
Insights on the Pathogenesis of Aneurysm through the Study of Hereditary Aortopathies


Abstract
:1. Introduction
2. Structure and Features of the Normal Aortic Wall
3. Modulation of Medial VSMC Phenotype
3.1. Modulation of VSMC Phenotype via Interaction with the ECM
3.2. Modulation of VSMC Phenotype by Soluble or Membrane-Bound Ligands
3.2.1. Angiotensin II Signaling via Angiotensin II Type 1 Receptor
3.2.2. TGF-β Signaling
3.2.3. Notch Signaling
4. Genes Associated with Syndromic and Non-Syndromic Hereditary Thoracic Aortic Aneurysm
4.1. Genes Coding for Components of the Extracellular Matrix
4.1.1. Fibrillin-1
4.1.2. Lysyl Oxidase
4.1.3. Fibulin-4
4.1.4. Microfibril-Associated Glycoprotein 2
4.1.5. Biglycan
4.1.6. Collagen Type III α 1 Chain
4.1.7. Tropoelastin
4.2. Genes Coding for Proteins Involved in Transduction of Mechanical Signals
4.2.1. Smooth Muscle Specific Contractile Proteins, α-SMA and SM-MHC
4.2.2. Myosin Light Chain Kinase and cGMP-Dependent Protein Kinase
4.2.3. Filamin-A
4.2.4. Ari-1
4.3. Genes Coding for Proteins Involved in Transduction of Biochemical Signals
4.3.1. Positive Regulators of the TGF-β Signaling Pathway
4.3.2. Negative Regulators of the TGF-β Signaling Pathway
4.3.3. Notch1
4.4. Genetic Variants Associated with Bicuspid Aortic Valve (BAV) with Aneurysm
5. Proposed Model of TAA Pathogenesis Based on the Function of Known Causal Variants
6. Adaptive and Maladaptive Responses in TAA: Implications for Therapy
6.1. Adaptive and Maladaptive Roles of “Aortic Stiffness”
6.2. Adaptive and Maladaptive Roles of TGF-β Signaling
6.3. Adaptive and Maladaptive Roles of Angiotensin II Signaling
7. Regional Heterogeneity within the Aorta
7.1. Developmental and Phenotypic Heterogeneity
7.2. Mechanical and Structural Heterogeneity
8. Summary and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Melvinsdottir, I.H.; Lund, S.H.; Agnarsson, B.A.; Sigvaldason, K.; Gudbjartsson, T.; Geirsson, A. The incidence and mortality of acute thoracic aortic dissection: Results from a whole nation study. Eur. J. Cardiothorac. Surg. 2016, 50, 1111–1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiratzka, L.F.; Bakris, G.L.; Beckman, J.A.; Bersin, R.M.; Carr, V.F.; Casey, D.E., Jr.; Eagle, K.A.; Hermann, L.K.; Isselbacher, E.M.; Kazerooni, E.A.; et al. 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM guidelines for the diagnosis and management of patients with Thoracic Aortic Disease: A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, American Association for Thoracic Surgery, American College of Radiology, American Stroke Association, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology, Society of Thoracic Surgeons, and Society for Vascular Medicine. Circulation 2010, 121, e266–e369. [Google Scholar] [PubMed] [Green Version]
- Quintana, R.A.; Taylor, W.R. Introduction to the Compendium on Aortic Aneurysms. Circ. Res. 2019, 124, 470–471. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.R.; Willett, W.C.; Rimm, E.B. Smoking, hypertension, alcohol consumption, and risk of abdominal aortic aneurysm in men. Am. J. Epidemiol. 2007, 165, 838–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinard, A.; Jones, G.T.; Milewicz, D.M. Genetics of Thoracic and Abdominal Aortic Diseases. Circ. Res. 2019, 124, 588–606. [Google Scholar] [CrossRef]
- Bossone, E.; Eagle, K.A. Epidemiology and management of aortic disease: Aortic aneurysms and acute aortic syndromes. Nat. Rev. Cardiol. 2020. [Google Scholar] [CrossRef]
- Vapnik, J.S.; Kim, J.B.; Isselbacher, E.M.; Ghoshhajra, B.B.; Cheng, Y.; Sundt, T.M., 3rd; MacGillivray, T.E.; Cambria, R.P.; Lindsay, M.E. Characteristics and Outcomes of Ascending Versus Descending Thoracic Aortic Aneurysms. Am. J. Cardiol. 2016, 117, 1683–1690. [Google Scholar] [CrossRef]
- Isselbacher, E.M. Thoracic and abdominal aortic aneurysms. Circulation 2005, 111, 816–828. [Google Scholar] [CrossRef] [Green Version]
- Saeyeldin, A.A.; Velasquez, C.A.; Mahmood, S.U.B.; Brownstein, A.J.; Zafar, M.A.; Ziganshin, B.A.; Elefteriades, J.A. Thoracic aortic aneurysm: Unlocking the “silent killer” secrets. Gen. Thorac. Cardiovasc. Surg. 2019, 67, 1–11. [Google Scholar] [CrossRef]
- LeMaire, S.A.; Russell, L. Epidemiology of thoracic aortic dissection. Nat. Rev. Cardiol. 2011, 8, 103–113. [Google Scholar] [CrossRef]
- den Hartog, A.W.; Franken, R.; Zwinderman, A.H.; Timmermans, J.; Scholte, A.J.; van den Berg, M.P.; de Waard, V.; Pals, G.; Mulder, B.J.; Groenink, M. The risk for type B aortic dissection in Marfan syndrome. J. Am. Coll. Cardiol. 2015, 65, 246–254. [Google Scholar] [CrossRef] [PubMed]
- LaBounty, T.M.; Eagle, K.A. Distal aorta: The next frontier in managing Marfan syndrome aortic disease. J. Am. Coll. Cardiol. 2015, 65, 255–256. [Google Scholar] [CrossRef] [PubMed]
- Mulder, B.J.M.; van de Laar, I.M.B.H.; De Backer, J. Heritable Thoracic Aortic Diseases: Syndromal and Isolated (F)TAAD. In Clinical Cardiogenetics; Baars, H.F., Doevendans, P.A.F.M., Houweling, A.C., van Tintelen, J.P., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 309–343. [Google Scholar]
- Chou, E.L.; Lindsay, M.E. The genetics of aortopathies: Hereditary thoracic aortic aneurysms and dissections. Am. J. Med. Genet. C. Semin. Med. Genet. 2020, 184, 136–148. [Google Scholar] [CrossRef] [PubMed]
- Wagenseil, J.E.; Mecham, R.P. Vascular extracellular matrix and arterial mechanics. Physiol. Rev. 2009, 89, 957–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, Y.; Wu, D.; Birukov, K.G. Mechanosensing and Mechanoregulation of Endothelial Cell Functions. Compr. Physiol. 2019, 9, 873–904. [Google Scholar]
- Cheng, J.K.; Wagenseil, J.E. Extracellular matrix and the mechanics of large artery development. Biomech. Model. Mechanobiol. 2012, 11, 1169–1186. [Google Scholar] [CrossRef]
- Davis, E.C. Smooth muscle cell to elastic lamina connections in developing mouse aorta. Role in aortic medial organization. Lab. Investig. 1993, 68, 89–99. [Google Scholar]
- Davis, E.C. Immunolocalization of microfibril and microfibril-associated proteins in the subendothelial matrix of the developing mouse aorta. J. Cell Sci. 1994, 107 Pt 3, 727–736. [Google Scholar]
- Barallobre-Barreiro, J.; Loeys, B.; Mayr, M.; Rienks, M.; Verstraeten, A.; Kovacic, J.C. Extracellular Matrix in Vascular Disease, Part 2/4: JACC Focus Seminar. J. Am. Coll. Cardiol. 2020, 75, 2189–2203. [Google Scholar] [CrossRef]
- Moiseeva, E.P. Adhesion receptors of vascular smooth muscle cells and their functions. Cardiovasc. Res. 2001, 52, 372–386. [Google Scholar] [CrossRef]
- Pohl, U. Connexins: Key Players in the Control of Vascular Plasticity and Function. Physiol. Rev. 2020, 100, 525–572. [Google Scholar] [CrossRef] [PubMed]
- Wagenseil, J.E.; Mecham, R.P. New insights into elastic fiber assembly. Birth Defects Res. C. Embryo. Today 2007, 81, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Sainz, J.; Al Haj Zen, A.; Caligiuri, G.; Demerens, C.; Urbain, D.; Lemitre, M.; Lafont, A. Isolation of “side population” progenitor cells from healthy arteries of adult mice. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 281–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passman, J.N.; Dong, X.R.; Wu, S.P.; Maguire, C.T.; Hogan, K.A.; Bautch, V.L.; Majesky, M.W. A sonic hedgehog signaling domain in the arterial adventitia supports resident Sca1+ smooth muscle progenitor cells. Proc. Natl. Acad. Sci. USA 2008, 105, 9349–9354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stenmark, K.R.; Yeager, M.E.; El Kasmi, K.C.; Nozik-Grayck, E.; Gerasimovskaya, E.V.; Li, M.; Riddle, S.R.; Frid, M.G. The adventitia: Essential regulator of vascular wall structure and function. Annu. Rev. Physiol. 2013, 75, 23–47. [Google Scholar] [CrossRef] [Green Version]
- Majesky, M.W. Adventitia and perivascular cells. Arterioscler. Thromb. Vasc. Biol. 2015, 35, e31–e35. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, J.D.; Schwartz, M.A.; Tellides, G.; Milewicz, D.M. Role of mechanotransduction in vascular biology: Focus on thoracic aortic aneurysms and dissections. Circ. Res. 2015, 116, 1448–1461. [Google Scholar] [CrossRef] [Green Version]
- Forte, A.; Della Corte, A.; De Feo, M.; Cerasuolo, F.; Cipollaro, M. Role of myofibroblasts in vascular remodelling: Focus on restenosis and aneurysm. Cardiovasc. Res. 2010, 88, 395–405. [Google Scholar] [CrossRef] [Green Version]
- Ritman, E.L.; Lerman, A. The dynamic vasa vasorum. Cardiovasc. Res. 2007, 75, 649–658. [Google Scholar] [CrossRef]
- Shen, Y.H.; LeMaire, S.A. Molecular pathogenesis of genetic and sporadic aortic aneurysms and dissections. Curr. Probl. Surg. 2017, 54, 95–155. [Google Scholar] [CrossRef]
- Sherifova, S.; Holzapfel, G.A. Biomechanics of aortic wall failure with a focus on dissection and aneurysm: A review. Acta Biomater. 2019, 99, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Malashicheva, A.; Kostina, D.; Kostina, A.; Irtyuga, O.; Voronkina, I.; Smagina, L.; Ignatieva, E.; Gavriliuk, N.; Uspensky, V.; Moiseeva, O.; et al. Phenotypic and Functional Changes of Endothelial and Smooth Muscle Cells in Thoracic Aortic Aneurysms. Int. J. Vasc. Med. 2016, 2016, 3107879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Billaud, M.; Hill, J.C.; Richards, T.D.; Gleason, T.G.; Phillippi, J.A. Medial Hypoxia and Adventitial Vasa Vasorum Remodeling in Human Ascending Aortic Aneurysm. Front. Cardiovasc. Med. 2018, 5, 124. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.H.; LeMaire, S.A.; Webb, N.R.; Cassis, L.A.; Daugherty, A.; Lu, H.S. Aortic Aneurysms and Dissections Series. Arterioscler. Thromb. Vasc. Biol. 2020, 40, e37–e46. [Google Scholar] [CrossRef]
- Cattell, M.A.; Hasleton, P.S.; Anderson, J.C. Glycosaminoglycan content is increased in dissecting aneurysms of human thoracic aorta. Clin. Chim. Acta 1994, 226, 29–46. [Google Scholar] [CrossRef]
