Ensemble-AMPPred: Robust AMP Prediction and Recognition Using the Ensemble Learning Method with a New Hybrid Feature for Differentiating AMPs


Abstract
:1. Introduction
2. Materials and Methods
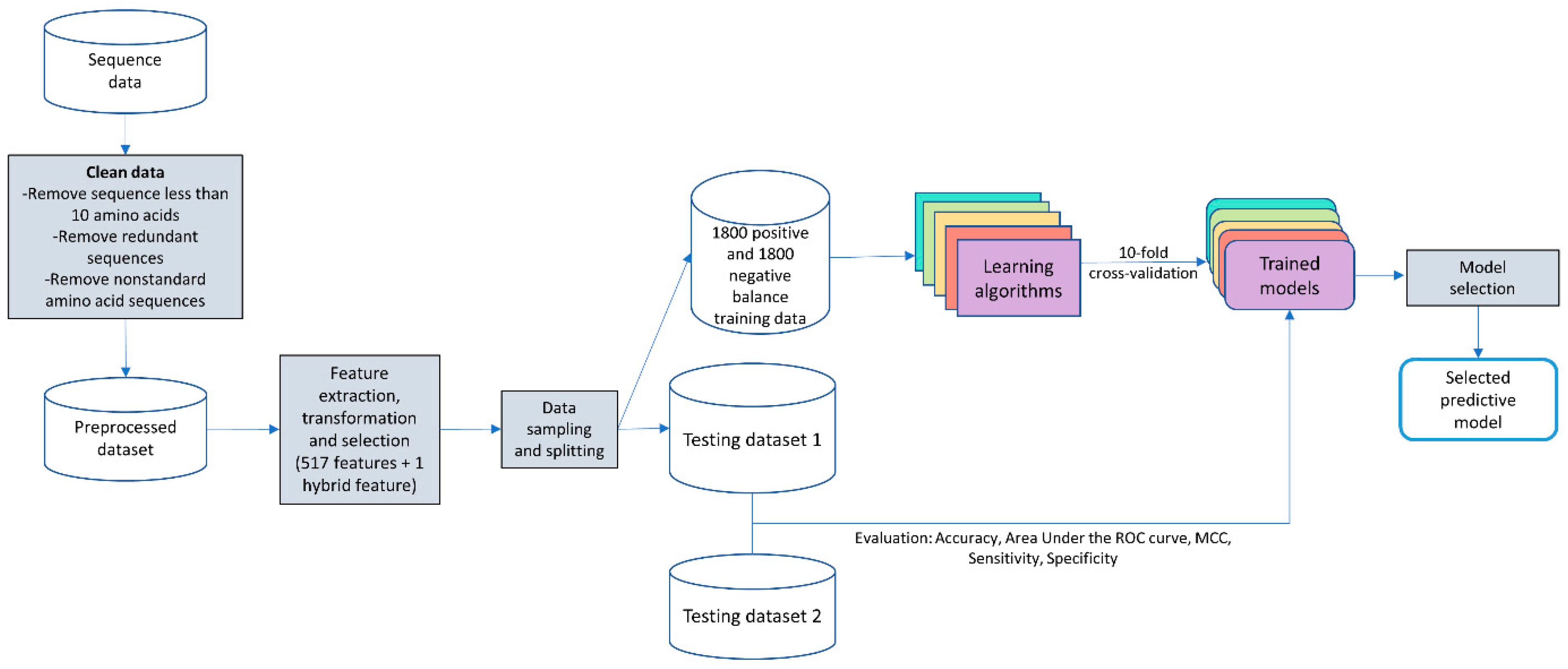
2.1. Workflow of Ensemble-AMPPred
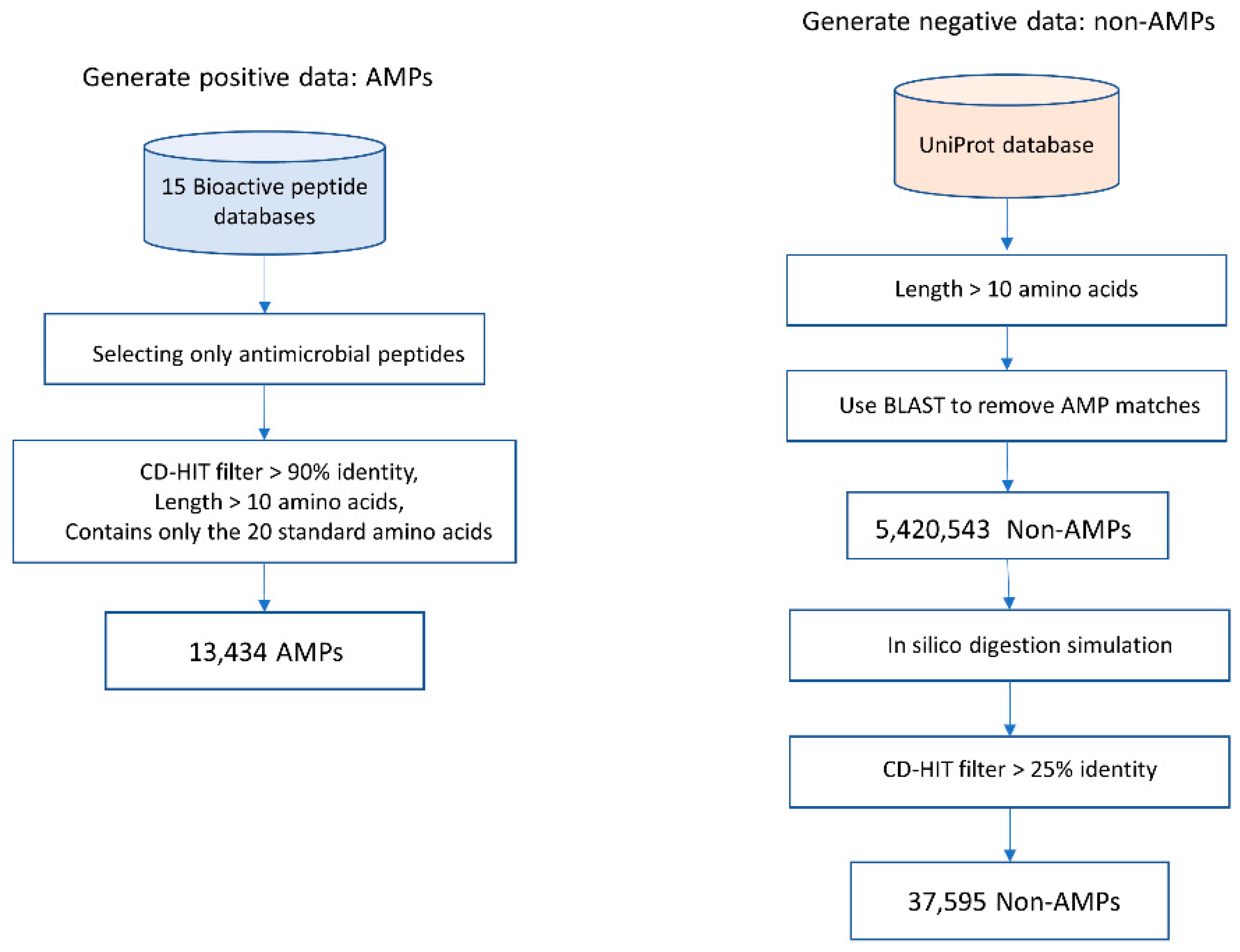
2.2. Dataset Preparation
- AMP data were collected from 15 public bioactive peptide databases, as listed in Table 2. Only peptides that have description-matched antimicrobial activities were selected. Peptide sequences with lengths <10 amino acids were discarded. To reduce data redundancy, we applied the Cluster Database at High Identity with Tolerance (CD-HIT) program [24] with threshold of 0.9 (90% sequence similarity). A total of 13,434 peptides were used as positive sequence data. Notably, lower sequence similarity thresholds (less than 50%) might reduce the sequence homology bias and could improve the model reliability [25]. Since AMPs are highly heterogeneous substances and there are likely various novel subtypes of AMPs that have not been discovered, using a threshold of 0.9 is applicable to identifying a novel AMP sequence.
- Currently, there is no database of experimentally verified non-AMP available. Therefore, we build negative data using the approach described in [13,16]. Negative data or non-AMP data were collected from the UniProt [26] database (February 2020) by choosing only proteins that do not contain functional information related to antimicrobial activity and do not have a secretory signal peptide position. The basic local alignment search tool (BLAST) was used to filter out AMP matches. Peptide sequences with lengths <10 amino acids were discarded. Then, the in silico enzymatic digestion simulation [27] was performed to digest polypeptides into digested peptide sequences. Then, the CD-HIT program [24] was used to remove peptide sequences with >25% identity. Therefore, a total of 37,595 peptides were designated as negative sequence data.
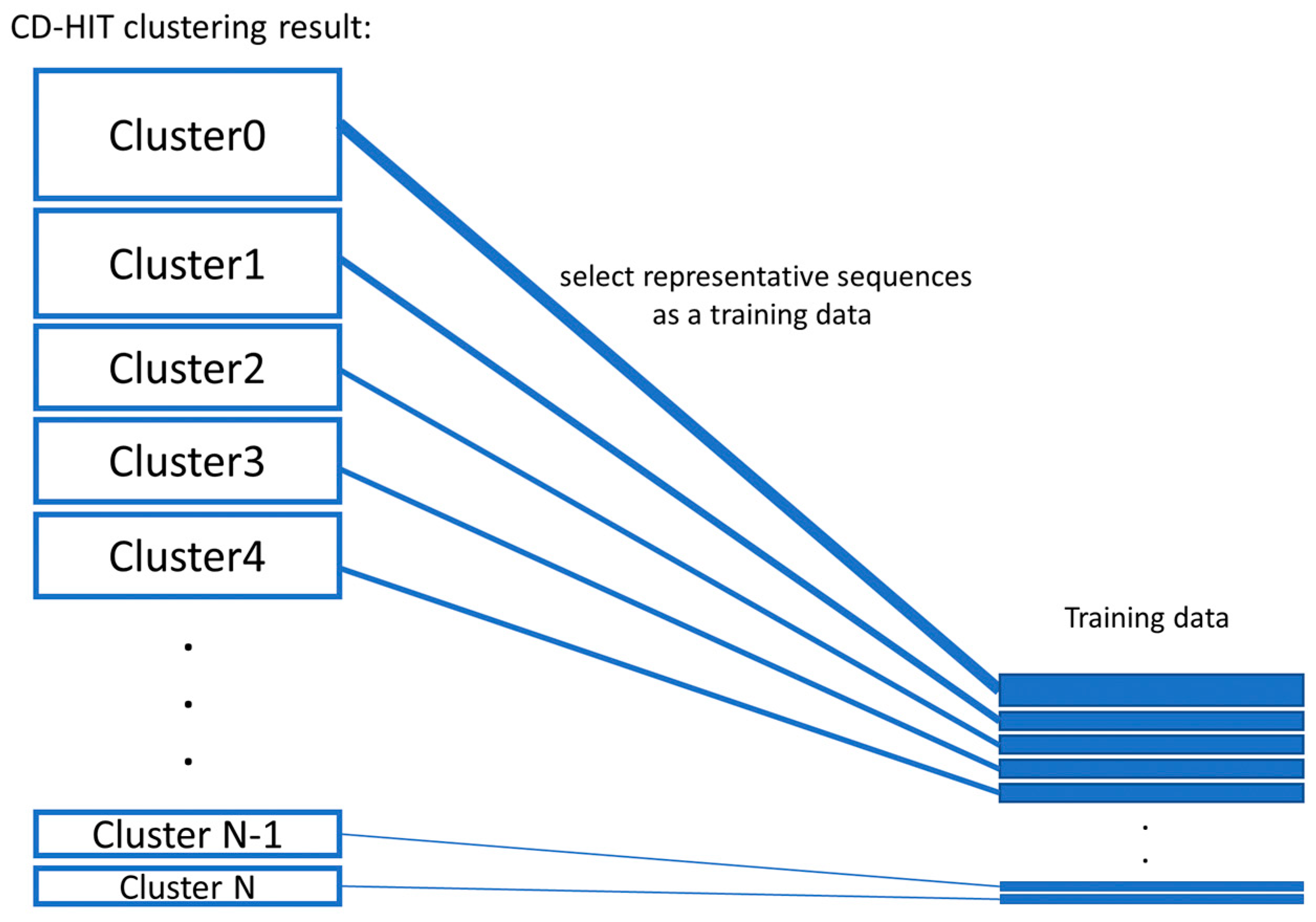
