Novel KCNH1 Mutations Associated with Epilepsy: Broadening the Phenotypic Spectrum of KCNH1-Associated Diseases

 , , and
, , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Human Subjects and DNA Collection from the Paraffin Samples
2.2. Exome Sequencing
2.3. Bioinformatical Analyses and 3D Structure Prediction
3. Results
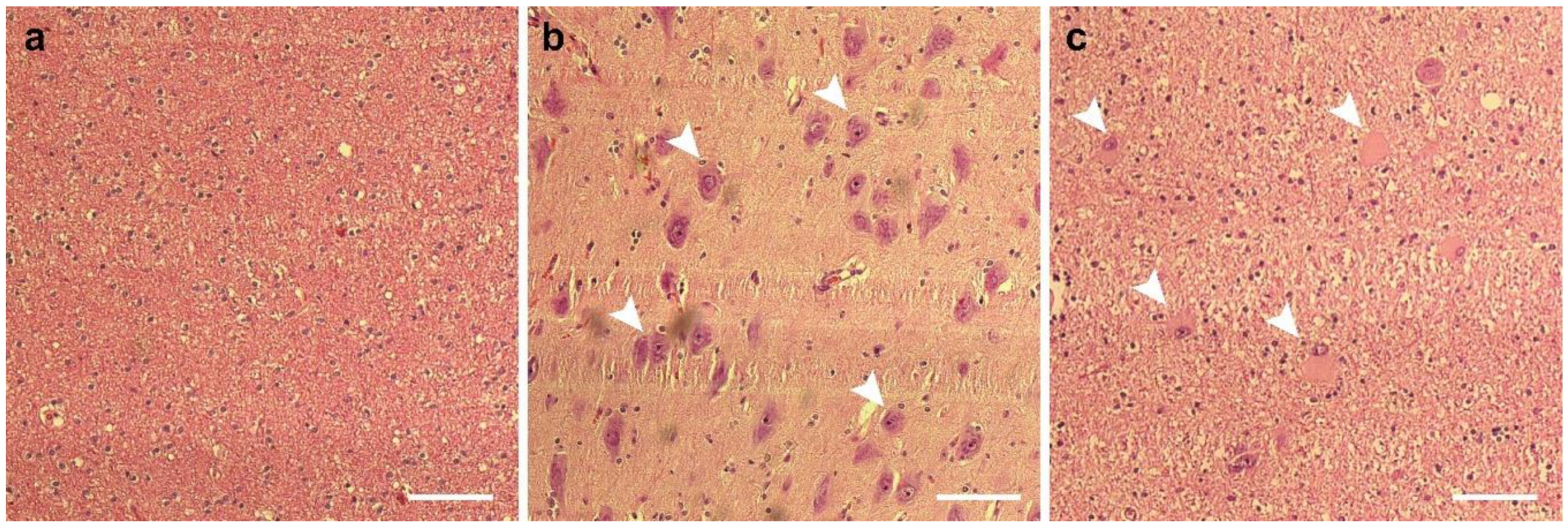
3.1. Case Reports
3.1.1. Case 1
3.1.2. Case 2
3.1.3. Case 3
3.1.4. Case 4
3.2. De Novo Missense Mutations, but Also Germline Missense Mutations with Reduced Penetrance and Somatic Missense Mutations in KCNH1 Are Associated with Epilepsy
3.3. Evolutionary Conservation of the Mutated Residue and Computational Prediction of the 3D Structure of KCNH1 p.Val713Glu
4. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kortüm, F.; Caputo, V.; Bauer, C.K.; Stella, L.; Ciolfi, A.; Alawi, M.; Bocchinfuso, G.; Flex, E.; Paolacci, S.; Dentici, M.L.; et al. Mutations in KCNH1 and ATP6V1B2 cause Zimmermann-Laband syndrome. Nat. Genet. 2015, 47, 661–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simons, C.; Rash, L.D.; Crawford, J.; Ma, L.; Cristofori-Armstrong, B.; Miller, D.; Ru, K.; Baillie, G.J.; Alanay, Y.; Jacquinet, A.; et al. Mutations in the voltage-gated potassium channel gene KCNH1 cause Temple-Baraitser syndrome and epilepsy. Nat. Genet. 2014, 47, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Mastrangelo, M.; Scheffer, I.E.; Bramswig, N.C.; Nair, L.D.V.; Myers, C.T.; Dentici, M.L.; Korenke, G.C.; Schoch, K.; Campeau, P.M.; White, S.M.; et al. Epilepsy in KCNH1-related syndromes. Epileptic Disord. 2016, 18, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Mégarbané, A.; Al-Ali, R.; Choucair, N.; Lek, M.; Wang, E.; Ladjimi, M.; Rose, C.M.; Hobeika, R.; Macary, Y.; Temanni, R.; et al. Temple-Baraitser Syndrome and Zimmermann-Laband Syndrome: One clinical entity? BMC Med. Genet. 2016, 17, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, S.A.; Dykes, D.D.; Polesky, H.F. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988, 16, 1215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudin, A.P.; Baron, G.; Zsurka, G.; Hampel, K.G.; Elger, C.E.; Grote, A.; Weber, Y.G.; Lerche, H.; Thiele, H.; Nürnberg, P.; et al. Homozygous mutation in TXNRD1 is associated with genetic generalized epilepsy. Free. Radic. Biol. Med. 2017, 106, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Varbank. Available online: https://varbank.ccg.uni-koeln.de/varbank2/ (accessed on 20 December 2020).