- Engle, J.; Safi, H.J.; Abbassi, O.; Iliopoulos, D.C.; Dorsay, D.; Cartwright, J., Jr.; Weilbaecher, D. Mucopolysaccharidosis presenting as pediatric multiple aortic aneurysm: First reported case. J. Vasc. Surg. 1997, 26, 704–710. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; LeMaire, S.A.; Chen, L.; Shen, Y.H.; Gan, Y.; Bartsch, H.; Carter, S.A.; Utama, B.; Ou, H.; Coselli, J.S.; et al. Increased collagen deposition and elevated expression of connective tissue growth factor in human thoracic aortic dissection. Circulation 2006, 114, I200–I205. [Google Scholar] [CrossRef] [Green Version]
- Halushka, M.K.; Angelini, A.; Bartoloni, G.; Basso, C.; Batoroeva, L.; Bruneval, P.; Buja, L.M.; Butany, J.; d’Amati, G.; Fallon, J.T.; et al. Consensus statement on surgical pathology of the aorta from the Society for Cardiovascular Pathology and the Association For European Cardiovascular Pathology: II. Noninflammatory degenerative diseases—Nomenclature and diagnostic criteria. Cardiovasc. Pathol. 2016, 25, 247–257. [Google Scholar] [CrossRef]
- Cikach, F.S.; Koch, C.D.; Mead, T.J.; Galatioto, J.; Willard, B.B.; Emerton, K.B.; Eagleton, M.J.; Blackstone, E.H.; Ramirez, F.; Roselli, E.E.; et al. Massive aggrecan and versican accumulation in thoracic aortic aneurysm and dissection. JCI Insight 2018, 3, e97167. [Google Scholar] [CrossRef] [Green Version]
- Michel, J.B.; Jondeau, G.; Milewicz, D.M. From genetics to response to injury: Vascular smooth muscle cells in aneurysms and dissections of the ascending aorta. Cardiovasc. Res. 2018, 114, 578–589. [Google Scholar] [CrossRef]
- Roccabianca, S.; Bellini, C.; Humphrey, J.D. Computational modelling suggests good, bad and ugly roles of glycosaminoglycans in arterial wall mechanics and mechanobiology. J. R Soc. Interface 2014, 11, 20140397. [Google Scholar] [CrossRef] [PubMed]
- Azeloglu, E.U.; Albro, M.B.; Thimmappa, V.A.; Ateshian, G.A.; Costa, K.D. Heterogeneous transmural proteoglycan distribution provides a mechanism for regulating residual stresses in the aorta. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H1197–H1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roccabianca, S.; Ateshian, G.A.; Humphrey, J.D. Biomechanical roles of medial pooling of glycosaminoglycans in thoracic aortic dissection. Biomech. Model. Mechanobiol. 2014, 13, 13–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, X.; Wanga, S.; Fellows, A.L.; Barallobre-Barreiro, J.; Lu, R.; Davaapil, H.; Franken, R.; Fava, M.; Baig, F.; Skroblin, P.; et al. Glycoproteomic Analysis of the Aortic Extracellular Matrix in Marfan Patients. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1859–1873. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, J.D. Possible mechanical roles of glycosaminoglycans in thoracic aortic dissection and associations with dysregulated transforming growth factor-β. J. Vasc. Res. 2013, 50, 1–10. [Google Scholar] [CrossRef] [Green Version]
- van de Pol, V.; Kurakula, K.; DeRuiter, M.C.; Goumans, M.J. Thoracic Aortic Aneurysm Development in Patients with Bicuspid Aortic Valve: What Is the Role of Endothelial Cells? Front. Physiol. 2017, 8, 938. [Google Scholar] [CrossRef] [Green Version]
- Tinajero, M.G.; Gotlieb, A.I. Recent Developments in Vascular Adventitial Pathobiology: The Dynamic Adventitia as a Complex Regulator of Vascular Disease. Am. J. Pathol. 2020, 190, 520–534. [Google Scholar] [CrossRef] [Green Version]
- Owens, G.K.; Kumar, M.S.; Wamhoff, B.R. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol. Rev. 2004, 84, 767–801. [Google Scholar] [CrossRef]
- Li, C.; Xu, Q. Mechanical stress-initiated signal transduction in vascular smooth muscle cells in vitro and in vivo. Cell Signal. 2007, 19, 881–891. [Google Scholar] [CrossRef]
- Mack, C.P. Signaling mechanisms that regulate smooth muscle cell differentiation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1495–1505. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.J.; Zhou, Y.; Chen, L.; Wang, Y.Q.; Wang, X.; Pi, Y.; Gao, C.Y.; Li, J.C.; Zhang, L.L. An overview of potential molecular mechanisms involved in VSMC phenotypic modulation. Histochem. Cell Biol. 2016, 145, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Isselbacher, E.M.; Lino Cardenas, C.L.; Lindsay, M.E. Hereditary Influence in Thoracic Aortic Aneurysm and Dissection. Circulation 2016, 133, 2516–2528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frismantiene, A.; Philippova, M.; Erne, P.; Resink, T.J. Smooth muscle cell-driven vascular diseases and molecular mechanisms of VSMC plasticity. Cell Signal. 2018, 52, 48–64. [Google Scholar] [CrossRef] [PubMed]
- Segura, A.M.; Luna, R.E.; Horiba, K.; Stetler-Stevenson, W.G.; McAllister, H.A., Jr.; Willerson, J.T.; Ferrans, V.J. Immunohistochemistry of matrix metalloproteinases and their inhibitors in thoracic aortic aneurysms and aortic valves of patients with Marfan’s syndrome. Circulation 1998, 98, II331–II337. [Google Scholar]
- Lesauskaite, V.; Tanganelli, P.; Sassi, C.; Neri, E.; Diciolla, F.; Ivanoviene, L.; Epistolato, M.C.; Lalinga, A.V.; Alessandrini, C.; Spina, D. Smooth muscle cells of the media in the dilatative pathology of ascending thoracic aorta: Morphology, immunoreactivity for osteopontin, matrix metalloproteinases, and their inhibitors. Hum. Pathol. 2001, 32, 1003–1011. [Google Scholar] [CrossRef]
- Goodall, S.; Porter, K.E.; Bell, P.R.; Thompson, M.M. Enhanced invasive properties exhibited by smooth muscle cells are associated with elevated production of MMP-2 in patients with aortic aneurysms. Eur. J. Vasc. Endovasc. Surg. 2002, 24, 72–80. [Google Scholar] [CrossRef] [Green Version]
- Longo, G.M.; Xiong, W.; Greiner, T.C.; Zhao, Y.; Fiotti, N.; Baxter, B.T. Matrix metalloproteinases 2 and 9 work in concert to produce aortic aneurysms. J. Clin. Investig. 2002, 110, 625–632. [Google Scholar] [CrossRef]
- LeMaire, S.A.; Wang, X.; Wilks, J.A.; Carter, S.A.; Wen, S.; Won, T.; Leonardelli, D.; Anand, G.; Conklin, L.D.; Wang, X.L.; et al. Matrix metalloproteinases in ascending aortic aneurysms: Bicuspid versus trileaflet aortic valves. J. Surg. Res. 2005, 123, 40–48. [Google Scholar] [CrossRef]
- Jones, J.A.; Barbour, J.R.; Lowry, A.S.; Bouges, S.; Beck, C.; McClister, D.M., Jr.; Mukherjee, R.; Ikonomidis, J.S. Spatiotemporal expression and localization of matrix metalloproteinas-9 in a murine model of thoracic aortic aneurysm. J. Vasc. Surg. 2006, 44, 1314–1321. [Google Scholar] [CrossRef] [Green Version]
- Barbour, J.R.; Spinale, F.G.; Ikonomidis, J.S. Proteinase systems and thoracic aortic aneurysm progression. J. Surg. Res. 2007, 139, 292–307. [Google Scholar] [CrossRef]
- Pannu, H.; Tran-Fadulu, V.; Papke, C.L.; Scherer, S.; Liu, Y.; Presley, C.; Guo, D.; Estrera, A.L.; Safi, H.J.; Brasier, A.R.; et al. MYH11 mutations result in a distinct vascular pathology driven by insulin-like growth factor 1 and angiotensin II. Hum. Mol. Genet. 2007, 16, 2453–2462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, Y.H.; Tian, C.; Liu, L.; Wang, L.; Chang, Q. Elevated expression of connective tissue growth factor, osteopontin and increased collagen content in human ascending thoracic aortic aneurysms. Vascular 2014, 22, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Meng, W.; Liu, S.; Li, D.; Liu, Z.; Yang, H.; Sun, B.; Liu, H. Expression of platelet-derived growth factor B is upregulated in patients with thoracic aortic dissection. J. Vasc. Surg. 2018, 68, 3S–13S. [Google Scholar] [CrossRef]
- Gomez, D.; Al Haj Zen, A.; Borges, L.F.; Philippe, M.; Gutierrez, P.S.; Jondeau, G.; Michel, J.B.; Vranckx, R. Syndromic and non-syndromic aneurysms of the human ascending aorta share activation of the Smad2 pathway. J. Pathol. 2009, 218, 131–142. [Google Scholar] [CrossRef] [PubMed]
- He, R.; Guo, D.C.; Sun, W.; Papke, C.L.; Duraisamy, S.; Estrera, A.L.; Safi, H.J.; Ahn, C.; Buja, L.M.; Arnett, F.C.; et al. Characterization of the inflammatory cells in ascending thoracic aortic aneurysms in patients with Marfan syndrome, familial thoracic aortic aneurysms, and sporadic aneurysms. J. Thorac. Cardiovasc. Surg. 2008, 136, 922–929. [Google Scholar] [CrossRef] [Green Version]
- van der Pluijm, I.; Burger, J.; van Heijningen, P.M.; A, I.J.; van Vliet, N.; Milanese, C.; Schoonderwoerd, K.; Sluiter, W.; Ringuette, L.J.; Dekkers, D.H.W.; et al. Decreased mitochondrial respiration in aneurysmal aortas of Fibulin-4 mutant mice is linked to PGC1A regulation. Cardiovasc. Res. 2018, 114, 1776–1793. [Google Scholar] [CrossRef] [Green Version]
- Parker, S.J.; Stotland, A.; MacFarlane, E.; Wilson, N.; Orosco, A.; Venkatraman, V.; Madrid, K.; Gottlieb, R.; Dietz, H.C.; Van Eyk, J.E. Proteomics reveals Rictor as a noncanonical TGF-β signaling target during aneurysm progression in Marfan mice. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1112–H1126. [Google Scholar] [CrossRef]
- Dinesh, N.E.H.; Reinhardt, D.P. Inflammation in thoracic aortic aneurysms. Herz 2019, 44, 138–146. [Google Scholar] [CrossRef]
- Gutierrez, P.S.; Piubelli, M.L.M.; Naal, K.G.; Dias, R.R.; Borges, L.F. Mitochondria in aneurysms and dissections of the human ascending aorta. Cardiovasc. Pathol. 2020, 47, 107207. [Google Scholar] [CrossRef]
- Humphrey, J.D.; Tellides, G. Central artery stiffness and thoracic aortopathy. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H169–H182. [Google Scholar] [CrossRef]
- Ngai, D.; Lino, M.; Bendeck, M.P. Cell-Matrix Interactions and Matricrine Signaling in the Pathogenesis of Vascular Calcification. Front. Cardiovasc. Med. 2018, 5, 174. [Google Scholar] [CrossRef] [PubMed]
- Clyman, R.I.; McDonald, K.A.; Kramer, R.H. Integrin receptors on aortic smooth muscle cells mediate adhesion to fibronectin, laminin, and collagen. Circ. Res. 1990, 67, 175–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, M.A. Integrins and extracellular matrix in mechanotransduction. Cold Spring Harb. Perspect. Biol. 2010, 2, a005066. [Google Scholar] [CrossRef] [PubMed]
- Ross, T.D.; Coon, B.G.; Yun, S.; Baeyens, N.; Tanaka, K.; Ouyang, M.; Schwartz, M.A. Integrins in mechanotransduction. Curr. Opin. Cell Biol. 2013, 25, 613–618. [Google Scholar] [CrossRef] [Green Version]
- Zeidan, A.; Nordstrom, I.; Albinsson, S.; Malmqvist, U.; Sward, K.; Hellstrand, P. Stretch-induced contractile differentiation of vascular smooth muscle: Sensitivity to actin polymerization inhibitors. Am. J. Physiol. Cell Physiol. 2003, 284, C1387–C1396. [Google Scholar] [CrossRef] [Green Version]