- Balanced training data were created by proportionate stratified random sampling to select peptide sequences to represent the positive and negative data. The stratified sampling was conducted by similarity clustering of sequence data into homogenous strata based on the CD-HIT clustering tool. The proportional stratified random sampling was performed with the following steps. (i) The sequences were clustered by using CD-HIT with a similarity threshold of 0.3. (ii) Representative sequences were selected from each cluster to use as training data, while the other remaining sequences that were not representative of the cluster will be used as testing data (results are shown in Supplementary File S1 the representative sequences are denoted with the * symbol at the end of the line; the non-representative sequences are displayed with the percentage of sequence similarity to the representative sequence of that cluster). We balanced the number of representative sequences based on the cluster size, as shown in Figure 3. Note that there is no testing sequence presented in the cluster with one sequence member. For some clusters that contain only 1 sequence, the sequence in that cluster will be used as a training set, and the testing sequence is not presented in that cluster. A summary of the sequence similarities between training and testing sequences in all clusters is presented in Supplementary File S2. The sequence similarity between training and testing sequences falls between 30% and 89.47%, with an average of 47.29%. Finally, the training data consists of 1800 peptide sequences of the AMP dataset and 1800 sequences of the non-AMP dataset to make an evenly balanced training dataset in order to reduce the likelihood of generating a predictive model biased toward the majority class.
- Two testing datasets (testing dataset 1 and testing dataset 2) were created. The first set of testing data was the remaining sequences of both positive data and negative data after training data preparation. Therefore, the first testing dataset consists of 11,634 positive sequence data or AMPs and 35,795 negative sequence data or non-AMPs. The second set of testing data was the benchmark dataset S from published works [13,16]. This dataset can be downloaded from the websites ([28,29]).
