 91, 1681–1684. [Google Scholar]
- Rets, A.V.; Reading, N.S.; Agarual, A.M. δ-Globin Chain Variants Associated with Decreased Hb A2 Levels: A National Reference Laboratory Experience. Hemoglobin 2020, 44, 438–441. [Google Scholar] [CrossRef] [PubMed]
- Khalil, M.S.; Marouf, S.; Element, D.; Timbs, A.; Gallienne, A.; Schuh, A.; Old, J.M.; Henderson, S. A study of δ-globin gene mutations in the UK population: Identification of three novel variants and development of a novel DNA test for Hb A’2. Hemoglobin 2014, 38, 201–206. [Google Scholar] [CrossRef]
- De Angioletti, M.; Lacerra, G.; Gaudiano, C.; Mastrolonardo, G.; Pagano, L.; Mastrullo, L.; Masciandaro, S.; Carestia, C. Epidemiology of the delta globin alleles in southern Italy shows complex molecular, genetic, and phenotypic features. Hum. Mutat. 2002, 20, 358–367. [Google Scholar] [CrossRef] [PubMed]
- Dodé, C.; Rochette, J.; Krishnamoorthy, R. Locus assignment of human alpha globin mutations by selective amplification and direct sequencing. Br. J. Haematol 1990, 76, 275–281. [Google Scholar] [CrossRef]
- Olds, R.J.; Sura, T.; Jachson, B.; Wonke, B.; Hoffbrand, A.V.; Thein, S.L. A novel delta 0 mutation in cis with Hb Knossos: A study of different genetic interactions in three Egyptian families. Br. J. Haematol. 1991, 78, 430–436. [Google Scholar] [CrossRef] [PubMed]
- HbVar. A Database of Human Hemoglobin Variants and Thalassemias. Available online: http://globin.cse.psu.edu/hbvar/menu.html (accessed on 31 October 2021).
- Den Dunnen, J.T.; Dalgelish, R.; Maglott, D.R.; Hart, R.K.; Greenblatt, M.S.; McGowan-Jordan, J.; Roux, A.-F.; Smith, T.; Antonarakis, S.E.; Taschner, P.E.M.; et al. Update: The HGVS recommendations for the description of sequence variants. Hum. Mutat. 2016, 37, 564–569. [Google Scholar] [CrossRef] [Green Version]
- Sansone, G.; Carrell, R.W.; Lehmann, H. Haemoglobin Genova: β28 (B10) leucine replaced by proline. Nature 1967, 214, 877–879. [Google Scholar] [CrossRef] [PubMed]
- Hall, G.W.; Thein, S.L.; Newland, A.C.; Chisholm, M.; Traeger-Synodinos, J.; Kanavakis, E.; Kattamis, C.; Higgs, D.R. A base substitution (T → C) in codon 29 of the alpha 2-globin gene causes alpha thalassaemia. Br. J. Haematol. 1993, 85, 546–552. [Google Scholar] [CrossRef]
- Galanello, R.; Ruggeri, R.; Addis, M.; Paglietti, E.; Cao, A. Hemoglobin A2 in iron deficient β-thalassemia heterozygotes. Hemoglobin 1981, 5, 613–618. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| δ Variant (Name) | Sex-Age | RBC (×1012/L) | Hb (g/dL) | MCV (fL) | MCH (pg) | Hb A2 (%) | Hb A2X (%) | Iron Level | HBD HGVS Nomenclature | Other Globin Mutations | Case |
|---|---|---|---|---|---|---|---|---|---|---|---|