- Albinsson, S.; Nordstrom, I.; Hellstrand, P. Stretch of the vascular wall induces smooth muscle differentiation by promoting actin polymerization. J. Biol. Chem. 2004, 279, 34849–34855. [Google Scholar] [CrossRef] [Green Version]
- Parsons, J.T.; Horwitz, A.R.; Schwartz, M.A. Cell adhesion: Integrating cytoskeletal dynamics and cellular tension. Nat. Rev. Mol. Cell Biol. 2010, 11, 633–643. [Google Scholar] [CrossRef]
- Hoffman, B.D.; Grashoff, C.; Schwartz, M.A. Dynamic molecular processes mediate cellular mechanotransduction. Nature 2011, 475, 316–323. [Google Scholar] [CrossRef]
- Bax, D.V.; Bernard, S.E.; Lomas, A.; Morgan, A.; Humphries, J.; Shuttleworth, C.A.; Humphries, M.J.; Kielty, C.M. Cell adhesion to fibrillin-1 molecules and microfibrils is mediated by α 5 β 1 and α v β 3 integrins. J. Biol. Chem. 2003, 278, 34605–34616. [Google Scholar] [CrossRef] [Green Version]
- Bezie, Y.; Lacolley, P.; Laurent, S.; Gabella, G. Connection of smooth muscle cells to elastic lamellae in aorta of spontaneously hypertensive rats. Hypertension 1998, 32, 166–169. [Google Scholar] [CrossRef] [Green Version]
- Karimi, A.; Milewicz, D.M. Structure of the Elastin-Contractile Units in the Thoracic Aorta and How Genes That Cause Thoracic Aortic Aneurysms and Dissections Disrupt This Structure. Can. J. Cardiol. 2016, 32, 26–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooke, B.S.; Bayes-Genis, A.; Li, D.Y. New insights into elastin and vascular disease. Trends. Cardiovasc. Med. 2003, 13, 176–181. [Google Scholar] [CrossRef]
- Karnik, S.K.; Brooke, B.S.; Bayes-Genis, A.; Sorensen, L.; Wythe, J.D.; Schwartz, R.S.; Keating, M.T.; Li, D.Y. A critical role for elastin signaling in vascular morphogenesis and disease. Development 2003, 130, 411–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duque Lasio, M.L.; Kozel, B.A. Elastin-driven genetic diseases. Matrix Biol. 2018, 71-72, 144–160. [Google Scholar] [CrossRef] [PubMed]
- Petsophonsakul, P.; Furmanik, M.; Forsythe, R.; Dweck, M.; Schurink, G.W.; Natour, E.; Reutelingsperger, C.; Jacobs, M.; Mees, B.; Schurgers, L. Role of Vascular Smooth Muscle Cell Phenotypic Switching and Calcification in Aortic Aneurysm Formation. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1351–1368. [Google Scholar] [CrossRef] [PubMed]
- Milewicz, D.M.; Trybus, K.M.; Guo, D.C.; Sweeney, H.L.; Regalado, E.; Kamm, K.; Stull, J.T. Altered Smooth Muscle Cell Force Generation as a Driver of Thoracic Aortic Aneurysms and Dissections. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 26–34. [Google Scholar] [CrossRef] [Green Version]
- Somlyo, A.P.; Somlyo, A.V. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: Modulated by G proteins, kinases, and myosin phosphatase. Physiol. Rev. 2003, 83, 1325–1358. [Google Scholar] [CrossRef] [Green Version]
- Gunst, S.J.; Zhang, W. Actin cytoskeletal dynamics in smooth muscle: A new paradigm for the regulation of smooth muscle contraction. Am. J. Physiol. Cell Physiol. 2008, 295, C576–C587. [Google Scholar] [CrossRef] [Green Version]
- Hill-Eubanks, D.C.; Werner, M.E.; Heppner, T.J.; Nelson, M.T. Calcium signaling in smooth muscle. Cold Spring Harb. Perspect. Biol. 2011, 3, a004549. [Google Scholar] [CrossRef]
- Burridge, K.; Guilluy, C. Focal adhesions, stress fibers and mechanical tension. Exp. Cell Res. 2016, 343, 14–20. [Google Scholar] [CrossRef] [Green Version]
- Fultz, M.E.; Li, C.; Geng, W.; Wright, G.L. Remodeling of the actin cytoskeleton in the contracting A7r5 smooth muscle cell. J. Muscle Res. Cell Motil. 2000, 21, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Cohen, D.M.; Kutscher, B.; Chen, H.; Murphy, D.B.; Craig, S.W. A conformational switch in vinculin drives formation and dynamics of a talin-vinculin complex at focal adhesions. J. Biol. Chem. 2006, 281, 16006–16015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- del Rio, A.; Perez-Jimenez, R.; Liu, R.; Roca-Cusachs, P.; Fernandez, J.M.; Sheetz, M.P. Stretching single talin rod molecules activates vinculin binding. Science 2009, 323, 638–641. [Google Scholar] [CrossRef]
- Friedland, J.C.; Lee, M.H.; Boettiger, D. Mechanically activated integrin switch controls alpha5beta1 function. Science 2009, 323, 642–644. [Google Scholar] [CrossRef] [PubMed]
- Pasapera, A.M.; Schneider, I.C.; Rericha, E.; Schlaepfer, D.D.; Waterman, C.M. Myosin II activity regulates vinculin recruitment to focal adhesions through FAK-mediated paxillin phosphorylation. J. Cell Biol. 2010, 188, 877–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinz, B.; Dugina, V.; Ballestrem, C.; Wehrle-Haller, B.; Chaponnier, C. α-smooth muscle actin is crucial for focal adhesion maturation in myofibroblasts. Mol. Biol. Cell 2003, 14, 2508–2519. [Google Scholar] [CrossRef] [Green Version]
- Tomasek, J.J.; Gabbiani, G.; Hinz, B.; Chaponnier, C.; Brown, R.A. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 2002, 3, 349–363. [Google Scholar] [CrossRef]
- Tamada, M.; Sheetz, M.P.; Sawada, Y. Activation of a signaling cascade by cytoskeleton stretch. Dev. Cell 2004, 7, 709–718. [Google Scholar] [CrossRef] [Green Version]
- Sawada, Y.; Tamada, M.; Dubin-Thaler, B.J.; Cherniavskaya, O.; Sakai, R.; Tanaka, S.; Sheetz, M.P. Force sensing by mechanical extension of the Src family kinase substrate p130Cas. Cell 2006, 127, 1015–1026. [Google Scholar] [CrossRef] [Green Version]
- Ruberti, J.W.; Hallab, N.J. Strain-controlled enzymatic cleavage of collagen in loaded matrix. Biochem. Biophys. Res. Commun. 2005, 336, 483–489. [Google Scholar] [CrossRef]
- Storch, U.; Mederos y Schnitzler, M.; Gudermann, T. G protein-mediated stretch reception. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H1241–H1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huelsz-Prince, G.; Belkin, A.M.; VanBavel, E.; Bakker, E.N. Activation of extracellular transglutaminase 2 by mechanical force in the arterial wall. J. Vasc. Res. 2013, 50, 383–395. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gilmore, T.D. Zyxin and paxillin proteins: Focal adhesion plaque LIM domain proteins go nuclear. Biochim. Biophys. Acta 2003, 1593, 115–120. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.T.; Chen, X.L.; Lim, Y.; Hanson, D.A.; Vo, T.T.; Howerton, K.; Larocque, N.; Fisher, S.J.; Schlaepfer, D.D.; Ilic, D. Nuclear FAK promotes cell proliferation and survival through FERM-enhanced p53 degradation. Mol. Cell 2008, 29, 9–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, K.; Kim, J.H.; Murphy, J.M.; Park, H.; Kim, S.J.; Rodriguez, Y.A.R.; Kong, H.; Choi, C.; Guan, J.L.; Taylor, J.M.; et al. Nuclear Focal Adhesion Kinase Controls Vascular Smooth Muscle Cell Proliferation and Neointimal Hyperplasia Through GATA4-Mediated Cyclin D1 Transcription. Circ. Res. 2019, 125, 152–166. [Google Scholar] [CrossRef] [PubMed]
- Elosegui-Artola, A.; Andreu, I.; Beedle, A.E.M.; Lezamiz, A.; Uroz, M.; Kosmalska, A.J.; Oria, R.; Kechagia, J.Z.; Rico-Lastres, P.; Le Roux, A.L.; et al. Force Triggers YAP Nuclear Entry by Regulating Transport across Nuclear Pores. Cell 2017, 171, 1397–1410. [Google Scholar] [CrossRef]
- Lu, S.; Weiser-Evans, M.C.M. Nuclear Focal Adhesion Kinase. Circ. Res. 2019, 125, 167–169. [Google Scholar] [CrossRef]
- Moujaber, O.; Stochaj, U. The Cytoskeleton as Regulator of Cell Signaling Pathways. Trends Biochem. Sci. 2020, 45, 96–107. [Google Scholar] [CrossRef]
- Kardassis, D.; Murphy, C.; Fotsis, T.; Moustakas, A.; Stournaras, C. Control of transforming growth factor β signal transduction by small GTPases. FEBS J. 2009, 276, 2947–2965. [Google Scholar] [CrossRef]
- Wang, N.; Tytell, J.D.; Ingber, D.E. Mechanotransduction at a distance: Mechanically coupling the extracellular matrix with the nucleus. Nat. Rev. Mol. Cell Biol. 2009, 10, 75–82. [Google Scholar] [CrossRef]
- Tapley, E.C.; Starr, D.A. Connecting the nucleus to the cytoskeleton by SUN-KASH bridges across the nuclear envelope. Curr. Opin. Cell Biol. 2013, 25, 57–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirby, T.J.; Lammerding, J. Emerging views of the nucleus as a cellular mechanosensor. Nat. Cell Biol. 2018, 20, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O. The extracellular matrix: Not just pretty fibrils. Science 2009, 326, 1216–1219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Clark, R.A.F. Fibronectin at select sites binds multiple growth factors and enhances their activity: Expansion of the collaborative ECM-GF paradigm. J. Invest. Dermatol. 2014, 134, 895–901. [Google Scholar] [CrossRef] [Green Version]
- Fouillade, C.; Monet-Lepretre, M.; Baron-Menguy, C.; Joutel, A. Notch signalling in smooth muscle cells during development and disease. Cardiovasc. Res. 2012, 95, 138–146. [Google Scholar] [CrossRef] [Green Version]
- van Dorst, D.C.H.; de Wagenaar, N.P.; van der Pluijm, I.; Roos-Hesselink, J.W.; Essers, J.; Danser, A.H.J. Transforming Growth Factor-β and the Renin-Angiotensin System in Syndromic Thoracic Aortic Aneurysms: Implications for Treatment. Cardiovasc. Drugs Ther. 2020. [Google Scholar] [CrossRef]
- Forrester, S.J.; Booz, G.W.; Sigmund, C.D.; Coffman, T.M.; Kawai, T.; Rizzo, V.; Scalia, R.; Eguchi, S. Angiotensin II Signal Transduction: An Update on Mechanisms of Physiology and Pathophysiology. Physiol. Rev. 2018, 98, 1627–1738. [Google Scholar] [CrossRef]
- Mehta, P.K.; Griendling, K.K. Angiotensin II cell signaling: Physiological and pathological effects in the cardiovascular system. Am. J. Physiol. Cell Physiol. 2007, 292, C82–C97. [Google Scholar] [CrossRef]
- Wu, C.H.; Mohammadmoradi, S.; Chen, J.Z.; Sawada, H.; Daugherty, A.; Lu, H.S. Renin-Angiotensin System and Cardiovascular Functions. Arterioscler. Thromb. Vasc. Biol. 2018, 38, e108–e116. [Google Scholar] [CrossRef] [Green Version]
- Yan, C.; Kim, D.; Aizawa, T.; Berk, B.C. Functional interplay between angiotensin II and nitric oxide: Cyclic GMP as a key mediator. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 26–36. [Google Scholar] [CrossRef] [Green Version]