- Sequences having less than 10 amino acids were removed from further analysis. The benchmark dataset S1 contains 1461 AMPs (classified into five functional types: antibacterial peptides, anticancer/tumor peptides, antifungal peptides, anti-HIV peptides, and antiviral peptides) and 2404 non-AMPs, and the dataset S2 contains 917 AMP sequences and 828 non-AMP sequences.
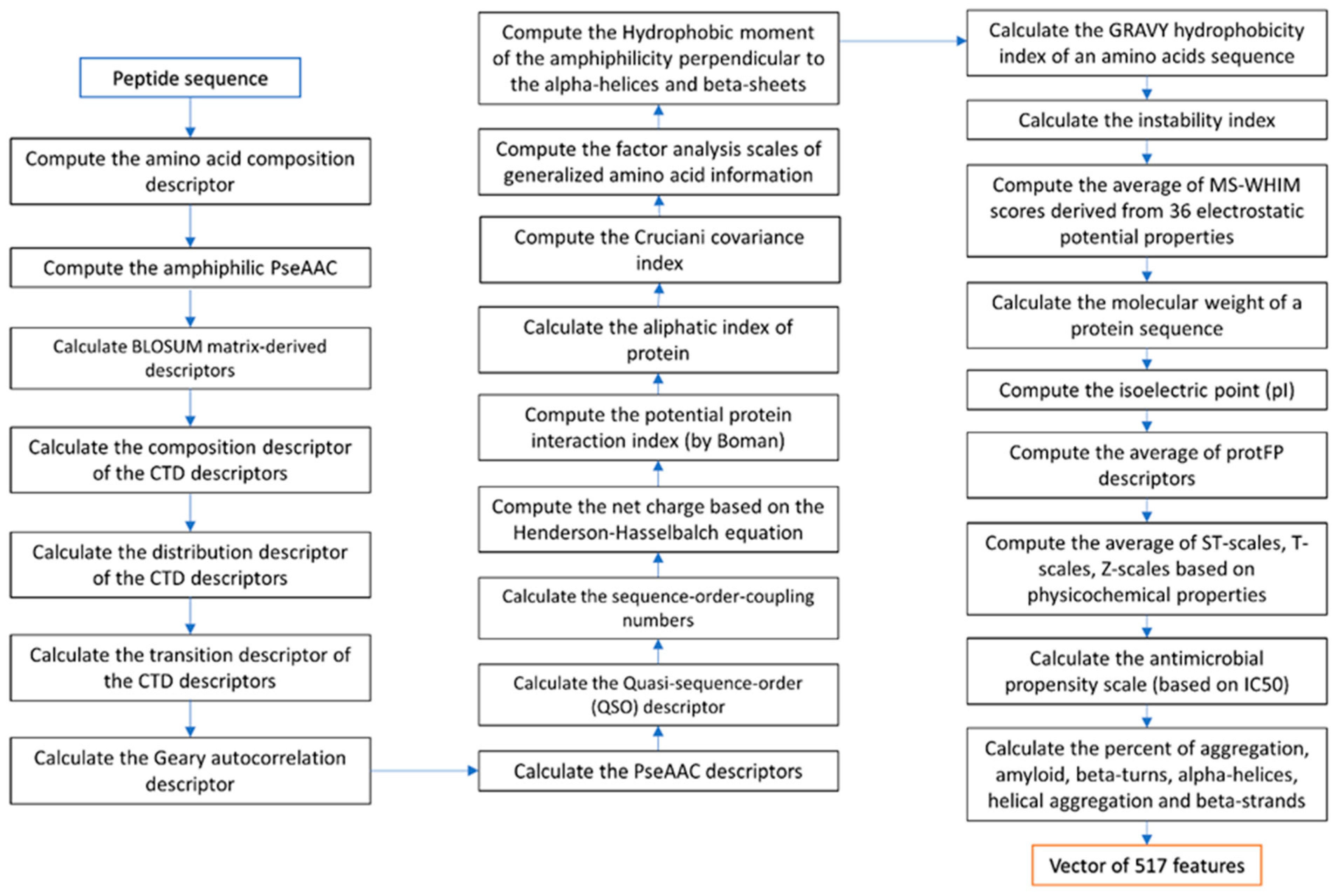
2.3. Feature Extraction and Feature Engineering
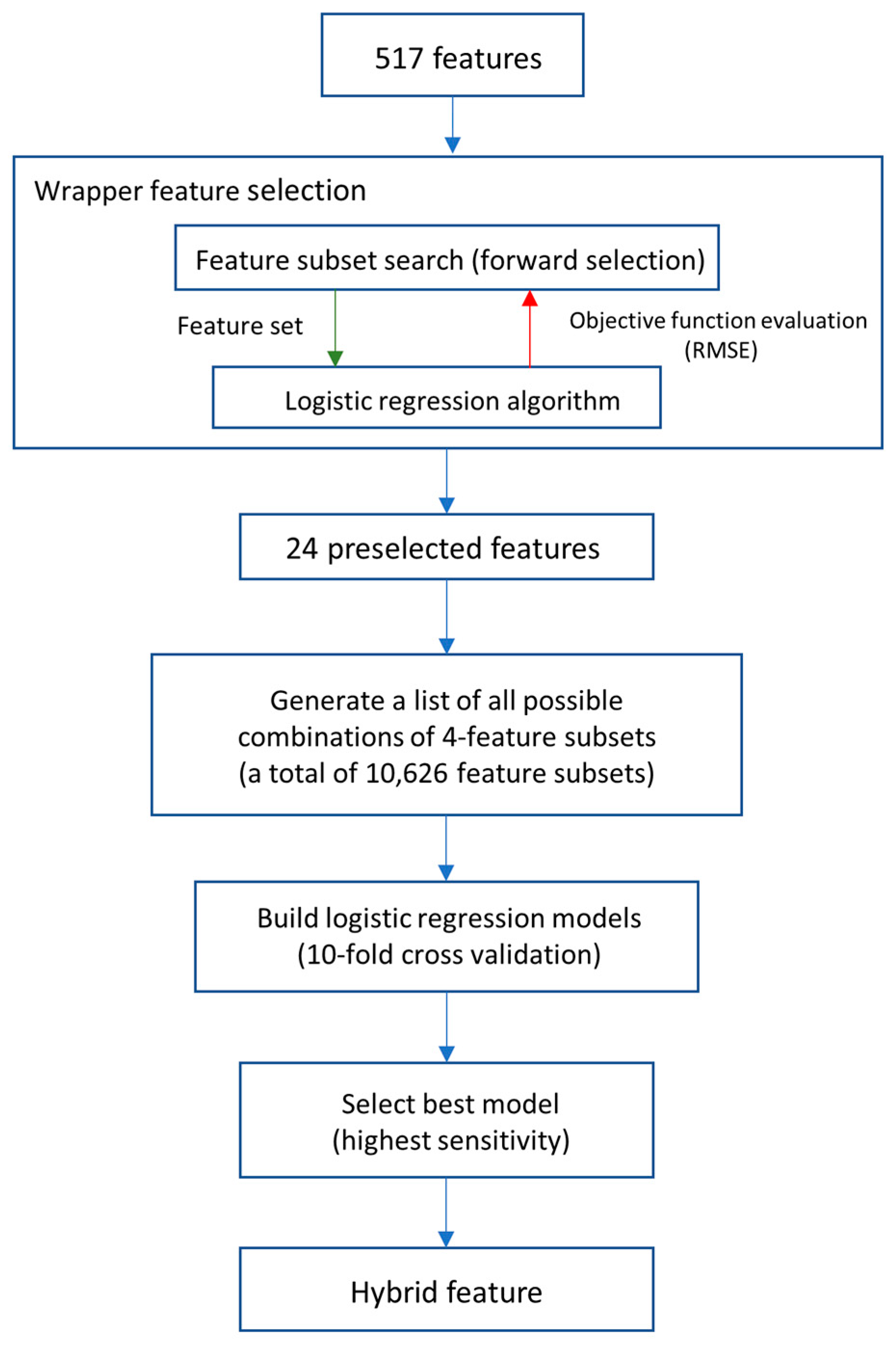
2.4. Feature Selection
2.5. Prediction Models
3. Results and Discussion
3.1. Informative Features Extracted from Peptides Affect the Performance the Most
3.2. Performance Comparison of Various Predictive Models
3.3. Ensemble Models have Better Performance than Single Models
3.4. Hybrid Feature to Improve the Sensitivity of the Predictive Performance
3.5. Comparison with Existing Prediction Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wu, Q.; Ke, H.; Li, D.; Wang, Q.; Fang, J.; Zhou, J. Recent progress in machine learning-based prediction of peptide activity for drug discovery. Curr. Top. Med. Chem. 2019, 19, 4–16. [Google Scholar] [CrossRef] [PubMed]
- Torres, M.D.T.; Sothiselvam, S.; Lu, T.K.; Fuente-Nunez, C. Peptide design principles for antimicrobial applications. J. Mol. Biol. 2019, 431, 3547–3567. [Google Scholar] [CrossRef] [PubMed]
- Torrent, M.; Di Tommaso, P.; Pulido, D.; Nogués, M.V.; Notredame, C.; Boix, E.; Andreu, D. AMPA: An automated web server for prediction of protein antimicrobial regions. Bioinformatics 2011, 28, 130–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, C.R.; Jhong, J.H.; Wang, Z.; Chen, S.; Wan, Y.; Horng, J.T.; Lee, T.Y. Characterization and identification of natural antimicrobial peptides on different organisms. Int. J. Mol. Sci. 2020, 21, 986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brogden, K.A. Antimicrobial peptides: Pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 2005, 3, 238. [Google Scholar] [CrossRef]