| δ130(-T) (Hb A2-Gaslini1) | F-35 | 4.66 | 12.0 | 81.0 | 27.0 | 1.6 | n.d. | Normal | HBD:c.391delT | 1 | |
| δ36(C2)Pro > Arg (Hb A2-Sanremo) | F-26 | 5.16 | 9.8 | 64.7 | 19.0 | 1.5 | n.d. | Low | HBD:c.110C > G | 2 | |
| M-58 | 4.69 | 14.1 | 87.6 | 30.1 | 1.5 | 0.5 | Normal | HBD:c.110C > G | 3 | ||
| δ3(NA3)Leu > Pro (Hb A2-Sile) | F-26 | 4.20 | 13.0 | 90.8 | 31.1 | 1.7(a) | n.d. | Normal | HBD:c.11T > C | 4 | |
| δ74(E18)Gly > Asp (Hb A2-Asti) | M-10 | 6.87 | 10.8 | 56.8 | 15.7 | 2.7 | n.d. | Normal | HBD:c.224G > A | HBB:c.118C > T (cd39 C > T) | 5 |
| M-45 | 5.73 | 17.3 | 91.1 | 30.2 | 1.0 | n.d. | n.d. | HBD:c.224G > A | HBA2:c.618C > A (Hb J Rovigo) | 6 | |
| δ144(HC1)Lys > Glu (Hb A2-Angola) | F-33 | 3.93 | 11.8 | 82.2 | 28.2 | 2.0 | n.d. | n.d. | HBD:c.433A > G | HBB:c.20A > T (HbS) | 7 |
| δ87(A4)Gln > Arg (Hb A2-Cremona) | F-40 | 4.43 | 12.7 | 86.8 | 28.7 | 1.5 | 1.0 | Normal | HBD:c.263A > G | 8 | |
| δ7(A4)Glu > Asp (Hb A2-Pordenone) | F-62 | 5.61 | 13.8 | 78.7 | 24.7 | 1.3 | 1.3 | Normal | HBD:c.24G > C | 9 | |
| F-80 | 4.91 | 14.1 | 90.4 | 28.7 | 1.6 | 1.4 | Normal | HBD:c.23A > C | 10 | ||
| δ7(A4)Glu > Ala (Hb A2-Udine) | M-54 | 5.86 | 14.0 | 65.6 | 23.8 | 2.8 | 2.2 | Normal | HBD:c.23A > C | HBB:c.118C > T (cd39 C > T) | 11 |
| F-26 | 5.28 | 13.3 | 78.8 | 25.1 | 1.8 | 1.2 | n.d. | HBD:c.23A > C | 12 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahmud, N.; Maffei, M.; Mogni, M.; Forni, G.L.; Pinto, V.M.; Barberio, G.; Ungari, S.; Maffè, A.; Curcio, C.; Zanolli, F.; et al. Hemoglobin A2 and Heterogeneous Diagnostic Relevance Observed in Eight New Variants of the Delta Globin Gene. Genes 2021, 12, 1821. https://doi.org/10.3390/genes12111821
Mahmud N, Maffei M, Mogni M, Forni GL, Pinto VM, Barberio G, Ungari S, Maffè A, Curcio C, Zanolli F, et al. Hemoglobin A2 and Heterogeneous Diagnostic Relevance Observed in Eight New Variants of the Delta Globin Gene. Genes. 2021; 12(11):1821. https://doi.org/10.3390/genes12111821
Chicago/Turabian StyleMahmud, Noraesah, Massimo Maffei, Massimo Mogni, Gian Luca Forni, Valeria Maria Pinto, Giuseppina Barberio, Silvana Ungari, Antonella Maffè, Cristina Curcio, Francesco Zanolli, and et al. 2021. "Hemoglobin A2 and Heterogeneous Diagnostic Relevance Observed in Eight New Variants of the Delta Globin Gene" Genes 12, no. 11: 1821. https://doi.org/10.3390/genes12111821
APA StyleMahmud, N., Maffei, M., Mogni, M., Forni, G. L., Pinto, V. M., Barberio, G., Ungari, S., Maffè, A., Curcio, C., Zanolli, F., Paventa, R., Carta, M., Caleffi, A., Mercadanti, M., Maoggi, S., Ivaldi, G., & Coviello, D. (2021). Hemoglobin A2 and Heterogeneous Diagnostic Relevance Observed in Eight New Variants of the Delta Globin Gene. Genes, 12(11), 1821. https://doi.org/10.3390/genes12111821