- Geisterfer, A.A.; Peach, M.J.; Owens, G.K. Angiotensin II induces hypertrophy, not hyperplasia, of cultured rat aortic smooth muscle cells. Circ. Res. 1988, 62, 749–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xi, X.P.; Graf, K.; Goetze, S.; Fleck, E.; Hsueh, W.A.; Law, R.E. Central role of the MAPK pathway in ang II-mediated DNA synthesis and migration in rat vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 73–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchesi, C.; Paradis, P.; Schiffrin, E.L. Role of the renin-angiotensin system in vascular inflammation. Trends Pharmacol Sci 2008, 29, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Linseman, D.A.; Benjamin, C.W.; Jones, D.A. Convergence of angiotensin II and platelet-derived growth factor receptor signaling cascades in vascular smooth muscle cells. J. Biol Chem 1995, 270, 12563–12568. [Google Scholar] [CrossRef] [Green Version]
- Ushio-Fukai, M.; Griendling, K.K.; Becker, P.L.; Hilenski, L.; Halleran, S.; Alexander, R.W. Epidermal growth factor receptor transactivation by angiotensin II requires reactive oxygen species in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 489–495. [Google Scholar] [CrossRef] [Green Version]
- Matsubara, H.; Moriguchi, Y.; Mori, Y.; Masaki, H.; Tsutsumi, Y.; Shibasaki, Y.; Uchiyama-Tanaka, Y.; Fujiyama, S.; Koyama, Y.; Nose-Fujiyama, A.; et al. Transactivation of EGF receptor induced by angiotensin II regulates fibronectin and TGF-β gene expression via transcriptional and post-transcriptional mechanisms. Mol. Cell Biochem. 2000, 212, 187–201. [Google Scholar] [CrossRef]
- Sanchez-Guerrero, E.; Midgley, V.C.; Khachigian, L.M. Angiotensin II induction of PDGF-C expression is mediated by AT1 receptor-dependent Egr-1 transactivation. Nucleic Acids Res. 2008, 36, 1941–1951. [Google Scholar] [CrossRef]
- Weigert, C.; Brodbeck, K.; Klopfer, K.; Haring, H.U.; Schleicher, E.D. Angiotensin II induces human TGF-β 1 promoter activation: Similarity to hyperglycaemia. Diabetologia 2002, 45, 890–898. [Google Scholar] [CrossRef] [Green Version]
- Shihab, F.S.; Bennett, W.M.; Tanner, A.M.; Andoh, T.F. Angiotensin II blockade decreases TGF-beta1 and matrix proteins in cyclosporine nephropathy. Kidney Int. 1997, 52, 660–673. [Google Scholar] [CrossRef] [Green Version]
- Gibbons, G.H.; Pratt, R.E.; Dzau, V.J. Vascular smooth muscle cell hypertrophy vs. hyperplasia. Autocrine transforming growth factor-β 1 expression determines growth response to angiotensin II. J. Clin. Investig. 1992, 90, 456–461. [Google Scholar] [CrossRef]
- Li, Q.; Muragaki, Y.; Hatamura, I.; Ueno, H.; Ooshima, A. Stretch-induced collagen synthesis in cultured smooth muscle cells from rabbit aortic media and a possible involvement of angiotensin II and transforming growth factor-β. J. Vasc. Res. 1998, 35, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Stanley, A.G.; Patel, H.; Knight, A.L.; Williams, B. Mechanical strain-induced human vascular matrix synthesis: The role of angiotensin II. J. Renin Angiotensin Aldosterone Syst. 2000, 1, 32–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kagami, S.; Border, W.A.; Miller, D.E.; Noble, N.A. Angiotensin II stimulates extracellular matrix protein synthesis through induction of transforming growth factor-β expression in rat glomerular mesangial cells. J. Clin. Investig. 1994, 93, 2431–2437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Q.; Stamenkovic, I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-β and promotes tumor invasion and angiogenesis. Genes Dev. 2000, 14, 163–176. [Google Scholar] [PubMed]
- Wang, C.; Chang, Q.; Sun, X.; Qian, X.; Liu, P.; Pei, H.; Guo, X.; Liu, W. Angiotensin II Induces an Increase in Matrix Metalloproteinase 2 Expression in Aortic Smooth Muscle Cells of Ascending Thoracic Aortic Aneurysms Through JNK, ERK1/2, and p38 MAPK Activation. J. Cardiovasc. Pharmacol. 2015, 66, 285–293. [Google Scholar] [CrossRef]
- Rakesh, K.; Yoo, B.; Kim, I.M.; Salazar, N.; Kim, K.S.; Rockman, H.A. β-Arrestin-biased agonism of the angiotensin receptor induced by mechanical stress. Sci. Signal. 2010, 3, ra46. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Hitomi, H.; Hosomi, N.; Lei, B.; Pelisch, N.; Nakano, D.; Kiyomoto, H.; Ma, H.; Nishiyama, A. Mechanical stretch potentiates angiotensin II-induced proliferation in spontaneously hypertensive rat vascular smooth muscle cells. Hypertens. Res. 2010, 33, 1250–1257. [Google Scholar] [CrossRef] [Green Version]
- Mederos y Schnitzler, M.; Storch, U.; Meibers, S.; Nurwakagari, P.; Breit, A.; Essin, K.; Gollasch, M.; Gudermann, T. Gq-coupled receptors as mechanosensors mediating myogenic vasoconstriction. EMBO J. 2008, 27, 3092–3103. [Google Scholar] [CrossRef]
- Schleifenbaum, J.; Kassmann, M.; Szijarto, I.A.; Hercule, H.C.; Tano, J.Y.; Weinert, S.; Heidenreich, M.; Pathan, A.R.; Anistan, Y.M.; Alenina, N.; et al. Stretch-activation of angiotensin II type 1a receptors contributes to the myogenic response of mouse mesenteric and renal arteries. Circ. Res. 2014, 115, 263–272. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Peters, A.; Papke, C.L.; Villamizar, C.; Ringuette, L.J.; Cao, J.; Wang, S.; Ma, S.; Gong, L.; Byanova, K.L.; et al. Loss of Smooth Muscle α-Actin Leads to NF-kappaB-Dependent Increased Sensitivity to Angiotensin II in Smooth Muscle Cells and Aortic Enlargement. Circ. Res. 2017, 120, 1903–1915. [Google Scholar] [CrossRef]
- MacFarlane, E.G.; Parker, S.J.; Shin, J.Y.; Ziegler, S.G.; Creamer, T.J.; Bagirzadeh, R.; Bedja, D.; Chen, Y.; Calderon, J.F.; Weissler, K.; et al. Lineage-specific events underlie aortic root aneurysm pathogenesis in Loeys-Dietz syndrome. J. Clin. Investig. 2019, 129, 659–675. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Yamashiro, Y.; Papke, C.L.; Ikeda, Y.; Lin, Y.; Patel, M.; Inagami, T.; Le, V.P.; Wagenseil, J.E.; Yanagisawa, H. Angiotensin-converting enzyme-induced activation of local angiotensin signaling is required for ascending aortic aneurysms in fibulin-4-deficient mice. Sci. Transl. Med. 2013, 5, 183ra158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, I.B.; Rifkin, D.B. Regulation of the Bioavailability of TGF-β and TGF-β-Related Proteins. Cold Spring Harb. Perspect. Biol. 2016, 8, a021907. [Google Scholar] [CrossRef] [PubMed]
- Heldin, C.H.; Moustakas, A. Signaling Receptors for TGF-β Family Members. Cold Spring Harb. Perspect. Biol. 2016, 8, a022053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hata, A.; Chen, Y.G. TGF-β Signaling from Receptors to Smads. Cold Spring Harb. Perspect. Biol. 2016, 8, a022061. [Google Scholar] [CrossRef] [PubMed]
- Itoh, S.; ten Dijke, P. Negative regulation of TGF-β receptor/Smad signal transduction. Curr Opin Cell Biol 2007, 19, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Luo, K.; Stroschein, S.L.; Wang, W.; Chen, D.; Martens, E.; Zhou, S.; Zhou, Q. The Ski oncoprotein interacts with the Smad proteins to repress TGFbeta signaling. Genes Dev. 1999, 13, 2196–2206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akiyoshi, S.; Inoue, H.; Hanai, J.; Kusanagi, K.; Nemoto, N.; Miyazono, K.; Kawabata, M. c-Ski acts as a transcriptional co-repressor in transforming growth factor-β signaling through interaction with smads. J. Biol. Chem. 1999, 274, 35269–35277. [Google Scholar] [CrossRef] [Green Version]
- Schmierer, B.; Hill, C.S. TGFbeta-SMAD signal transduction: Molecular specificity and functional flexibility. Nat. Rev. Mol. Cell Biol. 2007, 8, 970–982. [Google Scholar] [CrossRef]
- Tecalco-Cruz, A.C.; Rios-Lopez, D.G.; Vazquez-Victorio, G.; Rosales-Alvarez, R.E.; Macias-Silva, M. Transcriptional cofactors Ski and SnoN are major regulators of the TGF-β/Smad signaling pathway in health and disease. Signal. Transduct. Target. Ther. 2018, 3, 15. [Google Scholar] [CrossRef] [Green Version]
- Hiepen, C.; Mendez, P.L.; Knaus, P. It Takes Two to Tango: Endothelial TGFbeta/BMP Signaling Crosstalk with Mechanobiology. Cells 2020, 9, 1965. [Google Scholar] [CrossRef] [PubMed]
- Rys, J.P.; DuFort, C.C.; Monteiro, D.A.; Baird, M.A.; Oses-Prieto, J.A.; Chand, S.; Burlingame, A.L.; Davidson, M.W.; Alliston, T.N. Discrete spatial organization of TGFbeta receptors couples receptor multimerization and signaling to cellular tension. Elife 2015, 4, e09300. [Google Scholar] [CrossRef] [PubMed]
- O’Callaghan, C.J.; Williams, B. Mechanical strain-induced extracellular matrix production by human vascular smooth muscle cells: Role of TGF-β(1). Hypertension 2000, 36, 319–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munger, J.S.; Sheppard, D. Cross talk among TGF-β signaling pathways, integrins, and the extracellular matrix. Cold Spring Harb. Perspect. Biol. 2011, 3, a005017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hautmann, M.B.; Madsen, C.S.; Owens, G.K. A transforming growth factor β (TGFbeta) control element drives TGFbeta-induced stimulation of smooth muscle α-actin gene expression in concert with two CArG elements. J. Biol. Chem. 1997, 272, 10948–10956. [Google Scholar] [CrossRef] [Green Version]
- Adam, P.J.; Regan, C.P.; Hautmann, M.B.; Owens, G.K. Positive- and negative-acting Kruppel-like transcription factors bind a transforming growth factor β control element required for expression of the smooth muscle cell differentiation marker SM22alpha in vivo. J. Biol. Chem. 2000, 275, 37798–37806. [Google Scholar] [CrossRef] [Green Version]
- Hirschi, K.K.; Lai, L.; Belaguli, N.S.; Dean, D.A.; Schwartz, R.J.; Zimmer, W.E. Transforming growth factor-β induction of smooth muscle cell phenotpye requires transcriptional and post-transcriptional control of serum response factor. J. Biol. Chem. 2002, 277, 6287–6295. [Google Scholar] [CrossRef] [Green Version]
- Habashi, J.P.; Judge, D.P.; Holm, T.M.; Cohn, R.D.; Loeys, B.L.; Cooper, T.K.; Myers, L.; Klein, E.C.; Liu, G.; Calvi, C.; et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science 2006, 312, 117–121. [Google Scholar] [CrossRef] [Green Version]