- Yang, J.; Anishchenko, I.; Park, H.; Peng, Z.; Ovchinnikov, S.; Baker, D. Improved protein structure prediction using predicted interresidue orientations. Proc. Natl. Acad. Sci. USA 2020, 117, 1496–1503. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; MacArthur, M.W.; Thornton, J.M. PROCHECK: Validation of protein-structure coordinates. In International Tables for Crystallography; Crystallography of Biological Macromolecules; Springer: Heidelberg, Germany, 2012; Volume F, pp. 684–687. [Google Scholar] [CrossRef]
- von Voss, H.; Schulz, J.; Borggraefe, I.; Kutsche, K.; Wilke, C.; Peraud, A.; Fellinger, J.; Marschall, C.; Heinrich, U.; Klein, H.; et al. De novo variant in the KCNH1 gene associated with therapy refractory epilepsy. Paediatrische Praxis 2020, 93, 195–205. [Google Scholar]
- Baldassari, S.; Ribierre, T.; Marsan, E.; Adle-Biassette, H.; Ferrand-Sorbets, S.; Bulteau, C.; Dorison, N.; Fohlen, M.; Polivka, M.; Weckhuysen, S.; et al. Dissecting the genetic basis of focal cortical dysplasia: A large cohort study. Acta Neuropathol. 2019, 138, 885–900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blümcke, I.; Sarnat, H.B. Somatic mutations rather than viral infection classify focal cortical dysplasia type II as mTORopathy. Curr. Opin. Neurol. 2016, 29, 388–395. [Google Scholar] [CrossRef] [PubMed]
- GnomAD. Available online: https://gnomad.broadinstitute.org/gene/ENSG00000143473?dataset=gnomad_r2_1 (accessed on 20 December 2020).
- Alves, J.T.G.; Stühmer, W. Calmodulin Interaction with hEAG1 Visualized by FRET Microscopy. PLoS ONE 2010, 5, e10873. [Google Scholar] [CrossRef] [Green Version]
- Schönherr, R.; Löber, K.; Heinemann, S.H. Inhibition of human ether a go-go potassium channels by Ca2+/calmodulin. EMBO J. 2000, 19, 3263–3271. [Google Scholar] [CrossRef] [PubMed]
- Ziechner, U.; Schönherr, R.; Born, A.-K.; Gavrilova-Ruch, O.; Glaser, R.W.; Malešević, M.; Küllertz, G.; Heinemann, S.H. Inhibition of human ether a go-go potassium channels by Ca2+/calmodulin binding to the cytosolic N- and C-termini. FEBS J. 2006, 273, 1074–1086. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | KCNH1 Mutation | Inheritance | Phenotype |
|---|---|---|---|
| Case 1 | p.Lys199Arg | de novo | DEE |
| Case 2 | p.Arg535* | germline (pi) | GGE+ |
| Case 3 | p.Val713Glu | germline (mi) | GGE |
| Case 4 | p.Val713Glu | somatic | FCD IIb |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
von Wrede, R.; Jeub, M.; Ariöz, I.; Elger, C.E.; von Voss, H.; Klein, H.-G.; Becker, A.J.; Schoch, S.; Surges, R.; Kunz, W.S. Novel KCNH1 Mutations Associated with Epilepsy: Broadening the Phenotypic Spectrum of KCNH1-Associated Diseases. Genes 2021, 12, 132. https://doi.org/10.3390/genes12020132
von Wrede R, Jeub M, Ariöz I, Elger CE, von Voss H, Klein H-G, Becker AJ, Schoch S, Surges R, Kunz WS. Novel KCNH1 Mutations Associated with Epilepsy: Broadening the Phenotypic Spectrum of KCNH1-Associated Diseases. Genes. 2021; 12(2):132. https://doi.org/10.3390/genes12020132
Chicago/Turabian Stylevon Wrede, Randi, Monika Jeub, Idil Ariöz, Christian E. Elger, Hubertus von Voss, Hanns-Georg Klein, Albert J. Becker, Susanne Schoch, Rainer Surges, and Wolfram S. Kunz. 2021. "Novel KCNH1 Mutations Associated with Epilepsy: Broadening the Phenotypic Spectrum of KCNH1-Associated Diseases" Genes 12, no. 2: 132. https://doi.org/10.3390/genes12020132
APA Stylevon Wrede, R., Jeub, M., Ariöz, I., Elger, C. E., von Voss, H., Klein, H. -G., Becker, A. J., Schoch, S., Surges, R., & Kunz, W. S. (2021). Novel KCNH1 Mutations Associated with Epilepsy: Broadening the Phenotypic Spectrum of KCNH1-Associated Diseases. Genes, 12(2), 132. https://doi.org/10.3390/genes12020132