- Maria-Neto, S.; Almeida, K.C.; Macedo, M.L.; Franco, O.L. Understanding bacterial resistance to antimicrobial peptides: From the surface to deep inside. Biochim. Biophys. Acta 2015, 1848, 3078–3088. [Google Scholar] [CrossRef] [Green Version]
- Cardoso, M.H.; Orozco, R.Q.; Rezende, S.B.; Rodrigues, G.; Oshiro, K.G.N.; Cândido, E.S.; Franco, O.L. Computer-aided design of antimicrobial peptides: Are we generating effective drug candidates? Front. Microbiol. 2020, 10, 3097. [Google Scholar] [CrossRef]
- Lata, S.; Sharma, B.K.; Raghava, G.P.S. Analysis and prediction of antibacterial peptides. BMC Bioinform. 2007, 8, 263–272. [Google Scholar] [CrossRef] [Green Version]
- Meher, P.; Sahu, T.; Saini, V.; Rao, A.R. Predicting antimicrobial peptides with improved accuracy by incorporating the compositional, physico-chemical and structural features into Chou’s general PseAAC. Sci. Rep. 2017, 7, 42362. [Google Scholar] [CrossRef]
- Fjell, C.; Jenssen, H.; Hilpert, K.; Cheung, W.A.; Pante, N.; Hancock, R.E.; Cherkasov, A. Identification of novel antibacterial peptides by chemoinformatics and machine learning. J. Med. Chem. 2006, 52, 2006–2015. [Google Scholar] [CrossRef]
- Waghu, F.; Barai, R.; Gurung, P.; Thomas, S. CAMPR3: A database on sequences, structures and signatures of antimicrobial peptides. Nucleic Acids Res. 2016, 44, D1094–D1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lata, S.; Mishra, N.K.; Raghava, G.P. AntiBP2: Improved version of antibacterial peptide prediction. BMC Bioinform. 2010, 11, S1–S19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, X.; Wang, P.; Lin, W.Z.; Jia, J.H.; Chou, K.C. iAMP-2L: A two- level multi-label classifier for identifying antimicrobial peptides and their functional types. Anal. Biochem. 2013, 436, 168–177. [Google Scholar] [CrossRef]
- Pirtskhalava, M.; Gabrielian, A.; Cruz, P.; Griggs, H.L.; Squires, R.B.; Hurt, D.E.; Grigolava, M.; Chubinidze, M.; Gogoladze, G.; Vishnepolsky, B.; et al. DBAASP v.2: An enhanced database of structure and antimicrobial/cytotoxic activity of natural and synthetic peptides. Nucleic Acids Res. 2016, 44, D1104–D1112. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Xu, D. Imbalanced multi-label learning for identifying antimicrobial peptides and their functional types. Bioinformatics 2016, 32, 3745–3752. [Google Scholar] [CrossRef]