- Hanada, K.; Vermeij, M.; Garinis, G.A.; de Waard, M.C.; Kunen, M.G.; Myers, L.; Maas, A.; Duncker, D.J.; Meijers, C.; Dietz, H.C.; et al. Perturbations of vascular homeostasis and aortic valve abnormalities in fibulin-4 deficient mice. Circ. Res. 2007, 100, 738–746. [Google Scholar] [CrossRef] [Green Version]
- Gomez, D.; Coyet, A.; Ollivier, V.; Jeunemaitre, X.; Jondeau, G.; Michel, J.B.; Vranckx, R. Epigenetic control of vascular smooth muscle cells in Marfan and non-Marfan thoracic aortic aneurysms. Cardiovasc. Res. 2011, 89, 446–456. [Google Scholar] [CrossRef] [Green Version]
- Fukuda, H.; Aoki, H.; Yoshida, S.; Tobinaga, S.; Otsuka, H.; Shojima, T.; Takagi, K.; Fukumoto, Y.; Akashi, H.; Kato, S.; et al. Characterization of SMAD2 Activation in Human Thoracic Aortic Aneurysm. Ann. Vasc. Dis. 2018, 11, 112–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Low, E.L.; Baker, A.H.; Bradshaw, A.C. TGFbeta, smooth muscle cells and coronary artery disease: A review. Cell Signal. 2019, 53, 90–101. [Google Scholar] [CrossRef]
- Artavanis-Tsakonas, S.; Rand, M.D.; Lake, R.J. Notch signaling: Cell fate control and signal integration in development. Science 1999, 284, 770–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villa, N.; Walker, L.; Lindsell, C.E.; Gasson, J.; Iruela-Arispe, M.L.; Weinmaster, G. Vascular expression of Notch pathway receptors and ligands is restricted to arterial vessels. Mech. Dev. 2001, 108, 161–164. [Google Scholar] [CrossRef]
- Sweeney, C.; Morrow, D.; Birney, Y.A.; Coyle, S.; Hennessy, C.; Scheller, A.; Cummins, P.M.; Walls, D.; Redmond, E.M.; Cahill, P.A. Notch 1 and 3 receptor signaling modulates vascular smooth muscle cell growth, apoptosis, and migration via a CBF-1/RBP-Jk dependent pathway. FASEB J. 2004, 18, 1421–1423. [Google Scholar] [CrossRef]
- Liu, N.; Li, Y.; Chen, H.; Wei, W.; An, Y.; Zhu, G. RNA interference-mediated NOTCH3 knockdown induces phenotype switching of vascular smooth muscle cells in vitro. Int. J. Clin. Exp. Med. 2015, 8, 12674–12684. [Google Scholar]
- Baeten, J.T.; Lilly, B. Differential Regulation of NOTCH2 and NOTCH3 Contribute to Their Unique Functions in Vascular Smooth Muscle Cells. J. Biol. Chem. 2015, 290, 16226–16237. [Google Scholar] [CrossRef] [Green Version]
- Noseda, M.; Fu, Y.; Niessen, K.; Wong, F.; Chang, L.; McLean, G.; Karsan, A. Smooth Muscle α-actin is a direct target of Notch/CSL. Circ. Res. 2006, 98, 1468–1470. [Google Scholar] [CrossRef] [Green Version]
- Jin, S.; Hansson, E.M.; Tikka, S.; Lanner, F.; Sahlgren, C.; Farnebo, F.; Baumann, M.; Kalimo, H.; Lendahl, U. Notch signaling regulates platelet-derived growth factor receptor-β expression in vascular smooth muscle cells. Circ. Res. 2008, 102, 1483–1491. [Google Scholar] [CrossRef] [Green Version]
- Morrow, D.; Scheller, A.; Birney, Y.A.; Sweeney, C.; Guha, S.; Cummins, P.M.; Murphy, R.; Walls, D.; Redmond, E.M.; Cahill, P.A. Notch-mediated CBF-1/RBP-J{kappa}-dependent regulation of human vascular smooth muscle cell phenotype in vitro. Am. J. Physiol. Cell Physiol. 2005, 289, C1188–C1196. [Google Scholar] [CrossRef] [Green Version]
- Proweller, A.; Pear, W.S.; Parmacek, M.S. Notch signaling represses myocardin-induced smooth muscle cell differentiation. J. Biol. Chem. 2005, 280, 8994–9004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doi, H.; Iso, T.; Sato, H.; Yamazaki, M.; Matsui, H.; Tanaka, T.; Manabe, I.; Arai, M.; Nagai, R.; Kurabayashi, M. Jagged1-selective notch signaling induces smooth muscle differentiation via a RBP-Jkappa-dependent pathway. J. Biol. Chem. 2006, 281, 28555–28564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baeten, J.T.; Lilly, B. Notch Signaling in Vascular Smooth Muscle Cells. Adv. Pharmacol. 2017, 78, 351–382. [Google Scholar] [PubMed] [Green Version]
- Malashicheva, A.; Kostina, A.; Kostareva, A.; Irtyuga, O.; Gordeev, M.; Uspensky, V. Notch signaling in the pathogenesis of thoracic aortic aneurysms: A bridge between embryonic and adult states. Biochim Biophys Acta Mol. Basis Dis 2020, 1866, 165631. [Google Scholar] [CrossRef]
- Malashicheva, A.; Kostina, A.; Kostareva, A.; Irtyuga, O.; Uspensky, V. Corrigendum to “Notch signaling in the pathogenesis of thoracic aortic aneurysms: A bridge between embryonic and adult states” [Biochim. Biophys. Acta Mol. Basis Dis. 1866 (3) (Mar 1 2020) 165631]. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165732. [Google Scholar] [CrossRef]
- Yao, M.; Wang, X.; Wang, X.; Zhang, T.; Chi, Y.; Gao, F. The Notch pathway mediates the angiotensin II-induced synthesis of extracellular matrix components in podocytes. Int. J. Mol. Med. 2015, 36, 294–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blokzijl, A.; Dahlqvist, C.; Reissmann, E.; Falk, A.; Moliner, A.; Lendahl, U.; Ibanez, C.F. Cross-talk between the Notch and TGF-β signaling pathways mediated by interaction of the Notch intracellular domain with Smad3. J. Cell Biol. 2003, 163, 723–728. [Google Scholar] [CrossRef]
- Tang, Y.; Urs, S.; Boucher, J.; Bernaiche, T.; Venkatesh, D.; Spicer, D.B.; Vary, C.P.; Liaw, L. Notch and transforming growth factor-β (TGFbeta) signaling pathways cooperatively regulate vascular smooth muscle cell differentiation. J. Biol. Chem. 2010, 285, 17556–17563. [Google Scholar] [CrossRef] [Green Version]
- Zhao, N.; Koenig, S.N.; Trask, A.J.; Lin, C.H.; Hans, C.P.; Garg, V.; Lilly, B. MicroRNA miR145 regulates TGFBR2 expression and matrix synthesis in vascular smooth muscle cells. Circ. Res. 2015, 116, 23–34. [Google Scholar] [CrossRef] [Green Version]
- Kennard, S.; Liu, H.; Lilly, B. Transforming growth factor-β (TGF- 1) down-regulates Notch3 in fibroblasts to promote smooth muscle gene expression. J. Biol. Chem. 2008, 283, 1324–1333. [Google Scholar] [CrossRef] [Green Version]
- Ignatieva, E.; Kostina, D.; Irtyuga, O.; Uspensky, V.; Golovkin, A.; Gavriliuk, N.; Moiseeva, O.; Kostareva, A.; Malashicheva, A. Mechanisms of Smooth Muscle Cell Differentiation Are Distinctly Altered in Thoracic Aortic Aneurysms Associated with Bicuspid or Tricuspid Aortic Valves. Front. Physiol. 2017, 8, 536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiarini, A.; Onorati, F.; Marconi, M.; Pasquali, A.; Patuzzo, C.; Malashicheva, A.; Irtyega, O.; Faggian, G.; Pignatti, P.F.; Trabetti, E.; et al. Studies on sporadic non-syndromic thoracic aortic aneurysms: 1. Deregulation of Jagged/Notch 1 homeostasis and selection of synthetic/secretor phenotype smooth muscle cells. Eur. J. Prev. Cardiol. 2018, 25, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Perik, M.H.A.M.; Verstraeten, A.; Loeys, B.L. Pathophysiology and Principles of Management of Hereditary Aneurysmal Aortopathies. In Mechanisms of Vascular Disease: A Textbook for Vascular Specialists; Fitridge, R., Ed.; Springer International Publishing: Cham, Switzerland, 2020; pp. 293–316. [Google Scholar]
- Ostberg, N.P.; Zafar, M.A.; Ziganshin, B.A.; Elefteriades, J.A. The Genetics of Thoracic Aortic Aneurysms and Dissection: A Clinical Perspective. Biomolecules 2020, 10, 182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohde, S.; Zafar, M.A.; Ziganshin, B.A.; Elefteriades, J.A. Thoracic aortic aneurysm gene dictionary. Asian Cardiovasc. Thorac. Ann. 2020, 218492320943800. [Google Scholar]
- Renard, M.; Francis, C.; Ghosh, R.; Scott, A.F.; Witmer, P.D.; Ades, L.C.; Andelfinger, G.U.; Arnaud, P.; Boileau, C.; Callewaert, B.L.; et al. Clinical Validity of Genes for Heritable Thoracic Aortic Aneurysm and Dissection. J. Am. Coll. Cardiol. 2018, 72, 605–615. [Google Scholar] [CrossRef]
- Dietz, H.C.; Cutting, G.R.; Pyeritz, R.E.; Maslen, C.L.; Sakai, L.Y.; Corson, G.M.; Puffenberger, E.G.; Hamosh, A.; Nanthakumar, E.J.; Curristin, S.M.; et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature 1991, 352, 337–339. [Google Scholar] [CrossRef]
- Hucthagowder, V.; Sausgruber, N.; Kim, K.H.; Angle, B.; Marmorstein, L.Y.; Urban, Z. Fibulin-4: A novel gene for an autosomal recessive cutis laxa syndrome. Am. J. Hum. Genet. 2006, 78, 1075–1080. [Google Scholar] [CrossRef] [Green Version]
- Al-Hassnan, Z.N.; Almesned, A.R.; Tulbah, S.; Hakami, A.; Al-Omrani, A.; Al Sehly, A.; Mohammed, S.; Majid, S.; Meyer, B.; Al-Fayyadh, M. Recessively inherited severe aortic aneurysm caused by mutated EFEMP2. Am. J. Cardiol. 2012, 109, 1677–1680. [Google Scholar] [CrossRef]
- Schwarze, U.; Schievink, W.I.; Petty, E.; Jaff, M.R.; Babovic-Vuksanovic, D.; Cherry, K.J.; Pepin, M.; Byers, P.H. Haploinsufficiency for one COL3A1 allele of type III procollagen results in a phenotype similar to the vascular form of Ehlers-Danlos syndrome, Ehlers-Danlos syndrome type IV. Am. J. Hum. Genet. 2001, 69, 989–1001. [Google Scholar] [CrossRef] [Green Version]
- Pepin, M.; Schwarze, U.; Superti-Furga, A.; Byers, P.H. Clinical and genetic features of Ehlers-Danlos syndrome type IV, the vascular type. N. Engl. J. Med. 2000, 342, 673–680. [Google Scholar] [CrossRef]
- Superti-Furga, A.; Gugler, E.; Gitzelmann, R.; Steinmann, B. Ehlers-Danlos syndrome type IV: A multi-exon deletion in one of the two COL3A1 alleles affecting structure, stability, and processing of type III procollagen. J. Biol. Chem. 1988, 263, 6226–6232. [Google Scholar] [CrossRef]
- Kontusaari, S.; Tromp, G.; Kuivaniemi, H.; Ladda, R.L.; Prockop, D.J. Inheritance of an RNA splicing mutation (G+ 1 IVS20) in the type III procollagen gene (COL3A1) in a family having aortic aneurysms and easy bruisability: Phenotypic overlap between familial arterial aneurysms and Ehlers-Danlos syndrome type IV. Am. J. Hum. Genet. 1990, 47, 112–120. [Google Scholar] [PubMed]
- Meester, J.A.; Vandeweyer, G.; Pintelon, I.; Lammens, M.; Van Hoorick, L.; De Belder, S.; Waitzman, K.; Young, L.; Markham, L.W.; Vogt, J.; et al. Loss-of-function mutations in the X-linked biglycan gene cause a severe syndromic form of thoracic aortic aneurysms and dissections. Genet. Med. 2017, 19, 386–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbier, M.; Gross, M.S.; Aubart, M.; Hanna, N.; Kessler, K.; Guo, D.C.; Tosolini, L.; Ho-Tin-Noe, B.; Regalado, E.; Varret, M.; et al. MFAP5 loss-of-function mutations underscore the involvement of matrix alteration in the pathogenesis of familial thoracic aortic aneurysms and dissections. Am. J. Hum. Genet. 2014, 95, 736–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, V.S.; Halabi, C.M.; Hoffman, E.P.; Carmichael, N.; Leshchiner, I.; Lian, C.G.; Bierhals, A.J.; Vuzman, D.; Brigham Genomic, M.; Mecham, R.P.; et al. Loss of function mutation in LOX causes thoracic aortic aneurysm and dissection in humans. Proc. Natl. Acad. Sci. USA 2016, 113, 8759–8764. [Google Scholar] [CrossRef] [Green Version]
- Guo, D.C.; Regalado, E.S.; Gong, L.; Duan, X.; Santos-Cortez, R.L.; Arnaud, P.; Ren, Z.; Cai, B.; Hostetler, E.M.; Moran, R.; et al. LOX Mutations Predispose to Thoracic Aortic Aneurysms and Dissections. Circ. Res. 2016, 118, 928–934. [Google Scholar] [CrossRef] [Green Version]
- Szabo, Z.; Crepeau, M.W.; Mitchell, A.L.; Stephan, M.J.; Puntel, R.A.; Yin Loke, K.; Kirk, R.C.; Urban, Z. Aortic aneurysmal disease and cutis laxa caused by defects in the elastin gene. J. Med. Genet. 2006, 43, 255–258. [Google Scholar] [CrossRef] [Green Version]
- Callewaert, B.L.; Loeys, B.L.; Ficcadenti, A.; Vermeer, S.; Landgren, M.; Kroes, H.Y.; Yaron, Y.; Pope, M.; Foulds, N.; Boute, O.; et al. Comprehensive clinical and molecular assessment of 32 probands with congenital contractural arachnodactyly: Report of 14 novel mutations and review of the literature. Hum. Mutat. 2009, 30, 334–341. [Google Scholar] [CrossRef]
- Takeda, N.; Morita, H.; Fujita, D.; Inuzuka, R.; Taniguchi, Y.; Imai, Y.; Hirata, Y.; Komuro, I. Congenital contractural arachnodactyly complicated with aortic dilatation and dissection: Case report and review of literature. Am. J. Med. Genet. A 2015, 167A, 2382–2387. [Google Scholar] [CrossRef]
- Loeys, B.L.; Chen, J.; Neptune, E.R.; Judge, D.P.; Podowski, M.; Holm, T.; Meyers, J.; Leitch, C.C.; Katsanis, N.; Sharifi, N.; et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat. Genet. 2005, 37, 275–281. [Google Scholar] [CrossRef]
- van de Laar, I.M.; Oldenburg, R.A.; Pals, G.; Roos-Hesselink, J.W.; de Graaf, B.M.; Verhagen, J.M.; Hoedemaekers, Y.M.; Willemsen, R.; Severijnen, L.A.; Venselaar, H.; et al. Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat. Genet. 2011, 43, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Micha, D.; Guo, D.C.; Hilhorst-Hofstee, Y.; van Kooten, F.; Atmaja, D.; Overwater, E.; Cayami, F.K.; Regalado, E.S.; van Uffelen, R.; Venselaar, H.; et al. SMAD2 Mutations Are Associated with Arterial Aneurysms and Dissections. Hum. Mutat. 2015, 36, 1145–1149. [Google Scholar] [CrossRef]
- Lindsay, M.E.; Schepers, D.; Bolar, N.A.; Doyle, J.J.; Gallo, E.; Fert-Bober, J.; Kempers, M.J.; Fishman, E.K.; Chen, Y.; Myers, L.; et al. Loss-of-function mutations in TGFB2 cause a syndromic presentation of thoracic aortic aneurysm. Nat. Genet. 2012, 44, 922–927. [Google Scholar] [CrossRef] [PubMed]
- Boileau, C.; Guo, D.C.; Hanna, N.; Regalado, E.S.; Detaint, D.; Gong, L.; Varret, M.; Prakash, S.K.; Li, A.H.; d’Indy, H.; et al. TGFB2 mutations cause familial thoracic aortic aneurysms and dissections associated with mild systemic features of Marfan syndrome. Nat. Genet. 2012, 44, 916–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertoli-Avella, A.M.; Gillis, E.; Morisaki, H.; Verhagen, J.M.; de Graaf, B.M.; van de Beek, G.; Gallo, E.; Kruithof, B.P.; Venselaar, H.; Myers, L.A.; et al. Mutations in a TGF-β ligand, TGFB3, cause syndromic aortic aneurysms and dissections. J. Am. Coll. Cardiol. 2015, 65, 1324–1336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, X.Y.; Guo, D.C.; Regalado, E.S.; Shen, H.; University of Washington Center for Mendelian, G.; Coselli, J.S.; Estrera, A.L.; Safi, H.J.; Bamshad, M.J.; Nickerson, D.A.; et al. SMAD4 rare variants in individuals and families with thoracic aortic aneurysms and dissections. Eur. J. Hum. Genet. 2019, 27, 1054–1060. [Google Scholar] [CrossRef] [PubMed]
- Andrabi, S.; Bekheirnia, M.R.; Robbins-Furman, P.; Lewis, R.A.; Prior, T.W.; Potocki, L. SMAD4 mutation segregating in a family with juvenile polyposis, aortopathy, and mitral valve dysfunction. Am. J. Med. Genet. A 2011, 155A, 1165–1169. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.C.; Regalado, E.S.; Pinard, A.; Chen, J.; Lee, K.; Rigelsky, C.; Zilberberg, L.; Hostetler, E.M.; Aldred, M.; Wallace, S.E.; et al. LTBP3 Pathogenic Variants Predispose Individuals to Thoracic Aortic Aneurysms and Dissections. Am. J. Hum. Genet. 2018, 102, 706–712. [Google Scholar] [CrossRef] [Green Version]
- Doyle, A.J.; Doyle, J.J.; Bessling, S.L.; Maragh, S.; Lindsay, M.E.; Schepers, D.; Gillis, E.; Mortier, G.; Homfray, T.; Sauls, K.; et al. Mutations in the TGF-β repressor SKI cause Shprintzen-Goldberg syndrome with aortic aneurysm. Nat. Genet. 2012, 44, 1249–1254. [Google Scholar] [CrossRef] [Green Version]
- Carmignac, V.; Thevenon, J.; Ades, L.; Callewaert, B.; Julia, S.; Thauvin-Robinet, C.; Gueneau, L.; Courcet, J.B.; Lopez, E.; Holman, K.; et al. In-frame mutations in exon 1 of SKI cause dominant Shprintzen-Goldberg syndrome. Am. J. Hum. Genet. 2012, 91, 950–957. [Google Scholar] [CrossRef] [Green Version]
- Garg, V.; Muth, A.N.; Ransom, J.F.; Schluterman, M.K.; Barnes, R.; King, I.N.; Grossfeld, P.D.; Srivastava, D. Mutations in NOTCH1 cause aortic valve disease. Nature 2005, 437, 270–274. [Google Scholar] [CrossRef] [PubMed]
- McKellar, S.H.; Tester, D.J.; Yagubyan, M.; Majumdar, R.; Ackerman, M.J.; Sundt, T.M., 3rd. Novel NOTCH1 mutations in patients with bicuspid aortic valve disease and thoracic aortic aneurysms. J. Thorac. Cardiovasc. Surg. 2007, 134, 290–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loscalzo, M.L.; Goh, D.L.; Loeys, B.; Kent, K.C.; Spevak, P.J.; Dietz, H.C. Familial thoracic aortic dilation and bicommissural aortic valve: A prospective analysis of natural history and inheritance. Am. J. Med. Genet. A 2007, 143A, 1960–1967. [Google Scholar] [CrossRef] [PubMed]
- Debiec, R.; Hamby, S.E.; Jones, P.D.; Coolman, S.; Asiani, M.; Kharodia, S.; Skinner, G.J.; Samani, N.J.; Webb, T.R.; Bolger, A. Novel loss of function mutation in NOTCH1 in a family with bicuspid aortic valve, ventricular septal defect, thoracic aortic aneurysm, and aortic valve stenosis. Mol. Genet. Genomic Med. 2020, 8, e1437. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.C.; Pannu, H.; Tran-Fadulu, V.; Papke, C.L.; Yu, R.K.; Avidan, N.; Bourgeois, S.; Estrera, A.L.; Safi, H.J.; Sparks, E.; et al. Mutations in smooth muscle α-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat. Genet. 2007, 39, 1488–1493. [Google Scholar] [CrossRef]
- Zhu, L.; Vranckx, R.; Khau Van Kien, P.; Lalande, A.; Boisset, N.; Mathieu, F.; Wegman, M.; Glancy, L.; Gasc, J.M.; Brunotte, F.; et al. Mutations in myosin heavy chain 11 cause a syndrome associating thoracic aortic aneurysm/aortic dissection and patent ductus arteriosus. Nat. Genet. 2006, 38, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Guo, D.C.; Cao, J.; Gong, L.; Kamm, K.E.; Regalado, E.; Li, L.; Shete, S.; He, W.Q.; Zhu, M.S.; et al. Mutations in myosin light chain kinase cause familial aortic dissections. Am. J. Hum. Genet. 2010, 87, 701–707. [Google Scholar] [CrossRef] [Green Version]
- Guo, D.C.; Regalado, E.; Casteel, D.E.; Santos-Cortez, R.L.; Gong, L.; Kim, J.J.; Dyack, S.; Horne, S.G.; Chang, G.; Jondeau, G.; et al. Recurrent gain-of-function mutation in PRKG1 causes thoracic aortic aneurysms and acute aortic dissections. Am. J. Hum. Genet. 2013, 93, 398–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Wit, M.C.; de Coo, I.F.; Lequin, M.H.; Halley, D.J.; Roos-Hesselink, J.W.; Mancini, G.M. Combined cardiological and neurological abnormalities due to filamin A gene mutation. Clin. Res. Cardiol. 2011, 100, 45–50. [Google Scholar] [CrossRef] [Green Version]
- Reinstein, E.; Frentz, S.; Morgan, T.; Garcia-Minaur, S.; Leventer, R.J.; McGillivray, G.; Pariani, M.; van der Steen, A.; Pope, M.; Holder-Espinasse, M.; et al. Vascular and connective tissue anomalies associated with X-linked periventricular heterotopia due to mutations in Filamin A. Eur. J. Hum. Genet. 2013, 21, 494–502. [Google Scholar] [CrossRef]
- Nakamura, F.; Stossel, T.P.; Hartwig, J.H. The filamins: Organizers of cell structure and function. Cell Adh. Migr. 2011, 5, 160–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.H.; Choudhury, S.; Hirata, M.; Khalsa, S.; Chang, B.; Walsh, C.A. Thoracic aortic aneurysm in patients with loss of function Filamin A mutations: Clinical characterization, genetics, and recommendations. Am. J. Med. Genet. A 2018, 176, 337–350. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.L.; Haelterman, N.A.; Kwartler, C.S.; Regalado, E.S.; Lee, P.T.; Nagarkar-Jaiswal, S.; Guo, D.C.; Duraine, L.; Wangler, M.F.; University of Washington Center for Mendelian, G.; et al. Ari-1 Regulates Myonuclear Organization Together with Parkin and Is Associated with Aortic Aneurysms. Dev. Cell 2018, 45, 226–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coucke, P.J.; Willaert, A.; Wessels, M.W.; Callewaert, B.; Zoppi, N.; De Backer, J.; Fox, J.E.; Mancini, G.M.; Kambouris, M.; Gardella, R.; et al. Mutations in the facilitative glucose transporter GLUT10 alter angiogenesis and cause arterial tortuosity syndrome. Nat. Genet. 2006, 38, 452–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boel, A.; Burger, J.; Vanhomwegen, M.; Beyens, A.; Renard, M.; Barnhoorn, S.; Casteleyn, C.; Reinhardt, D.P.; Descamps, B.; Vanhove, C.; et al. Slc2a10 knock-out mice deficient in ascorbic acid synthesis recapitulate aspects of arterial tortuosity syndrome and display mitochondrial respiration defects. Hum. Mol. Genet. 2020, 29, 1476–1488. [Google Scholar] [CrossRef]
- Willaert, A.; Khatri, S.; Callewaert, B.L.; Coucke, P.J.; Crosby, S.D.; Lee, J.G.; Davis, E.C.; Shiva, S.; Tsang, M.; De Paepe, A.; et al. GLUT10 is required for the development of the cardiovascular system and the notochord and connects mitochondrial function to TGFbeta signaling. Hum. Mol. Genet. 2012, 21, 1248–1259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vermeer, A.M.C.; Lodder, E.M.; Thomas, D.; Duijkers, F.A.M.; Marcelis, C.; van Gorselen, E.O.F.; Fortner, P.; Buss, S.J.; Mereles, D.; Katus, H.A.; et al. Dilation of the Aorta Ascendens Forms Part of the Clinical Spectrum of HCN4 Mutations. J. Am. Coll Cardiol. 2016, 67, 2313–2315. [Google Scholar] [CrossRef]