- Veltri, D.; Kamath, U.; Shehu, A. Deep learning improves antimicrobial peptide recognition. Bioinformatics 2018, 34, 2740–2747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabere, M.N.; Noble, W.S. Empirical comparison of web-based antimicrobial peptide prediction tools. Bioinformatics 2017, 33, 1921–1929. [Google Scholar] [CrossRef]
- Polikar, R. Ensemble based systems in decision making. IEEE Circuits Syst. Mag. 2006, 6, 21–45. [Google Scholar]
- Breiman, L. Bagging predictors. Mach. Learn. 1996, 26, 123–140. [Google Scholar] [CrossRef] [Green Version]
- Freund, Y. Boosting a weak learning algorithm by majority. Inf. Comput. 1995, 121, 256–285. [Google Scholar] [CrossRef]
- Dietterich, T.G. An experimental comparison of three methods for constructing ensembles of decision trees: Bagging, boosting, and randomization. Mach. Learn. 2000, 40, 139. [Google Scholar] [CrossRef]
- Wolpert, D.; Macready, W. No free lunch theorems for optimization. IEEE Trans. Evol. Comput. 1997, 1, 67–82. [Google Scholar] [CrossRef] [Green Version]
- Kuncheva, L. Combining Pattern Classifiers: Methods and Algorithms, 2nd ed.; Wiley: Hoboken, NJ, USA, 2014; Volume 8, pp. 263–272. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boopathi, V.; Subramaniyam, S.; Malik, A.; Lee, G.; Manavalan, B.; Yang, D. mACPpred: A support vector machine-based meta-Predictor for identification of anticancer peptides. Int. J. Mol. Sci. 2019, 20, 1964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The UniProt Consortium. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anekthanakul, K.; Hongsthong, A.; Senachak, J.; Ruengjitchatchawalya, M. SpirPep: An in-silico digestion-based platform to assist bioactive peptides discovery from a genome-wide database. BMC Bioinform. 2018, 19, 149. [Google Scholar] [CrossRef] [Green Version]
- Available online: http://www.jci-bioinfo.cn/iAMP/data.html (accessed on 17 February 2020).
- Available online: https://www.dveltri.com/ascan/v2/data/AMP_Scan2_Feb2020_Dataset.zip (accessed on 17 February 2020).