- Hanania, H.L.; Regalado, E.S.; Guo, D.C.; Xu, L.; Demo, E.; Sallee, D.; Milewicz, D.M. Do HCN4 Variants Predispose to Thoracic Aortic Aneurysms and Dissections? Circ. Genom. Precis. Med. 2019, 12, e002626. [Google Scholar] [CrossRef]
- Guo, D.C.; Gong, L.; Regalado, E.S.; Santos-Cortez, R.L.; Zhao, R.; Cai, B.; Veeraraghavan, S.; Prakash, S.K.; Johnson, R.J.; Muilenburg, A.; et al. MAT2A mutations predispose individuals to thoracic aortic aneurysms. Am. J. Hum. Genet. 2015, 96, 170–177. [Google Scholar] [CrossRef] [Green Version]
- Kuang, S.Q.; Medina-Martinez, O.; Guo, D.C.; Gong, L.; Regalado, E.S.; Reynolds, C.L.; Boileau, C.; Jondeau, G.; Prakash, S.K.; Kwartler, C.S.; et al. FOXE3 mutations predispose to thoracic aortic aneurysms and dissections. J. Clin. Investig. 2016, 126, 948–961. [Google Scholar] [CrossRef] [Green Version]
- Sherratt, M.J.; Wess, T.J.; Baldock, C.; Ashworth, J.; Purslow, P.P.; Shuttleworth, C.A.; Kielty, C.M. Fibrillin-rich microfibrils of the extracellular matrix: Ultrastructure and assembly. Micron 2001, 32, 185–200. [Google Scholar] [CrossRef]
- Dietz, H.C.; Saraiva, J.M.; Pyeritz, R.E.; Cutting, G.R.; Francomano, C.A. Clustering of fibrillin (FBN1) missense mutations in Marfan syndrome patients at cysteine residues in EGF-like domains. Hum. Mutat. 1992, 1, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Loeys, B.; De Backer, J.; Van Acker, P.; Wettinck, K.; Pals, G.; Nuytinck, L.; Coucke, P.; De Paepe, A. Comprehensive molecular screening of the FBN1 gene favors locus homogeneity of classical Marfan syndrome. Hum. Mutat. 2004, 24, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Robinson, P.N.; Arteaga-Solis, E.; Baldock, C.; Collod-Beroud, G.; Booms, P.; De Paepe, A.; Dietz, H.C.; Guo, G.; Handford, P.A.; Judge, D.P.; et al. The molecular genetics of Marfan syndrome and related disorders. J. Med. Genet. 2006, 43, 769–787. [Google Scholar] [CrossRef] [Green Version]
- Milewicz, D.M.; Pyeritz, R.E.; Crawford, E.S.; Byers, P.H. Marfan syndrome: Defective synthesis, secretion, and extracellular matrix formation of fibrillin by cultured dermal fibroblasts. J. Clin. Investig. 1992, 89, 79–86. [Google Scholar] [CrossRef] [Green Version]
- Dietz, H.C.; Loeys, B.; Carta, L.; Ramirez, F. Recent progress towards a molecular understanding of Marfan syndrome. Am. J. Med. Genet. C. Semin. Med. Genet. 2005, 139C, 4–9. [Google Scholar] [CrossRef]
- Bunton, T.E.; Biery, N.J.; Myers, L.; Gayraud, B.; Ramirez, F.; Dietz, H.C. Phenotypic alteration of vascular smooth muscle cells precedes elastolysis in a mouse model of Marfan syndrome. Circ. Res. 2001, 88, 37–43. [Google Scholar] [CrossRef] [Green Version]
- Neptune, E.R.; Frischmeyer, P.A.; Arking, D.E.; Myers, L.; Bunton, T.E.; Gayraud, B.; Ramirez, F.; Sakai, L.Y.; Dietz, H.C. Dysregulation of TGF-β activation contributes to pathogenesis in Marfan syndrome. Nat. Genet. 2003, 33, 407–411. [Google Scholar] [CrossRef]
- Isogai, Z.; Ono, R.N.; Ushiro, S.; Keene, D.R.; Chen, Y.; Mazzieri, R.; Charbonneau, N.L.; Reinhardt, D.P.; Rifkin, D.B.; Sakai, L.Y. Latent transforming growth factor β-binding protein 1 interacts with fibrillin and is a microfibril-associated protein. J. Biol. Chem. 2003, 278, 2750–2757. [Google Scholar] [CrossRef] [Green Version]
- Judge, D.P.; Biery, N.J.; Keene, D.R.; Geubtner, J.; Myers, L.; Huso, D.L.; Sakai, L.Y.; Dietz, H.C. Evidence for a critical contribution of haploinsufficiency in the complex pathogenesis of Marfan syndrome. J. Clin. Investig. 2004, 114, 172–181. [Google Scholar] [CrossRef] [Green Version]
- Charbonneau, N.L.; Carlson, E.J.; Tufa, S.; Sengle, G.; Manalo, E.C.; Carlberg, V.M.; Ramirez, F.; Keene, D.R.; Sakai, L.Y. In vivo studies of mutant fibrillin-1 microfibrils. J Biol. Chem. 2010, 285, 24943–24955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, A.W.; Au Yeung, K.; Sandor, G.G.; Judge, D.P.; Dietz, H.C.; van Breemen, C. Loss of elastic fiber integrity and reduction of vascular smooth muscle contraction resulting from the upregulated activities of matrix metalloproteinase-2 and -9 in the thoracic aortic aneurysm in Marfan syndrome. Circ. Res. 2007, 101, 512–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sengle, G.; Sakai, L.Y. The fibrillin microfibril scaffold: A niche for growth factors and mechanosensation? Matrix Biol. 2015, 47, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Maki, J.M.; Rasanen, J.; Tikkanen, H.; Sormunen, R.; Makikallio, K.; Kivirikko, K.I.; Soininen, R. Inactivation of the lysyl oxidase gene Lox leads to aortic aneurysms, cardiovascular dysfunction, and perinatal death in mice. Circulation 2002, 106, 2503–2509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kagan, H.M.; Li, W. Lysyl oxidase: Properties, specificity, and biological roles inside and outside of the cell. J. Cell Biochem. 2003, 88, 660–672. [Google Scholar] [CrossRef]
- Papke, C.L.; Yanagisawa, H. Fibulin-4 and fibulin-5 in elastogenesis and beyond: Insights from mouse and human studies. Matrix Biol. 2014, 37, 142–149. [Google Scholar] [CrossRef]
- Csiszar, K. Lysyl oxidases: A novel multifunctional amine oxidase family. Prog. Nucleic Acid Res. Mol. Biol. 2001, 70, 1–32. [Google Scholar]
- Lucero, H.A.; Ravid, K.; Grimsby, J.L.; Rich, C.B.; DiCamillo, S.J.; Maki, J.M.; Myllyharju, J.; Kagan, H.M. Lysyl oxidase oxidizes cell membrane proteins and enhances the chemotactic response of vascular smooth muscle cells. J. Biol. Chem. 2008, 283, 24103–24117. [Google Scholar] [CrossRef] [Green Version]
- Laczko, R.; Csiszar, K. Lysyl Oxidase (LOX): Functional Contributions to Signaling Pathways. Biomolecules 2020, 10, 1093. [Google Scholar] [CrossRef]
- Atsawasuwan, P.; Mochida, Y.; Katafuchi, M.; Kaku, M.; Fong, K.S.; Csiszar, K.; Yamauchi, M. Lysyl oxidase binds transforming growth factor-β and regulates its signaling via amine oxidase activity. J. Biol. Chem. 2008, 283, 34229–34240. [Google Scholar] [CrossRef] [Green Version]
- Kutchuk, L.; Laitala, A.; Soueid-Bomgarten, S.; Shentzer, P.; Rosendahl, A.H.; Eilot, S.; Grossman, M.; Sagi, I.; Sormunen, R.; Myllyharju, J.; et al. Muscle composition is regulated by a Lox-TGFbeta feedback loop. Development 2015, 142, 983–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schips, T.G.; Vanhoutte, D. Marfan syndrome and aortic aneurysm: Lysyl oxidases to the rescue? J. Mol. Cell Cardiol. 2015, 86, 9–11. [Google Scholar] [CrossRef] [PubMed]
- Busnadiego, O.; Gorbenko Del Blanco, D.; Gonzalez-Santamaria, J.; Habashi, J.P.; Calderon, J.F.; Sandoval, P.; Bedja, D.; Guinea-Viniegra, J.; Lopez-Cabrera, M.; Rosell-Garcia, T.; et al. Elevated expression levels of lysyl oxidases protect against aortic aneurysm progression in Marfan syndrome. J. Mol. Cell Cardiol. 2015, 85, 48–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horiguchi, M.; Inoue, T.; Ohbayashi, T.; Hirai, M.; Noda, K.; Marmorstein, L.Y.; Yabe, D.; Takagi, K.; Akama, T.O.; Kita, T.; et al. Fibulin-4 conducts proper elastogenesis via interaction with cross-linking enzyme lysyl oxidase. Proc. Natl. Acad. Sci. USA 2009, 106, 19029–19034. [Google Scholar] [CrossRef] [Green Version]
- Noda, K.; Kitagawa, K.; Miki, T.; Horiguchi, M.; Akama, T.O.; Taniguchi, T.; Taniguchi, H.; Takahashi, K.; Ogra, Y.; Mecham, R.P.; et al. A matricellular protein fibulin-4 is essential for the activation of lysyl oxidase. Sci. Adv. 2020, 6, sciadv.abc1404. [Google Scholar] [CrossRef]
- Huang, J.; Davis, E.C.; Chapman, S.L.; Budatha, M.; Marmorstein, L.Y.; Word, R.A.; Yanagisawa, H. Fibulin-4 deficiency results in ascending aortic aneurysms: A potential link between abnormal smooth muscle cell phenotype and aneurysm progression. Circ. Res. 2010, 106, 583–592. [Google Scholar] [CrossRef] [Green Version]
- Halabi, C.M.; Broekelmann, T.J.; Lin, M.; Lee, V.S.; Chu, M.L.; Mecham, R.P. Fibulin-4 is essential for maintaining arterial wall integrity in conduit but not muscular arteries. Sci Adv. 2017, 3, e1602532. [Google Scholar] [CrossRef] [Green Version]
- Yamashiro, Y.; Papke, C.L.; Kim, J.; Ringuette, L.J.; Zhang, Q.J.; Liu, Z.P.; Mirzaei, H.; Wagenseil, J.E.; Davis, E.C.; Yanagisawa, H. Abnormal mechanosensing and cofilin activation promote the progression of ascending aortic aneurysms in mice. Sci. Signal. 2015, 8, ra105. [Google Scholar] [CrossRef] [Green Version]
- Yamashiro, Y.; Thang, B.Q.; Shin, S.J.; Lino, C.A.; Nakamura, T.; Kim, J.; Sugiyama, K.; Tokunaga, C.; Sakamoto, H.; Osaka, M.; et al. Role of Thrombospondin-1 in Mechanotransduction and Development of Thoracic Aortic Aneurysm in Mouse and Humans. Circ. Res. 2018, 123, 660–672. [Google Scholar] [CrossRef]
- Miyamoto, A.; Lau, R.; Hein, P.W.; Shipley, J.M.; Weinmaster, G. Microfibrillar proteins MAGP-1 and MAGP-2 induce Notch1 extracellular domain dissociation and receptor activation. J. Biol. Chem. 2006, 281, 10089–10097. [Google Scholar] [CrossRef] [Green Version]
- Combs, M.D.; Knutsen, R.H.; Broekelmann, T.J.; Toennies, H.M.; Brett, T.J.; Miller, C.A.; Kober, D.L.; Craft, C.S.; Atkinson, J.J.; Shipley, J.M.; et al. Microfibril-associated glycoprotein 2 (MAGP2) loss of function has pleiotropic effects in vivo. J. Biol. Chem. 2013, 288, 28869–28880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craft, C.S.; Broekelmann, T.J.; Mecham, R.P. Microfibril-associated glycoproteins MAGP-1 and MAGP-2 in disease. Matrix Biol. 2018, 71-72, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Iozzo, R.V. The biology of the small leucine-rich proteoglycans. Functional network of interactive proteins. J. Biol Chem. 1999, 274, 18843–18846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reinboth, B.; Hanssen, E.; Cleary, E.G.; Gibson, M.A. Molecular interactions of biglycan and decorin with elastic fiber components: Biglycan forms a ternary complex with tropoelastin and microfibril-associated glycoprotein 1. J. Biol Chem. 2002, 277, 3950–3957. [Google Scholar] [CrossRef] [Green Version]