- Wang, G.; Li, X.; Wang, Z. APD2: The updated antimicrobial peptide database and its application in peptide design. Nucleic Acids Res. 2009, 37, D933–D937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammami, R.; Zouhir, A.; Lay, C.; Hamida, J.; Fliss, I. BACTIBASE second release: A database and tool platform for bacteriocin characterization. BMC Microbiol. 2010, 10, 22. [Google Scholar] [CrossRef] [Green Version]
- Heel, A.; Jong, A.; Montalbán-López, M.; Kok, J.; Kuipers, O. BAGEL3: Automated identification of genes encoding bacteriocins and (non-)bactericidal posttranslationally modified peptides. Nucleic Acids Res. 2013, 41, W448–W453. [Google Scholar] [CrossRef]
- Thomas, S.; Karnik, S.; Barai, R.S.; Jayaraman, V.K.; Idicula-Thomas, S. CAMP: A useful resource for research on antimicrobial peptides. Nucleic Acids Res. 2010, 38, D774–D780. [Google Scholar] [CrossRef] [Green Version]
- Kang, X.; Dong, F.; Shi, C.; Liu, S.; Sun, J.; Chen, J.; Li, H.; Xu, H.; Lao, X.; Zheng, H. DRAMP 2.0, an updated data repository of antimicrobial peptides. Sci. Data 2019, 6, 148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seebah, S.; Suresh, A.; Zhuo, S.; Choong, Y.; Chua, H.; Chuon, D.; Beuerman, R.; Verma, C. Defensins knowledgebase: A manually curated database and information source focused on the defensins family of antimicrobial peptides. Nucleic Acids Res. 2007, 35, D265–D268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zamyatnin, A.A.; Borchikov, A.S.; Vladimirov, M.G.; Voronina, O.L. The EROP-Moscow oligopeptide database. Nucleic Acids Res. 2006, 34, D261–D266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gueguen, Y.; Garnier, J.; Robert, L.; Lefranc, M.P.; Mougenot, I.; de Lorgeril, J.; Janech, M.; Gross, P.S.; Warr, G.W.; Cuthbertson, B.; et al. Penbase, the shrimp antimicrobial peptide penaeidin database: Sequence-based classification and recommended nomenclature. Dev. Comp. Immunol. 2006, 30, 283–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Wu, H.; Lu, H.; Li, G.; Huang, Q. LAMP: A database linking antimicrobial peptides. PLoS ONE 2013, 8, e66557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammami, R.; Hamida, J.; Vergoten, G.; Fliss, I. PhytAMP: A database dedicated to antimicrobial plant peptides. Nucleic Acids Res. 2009, 37, D963–D968. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Chen, Z. RAPD: A database of recombinantly-produced antimicrobial peptides. FEMS Microbiol. Lett. 2008, 289, 126–129. [Google Scholar] [CrossRef] [Green Version]
- Minkiewicz, P.; Iwaniak, A.; Darewicz, M. BIOPEP-UWM database of bioactive peptides: Current opportunities. Int. J. Mol. Sci. 2019, 20, 5978. [Google Scholar] [CrossRef] [Green Version]
- Tyagi, A.; Tuknait, A.; Anand, P.; Gupta, S.; Sharma, M.; Mathur, D.; Joshi, A.; Singh, S.; Gautam, A.; Raghava, G. CancerPPD: A database of anticancer peptides and proteins. Nucleic Acids Res. 2015, 43, D837–D843. [Google Scholar] [CrossRef] [Green Version]
- Mehta, D.; Anand, P.; Kumar, V.; Joshi, A.; Mathur, D.; Singh, S.; Tuknait, A.; Chaudhary, K.; Gautam, S.; Gautam, A.; et al. ParaPep: A web resource for experimentally validated antiparasitic peptide sequences and their structures. Database 2014, 2014. [Google Scholar] [CrossRef]
- Xiao, N.; Cao, D.S.; Zhu, M.F.; Xu, Q.S. protr/ProtrWeb: R package and web server for generating various numerical representation schemes of protein sequences. Bioinformatics 2015, 31, 1857–1859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2012; ISBN 3-900051-07-0. [Google Scholar]
- Osorio, D.; Rondon-Villarreal, P.; Torres, R. Peptides: A package for data mining of antimicrobial peptides. R J. 2015, 7, 4–14. [Google Scholar] [CrossRef]
- Torrent, M.; Nogués, V.M.; Boix, E. A theoretical approach to spot active regions in antimicrobial proteins. BMC Bioinform. 2009, 10, 373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Escamilla, A.M.; Rousseau, F.; Schymkowitz, J.; Serrano, L. Prediction of sequence-dependent and mutational effects on the aggregation of peptides and proteins. Nat. Biotech. 2004, 22, 1302–1306. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Liu, F.; Wang, X.; Chen, J.; Fang, L.; Chou, K. Pse-in-One: A web server for generating various modes of pseudo components of DNA, RNA, and protein sequences. Nucleic Acids Res. 2015, 43, W65–W71. [Google Scholar] [CrossRef] [Green Version]
- Chou, K.C. Using amphiphilic pseudo amino acid composition to predict enzyme subfamily classes. Bioinformatics 2005, 21, 10–19. [Google Scholar] [CrossRef]
- Chou, K.C. Some remarks on protein attribute prediction and pseudo amino acid composition (50th anniversary year review). J. Theor. Biol. 2011, 273, 236–247. [Google Scholar] [CrossRef]
- Zhao, W.; Wang, L.; Zhang, T.; Zhao, Z.; Du, P. A brief review on software tools in generating Chou’s pseudo-factor representations for all types of biological sequences. Protein Pept. Lett. 2018, 25, 822–829. [Google Scholar] [CrossRef]
- Chang, C.; Lin, C. LIBSVM: A library for support vector machines. ACM Trans. Intell. Syst. Technol. 2011, 2, 21–27. [Google Scholar] [CrossRef]
- Hall, M.; Frank, E.; Holmes, G.; Pfahringer, B.; Reutemann, P.; Witten, I.H. The WEKA data mining software: An update. SIGKDD Explor. Newsl. 2009, 11, 10–18. [Google Scholar] [CrossRef]
- Hall, M.A.; Holmes, G. Benchmarking attribute selection techniques for discrete class data mining. IEEE Trans. Knowl. Data Eng. 2003, 15, 1437–1447. [Google Scholar] [CrossRef] [Green Version]
- Leevy, J.; Khoshgoftaar, T.; Bauder, R.; Seliya, N. A survey on addressing high-class imbalance in big data. J. Big Data 2008, 5, 42. [Google Scholar] [CrossRef]
- Bauder, R.A.; Khoshgoftaar, T.M. The effects of varying class distribution on learner behavior for medicare fraud detection with imbalanced big data. Health Inf. Sci. Syst. 2018, 6, 9. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Shamsuddin, S.M.; Ralescu, A.L. Classification with class imbalance problem: A review. Int. J. Adv. Soft Comput. Appl. 2015, 7, 176–204. [Google Scholar]
- Ensemble AMPPred. Available online: http://ncrna-pred.com/Hybrid_AMPPred.htm (accessed on 17 February 2020).
- Li, L.; Kuang, H.; Zhang, Y.; Zhou, Y.; Wang, K.; Wan, Y. Prediction of eukaryotic protein subcellular multi-localisation with a combined KNN-SVM ensemble classifier. J. Comput. Biol. Bioinform. Res. 2011, 3, 15–24. [Google Scholar]
- Wang, T.; Yang, J. Using the nonlinear dimensionality reduction method for the prediction of subcellular localization of gram-negative bacterial proteins. Mol. Divers. 2009, 13, 475–481. [Google Scholar] [CrossRef]
- Wang, G.; Li, X.; Wang, Z. APD3: The antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 2016, 44, D1087–D1093. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Program Name | Techniques | Features | References |
|---|---|---|---|
| AMPer | Random Forests | Profile hidden Markov model (HMM) score | [10] |
| CAMP-SVM | Support Vector Machine | Sequence composition, physicochemical properties, and structural characteristics of amino acids | [11] |
| CAMP-RF | Random Forests | Sequence composition, physicochemical properties, and structural characteristics of amino acids | [11] |
| CAMP-DA | Discriminant Analysis | Sequence composition, physicochemical properties, and structural characteristics of amino acids | [11] |
| AntiBP | Support Vector Machine | N-terminal and C-terminal residues | [8] |
| AntiBP2 | Support Vector Machine | N-terminal and C-terminal residues | [12] |
| AMPA | Antimicrobial propensity scale threshold | Antimicrobial index based on IC50 value | [3] |
| iAMP-2 L | fuzzy K-nearest neighbor | Pseudo amino acid composition (PseAAC) incorporating five physicochemical properties | [13] |
| DBAASP | Cutoff discriminator | Physicochemical characteristics of peptides: hydrophobic moment, charge density and depth-dependent potential | [14] |
| MLAMP | ML-SMOTE | PseAAC with the gray model (GM) | [15] |
| iAMPpred | Support Vector Machine | PseAAC, normalized amino acid compositions, structural features (α-helix, β-sheet and turn structure propensity), isoelectric point, hydrophobicity, and net charge | [9] |
| AMPscanner | Deep Learning | Numerical matrix from deep neural network (DNN) | [16] |
| Database Name | Reference | Biological Function | Last Updated |
|---|---|---|---|
| The Antimicrobial Peptide Database (APD) | [30] | Antimicrobial | 2020 |
| Database Dedicated to Bacteriocin (BACTIBASE) | [31] | Antibacterial | May 2019 |
| Prediction of Bacteriocins In Prokaryotes (BAGEL3) | [32] | Antibacterial | Jan 2019 |
| Collection of Antimicrobial Peptides (CAMP) | [33] | Antimicrobial | Apr 2019 |
| Data repository of antimicrobial peptides (DRAMP) | [34] | Antimicrobial | Sep 2020 |
| Defensins Knowledgebase | [35] | Defensin, antimicrobial | Jun 2019 |
| Endogenous Regulatory OligoPeptide knowledgebase | [36] | Neuropeptide, Antimicrobial | Dec 2019 |
| The Shrimp Antimicrobial Peptide Penaeidin Database (PenBase) | [37] | Antimicrobial | Jul 2008 (Not available now) |
| A Database Linking Antimicrobial Peptides (LAMP) | [38] | Antimicrobial | Dec 2016 |
| A Database Dedicated to Antimicrobial Plant Peptides (PhytAMP) | [39] | Antimicrobial | Jan 2012 |
| Recombinantly produced Antimicrobial Peptides Database (RAPD) | [40] | Antimicrobial | Mar 2010 (Not available now) |
| Database of Antimicrobial Activity and Structure of Peptides (DBAASP) | [14] | Antimicrobial | Nov 2017 (Not available now) |
| BIOPEP-UWM database (BIOPEP) | [41] | Antimicrobial | N/A |
| A database of anticancer peptides and proteins (CancerPPD) | [42] | Anticancer | N/A |
| A database of Antiparasitic peptides (ParaPep) | [43] | Antiparasitic | N/A |
| Single Model | Training Accuracy | Training AUC | Training MCC | Testing Dataset 1 | Testing Dataset 2 | ||||
|---|---|---|---|---|---|---|---|---|---|
| AMP (11,634) | Non_AMP (35,795) | AMP_S1 (1461) | AMP_S2 (917) | Non_AMP_S1 (2404) | Non_AMP_S2 (828) | ||||
| MLP | 81.92% | 0.879 | 0.629 | 10,224 | 33,116 | 1411 | 907 | 1613 | 689 |
| 87.88% | 92.51% | 96.58% | 98.91% | 67.07% | 83.11% | ||||
| SVM | 85.89% | 0.867 | 0.598 | 10,684 | 34,283 | 1437 | 915 | 1870 | 799 |
| 91.83% | 95.78% | 98.36% | 99.78% | 77.75% | 96.38% | ||||
| KNN | 85.37% | 0.906 | 0.701 | 10,886 | 33,448 | 1400 | 908 | 1951 | 815 |
| 93.57% | 93.44% | 95.82% | 99.01% | 81.12% | 98.31% | ||||
| RBF | 83.04% | 0.902 | 0.642 | 10,161 | 33,665 | 1420 | 912 | 1668 | 732 |
| 87.34% | 94.05% | 97.19% | 99.45% | 69.36% | 88.30% | ||||
| LDA | 83.57% | 0.901 | 0.675 | 9984 | 33,767 | 1377 | 900 | 1773 | 762 |
| 85.82% | 94.33% | 94.25% | 98.15% | 73.72% | 91.92% | ||||
| NB | 74.00% | 0.786 | 0.516 | 7426 | 34,629 | 1350 | 880 | 1786 | 782 |
| 63.83% | 96.74% | 92.40% | 95.97% | 74.26% | 94.33% | ||||
| DT | 80.61% | 0.797 | 0.603 | 10,483 | 29,020 | 1374 | 880 | 1824 | 777 |
| 90.11% | 81.07% | 94.05% | 95.97% | 75.84% | 93.73% | ||||
| DL | 85.36% | 0.900 | 0.661 | 11,063 | 34,180 | 1405 | 900 | 1967 | 819 |
| 95.09% | 95.49% | 96.17% | 98.15% | 81.79% | 98.79% | ||||
| CAMP [11] | - | - | - | 8657 | 20,453 | 1375 | 893 | 1692 | 615 |
| 74.41% | 57.14% | 94.11% | 97.38% | 70.38% | 74.28% | ||||
| AMPScanner [16] | - | - | - | 10,557 | 22,511 | 1420 | 909 | 1491 | 516 |
| 90.74% | 62.89% | 97.19% | 99.13% | 62.02% | 62.32% | ||||
| Ensemble Model | Training Accuracy | Training AUC | Training MCC | Testing Dataset 1 | Testing Dataset 2 | ||||
| AMP (11,634) | Non_AMP (35,795) | AMP_S1 (1461) | AMP_S2 (917) | Non_AMP_S1 (2404) | Non_AMP_S2 (828) | ||||
| RF | 86.45% | 0.936 | 0.730 | 11,115 | 34,314 | 1447 | 915 | 1895 | 818 |
| 95.54% | 95.86% | 99.04% | 99.78% | 78.79% | 98.67% | ||||
| MaxProbVote (RF, KNN) | 87.41% | 0.925 | 0.749 | 11,254 | 33,992 | 1441 | 913 | 1965 | 824 |
| 96.73% | 94.96% | 98.63% | 99.56% | 81.70% | 99.40% | ||||
| Majority voting (RF, KNN, SVM) | 86.05% | 0.892 | 0.767 | 11,094 | 34,598 | 1441 | 916 | 2006 | 822 |
| 95.36% | 96.65% | 98.63% | 99.89% | 83.41% | 99.16% | ||||
| XGBoost | 85.68% | 0.924 | 0.734 | 11,156 | 33,421 | 1439 | 910 | 1920 | 809 |
| 95.89% | 93.37% | 98.50% | 99.24% | 79.88% | 97.71% | ||||
| AdaBoost | 82.52% | 0.910 | 0.668 | 9899 | 33,560 | 1404 | 905 | 1664 | 711 |
| 85.09% | 93.76% | 96.09% | 98.69% | 69.19% | 85.77% | ||||
| Ensemble model with hybrid feature | Training Accuracy | Training AUC | Training MCC | Testing Dataset 1 | Testing Dataset 2 | ||||
| AMP (11,634) | Non_AMP (35,795) | AMP_S1 (1461) | AMP_S2 (917) | Non_AMP_S1 (2404) | Non_AMP_S2 (828) | ||||
| RF | 86.92% | 0.939 | 0.744 | 11,127 | 34,447 | 1451 | 915 | 1904 | 822 |
| 95.64% | 96.23% | 99.32% | 99.78% | 79.17% | 99.15% | ||||
| MaxProbVote (RF, KNN) | 88.13% | 0.946 | 0.764 | 11,344 | 33,866 | 1448 | 915 | 2042 | 825 |
| 97.51% | 94.61% | 99.11% | 99.78% | 84.94% | 99.63% | ||||
| Majority voting (RF, KNN, SVM) | 88.63% | 0.927 | 0.773 | 11,295 | 34,516 | 1448 | 915 | 1993 | 827 |
| 97.09% | 96.43% | 99.11% | 99.78% | 82.87% | 99.76% | ||||
| XGBoost | 87.44% | 0.941 | 0.749 | 11,118 | 33,594 | 1440 | 911 | 1951 | 818 |
| 95.57% | 93.85% | 98.56% | 99.35% | 81.16% | 98.79% | ||||
| AdaBoost | 83.19% | 0.917 | 0.693 | 10,866 | 33,917 | 1436 | 909 | 1681 | 729 |
| 93.39% | 94.75% | 98.29% | 99.12% | 69.89% | 87.94% | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lertampaiporn, S.; Vorapreeda, T.; Hongsthong, A.; Thammarongtham, C. Ensemble-AMPPred: Robust AMP Prediction and Recognition Using the Ensemble Learning Method with a New Hybrid Feature for Differentiating AMPs. Genes 2021, 12, 137. https://doi.org/10.3390/genes12020137
Lertampaiporn S, Vorapreeda T, Hongsthong A, Thammarongtham C. Ensemble-AMPPred: Robust AMP Prediction and Recognition Using the Ensemble Learning Method with a New Hybrid Feature for Differentiating AMPs. Genes. 2021; 12(2):137. https://doi.org/10.3390/genes12020137
Chicago/Turabian StyleLertampaiporn, Supatcha, Tayvich Vorapreeda, Apiradee Hongsthong, and Chinae Thammarongtham. 2021. "Ensemble-AMPPred: Robust AMP Prediction and Recognition Using the Ensemble Learning Method with a New Hybrid Feature for Differentiating AMPs" Genes 12, no. 2: 137. https://doi.org/10.3390/genes12020137
APA StyleLertampaiporn, S., Vorapreeda, T., Hongsthong, A., & Thammarongtham, C. (2021). Ensemble-AMPPred: Robust AMP Prediction and Recognition Using the Ensemble Learning Method with a New Hybrid Feature for Differentiating AMPs. Genes, 12(2), 137. https://doi.org/10.3390/genes12020137