- Kinsella, M.G.; Bressler, S.L.; Wight, T.N. The regulated synthesis of versican, decorin, and biglycan: Extracellular matrix proteoglycans that influence cellular phenotype. Crit. Rev. Eukaryot Gene Expr. 2004, 14, 203–234. [Google Scholar] [CrossRef]
- Douglas, T.; Heinemann, S.; Bierbaum, S.; Scharnweber, D.; Worch, H. Fibrillogenesis of collagen types I, II, and III with small leucine-rich proteoglycans decorin and biglycan. Biomacromolecules 2006, 7, 2388–2393. [Google Scholar] [CrossRef]
- Powell, J.T.; Lanne, T. Through thick and thin collagen fibrils, stress, and aortic rupture: Another piece in the jigsaw. Circulation 2007, 115, 2687–2688. [Google Scholar] [CrossRef] [Green Version]
- Heegaard, A.M.; Corsi, A.; Danielsen, C.C.; Nielsen, K.L.; Jorgensen, H.L.; Riminucci, M.; Young, M.F.; Bianco, P. Biglycan deficiency causes spontaneous aortic dissection and rupture in mice. Circulation 2007, 115, 2731–2738. [Google Scholar] [CrossRef] [Green Version]
- Hildebrand, A.; Romaris, M.; Rasmussen, L.M.; Heinegard, D.; Twardzik, D.R.; Border, W.A.; Ruoslahti, E. Interaction of the small interstitial proteoglycans biglycan, decorin and fibromodulin with transforming growth factor β. Biochem. J. 1994, 302, 527–534. [Google Scholar] [CrossRef]
- Kolb, M.; Margetts, P.J.; Sime, P.J.; Gauldie, J. Proteoglycans decorin and biglycan differentially modulate TGF-β-mediated fibrotic responses in the lung. Am. J. Physiol. Lung Cell Mol. Physiol. 2001, 280, L1327–L1334. [Google Scholar] [CrossRef]
- de Figueiredo Borges, L.; Jaldin, R.G.; Dias, R.R.; Stolf, N.A.; Michel, J.B.; Gutierrez, P.S. Collagen is reduced and disrupted in human aneurysms and dissections of ascending aorta. Hum. Pathol. 2008, 39, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Bowen, C.J.; Calderon Giadrosic, J.F.; Burger, Z.; Rykiel, G.; Davis, E.C.; Helmers, M.R.; Benke, K.; Gallo MacFarlane, E.; Dietz, H.C. Targetable cellular signaling events mediate vascular pathology in vascular Ehlers-Danlos syndrome. J. Clin. Investig. 2020, 130, 686–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graul-Neumann, L.M.; Hausser, I.; Essayie, M.; Rauch, A.; Kraus, C. Highly variable cutis laxa resulting from a dominant splicing mutation of the elastin gene. Am. J. Med. Genet. A 2008, 146A, 977–983. [Google Scholar] [CrossRef] [PubMed]
- Cocciolone, A.J.; Hawes, J.Z.; Staiculescu, M.C.; Johnson, E.O.; Murshed, M.; Wagenseil, J.E. Elastin, arterial mechanics, and cardiovascular disease. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H189–H205. [Google Scholar] [CrossRef] [PubMed]
- Callewaert, B.; Renard, M.; Hucthagowder, V.; Albrecht, B.; Hausser, I.; Blair, E.; Dias, C.; Albino, A.; Wachi, H.; Sato, F.; et al. New insights into the pathogenesis of autosomal-dominant cutis laxa with report of five ELN mutations. Hum. Mutat. 2011, 32, 445–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tassabehji, M.; Metcalfe, K.; Hurst, J.; Ashcroft, G.S.; Kielty, C.; Wilmot, C.; Donnai, D.; Read, A.P.; Jones, C.J. An elastin gene mutation producing abnormal tropoelastin and abnormal elastic fibres in a patient with autosomal dominant cutis laxa. Hum. Mol. Genet. 1998, 7, 1021–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curran, M.E.; Atkinson, D.L.; Ewart, A.K.; Morris, C.A.; Leppert, M.F.; Keating, M.T. The elastin gene is disrupted by a translocation associated with supravalvular aortic stenosis. Cell 1993, 73, 159–168. [Google Scholar] [CrossRef]
- Ewart, A.K.; Morris, C.A.; Atkinson, D.; Jin, W.; Sternes, K.; Spallone, P.; Stock, A.D.; Leppert, M.; Keating, M.T. Hemizygosity at the elastin locus in a developmental disorder, Williams syndrome. Nat. Genet. 1993, 5, 11–16. [Google Scholar] [CrossRef]
- Ewart, A.K.; Jin, W.; Atkinson, D.; Morris, C.A.; Keating, M.T. Supravalvular aortic stenosis associated with a deletion disrupting the elastin gene. J. Clin. Investig. 1994, 93, 1071–1077. [Google Scholar] [CrossRef] [Green Version]
- Tassabehji, M.; Metcalfe, K.; Donnai, D.; Hurst, J.; Reardon, W.; Burch, M.; Read, A.P. Elastin: Genomic structure and point mutations in patients with supravalvular aortic stenosis. Hum. Mol. Genet. 1997, 6, 1029–1036. [Google Scholar] [CrossRef] [Green Version]
- Li, D.Y.; Brooke, B.; Davis, E.C.; Mecham, R.P.; Sorensen, L.K.; Boak, B.B.; Eichwald, E.; Keating, M.T. Elastin is an essential determinant of arterial morphogenesis. Nature 1998, 393, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Koch, A.; Buheitel, G.; Hofbeck, M.; Rauch, A.; Kraus, C.; Tassabehji, M.; Singer, H. Spectrum of arterial obstructions caused by one elastin gene point mutation. Eur. J. Pediatr. 2003, 162, 53–54. [Google Scholar] [CrossRef] [PubMed]
- Micale, L.; Turturo, M.G.; Fusco, C.; Augello, B.; Jurado, L.A.; Izzi, C.; Digilio, M.C.; Milani, D.; Lapi, E.; Zelante, L.; et al. Identification and characterization of seven novel mutations of elastin gene in a cohort of patients affected by supravalvular aortic stenosis. Eur. J. Hum. Genet. 2010, 18, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.J.; Staiculescu, M.C.; Hawes, J.Z.; Cocciolone, A.J.; Hunkins, B.M.; Roth, R.A.; Lin, C.Y.; Mecham, R.P.; Wagenseil, J.E. Heterogeneous Cellular Contributions to Elastic Laminae Formation in Arterial Wall Development. Circ. Res. 2019, 125, 1006–1018. [Google Scholar] [CrossRef]
- Li, D.Y.; Faury, G.; Taylor, D.G.; Davis, E.C.; Boyle, W.A.; Mecham, R.P.; Stenzel, P.; Boak, B.; Keating, M.T. Novel arterial pathology in mice and humans hemizygous for elastin. J. Clin. Investig. 1998, 102, 1783–1787. [Google Scholar] [CrossRef] [Green Version]
- Faury, G.; Pezet, M.; Knutsen, R.H.; Boyle, W.A.; Heximer, S.P.; McLean, S.E.; Minkes, R.K.; Blumer, K.J.; Kovacs, A.; Kelly, D.P.; et al. Developmental adaptation of the mouse cardiovascular system to elastin haploinsufficiency. J. Clin. Investig. 2003, 112, 1419–1428. [Google Scholar] [CrossRef]
- Yanagisawa, H.; Davis, E.C.; Starcher, B.C.; Ouchi, T.; Yanagisawa, M.; Richardson, J.A.; Olson, E.N. Fibulin-5 is an elastin-binding protein essential for elastic fibre development in vivo. Nature 2002, 415, 168–171. [Google Scholar] [CrossRef]
- Nakamura, T.; Lozano, P.R.; Ikeda, Y.; Iwanaga, Y.; Hinek, A.; Minamisawa, S.; Cheng, C.F.; Kobuke, K.; Dalton, N.; Takada, Y.; et al. Fibulin-5/DANCE is essential for elastogenesis in vivo. Nature 2002, 415, 171–175. [Google Scholar] [CrossRef]
- Hu, Q.; Loeys, B.L.; Coucke, P.J.; De Paepe, A.; Mecham, R.P.; Choi, J.; Davis, E.C.; Urban, Z. Fibulin-5 mutations: Mechanisms of impaired elastic fiber formation in recessive cutis laxa. Hum. Mol. Genet. 2006, 15, 3379–3386. [Google Scholar] [CrossRef] [Green Version]
- Loeys, B.; Van Maldergem, L.; Mortier, G.; Coucke, P.; Gerniers, S.; Naeyaert, J.M.; De Paepe, A. Homozygosity for a missense mutation in fibulin-5 (FBLN5) results in a severe form of cutis laxa. Hum. Mol. Genet. 2002, 11, 2113–2118. [Google Scholar] [CrossRef]
- Yanagisawa, H.; Wagenseil, J. Elastic fibers and biomechanics of the aorta: Insights from mouse studies. Matrix Biol. 2020, 85-86, 160–172. [Google Scholar] [CrossRef] [PubMed]
- Morisaki, H.; Akutsu, K.; Ogino, H.; Kondo, N.; Yamanaka, I.; Tsutsumi, Y.; Yoshimuta, T.; Okajima, T.; Matsuda, H.; Minatoya, K.; et al. Mutation of ACTA2 gene as an important cause of familial and nonfamilial nonsyndromatic thoracic aortic aneurysm and/or dissection (TAAD). Hum. Mutat. 2009, 30, 1406–1411. [Google Scholar] [CrossRef] [PubMed]
- Disabella, E.; Grasso, M.; Gambarin, F.I.; Narula, N.; Dore, R.; Favalli, V.; Serio, A.; Antoniazzi, E.; Mosconi, M.; Pasotti, M.; et al. Risk of dissection in thoracic aneurysms associated with mutations of smooth muscle α-actin 2 (ACTA2). Heart 2011, 97, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Fagnant, P.M.; Bookwalter, C.S.; Joel, P.; Trybus, K.M. Vascular disease-causing mutation R258C in ACTA2 disrupts actin dynamics and interaction with myosin. Proc. Natl. Acad. Sci. USA 2015, 112, E4168–E4177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luyckx, I.; Proost, D.; Hendriks, J.M.; Saenen, J.; Van Craenenbroeck, E.M.; Vermeulen, T.; Peeters, N.; Wuyts, W.; Rodrigus, I.; Verstraeten, A.; et al. Two novel MYLK nonsense mutations causing thoracic aortic aneurysms/dissections in patients without apparent family history. Clin. Genet. 2017. [Google Scholar] [CrossRef] [PubMed]
- Shalata, A.; Mahroom, M.; Milewicz, D.M.; Limin, G.; Kassum, F.; Badarna, K.; Tarabeih, N.; Assy, N.; Fell, R.; Cohen, H.; et al. Fatal thoracic aortic aneurysm and dissection in a large family with a novel MYLK gene mutation: Delineation of the clinical phenotype. Orphanet J. Rare Dis. 2018, 13, 41. [Google Scholar] [CrossRef] [Green Version]
- Wallace, S.E.; Regalado, E.S.; Gong, L.; Janda, A.L.; Guo, D.C.; Russo, C.F.; Kulmacz, R.J.; Hanna, N.; Jondeau, G.; Boileau, C.; et al. MYLK pathogenic variants aortic disease presentation, pregnancy risk, and characterization of pathogenic missense variants. Genet. Med. 2019, 21, 144–151. [Google Scholar] [CrossRef] [Green Version]
- Sheen, V.L.; Jansen, A.; Chen, M.H.; Parrini, E.; Morgan, T.; Ravenscroft, R.; Ganesh, V.; Underwood, T.; Wiley, J.; Leventer, R.; et al. Filamin A mutations cause periventricular heterotopia with Ehlers-Danlos syndrome. Neurology 2005, 64, 254–262. [Google Scholar] [CrossRef]
- Kim, H.; McCulloch, C.A. Filamin A mediates interactions between cytoskeletal proteins that control cell adhesion. FEBS Lett. 2011, 585, 18–22. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Walsh, C.A. The many faces of filamin: A versatile molecular scaffold for cell motility and signalling. Nat. Cell Biol. 2004, 6, 1034–1038. [Google Scholar] [CrossRef]
- Stossel, T.P.; Condeelis, J.; Cooley, L.; Hartwig, J.H.; Noegel, A.; Schleicher, M.; Shapiro, S.S. Filamins as integrators of cell mechanics and signalling. Nat. Rev. Mol. Cell Biol. 2001, 2, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, A.; Masuda, Y.; Ohta, Y.; Ikeda, K.; Watanabe, K. Filamin associates with Smads and regulates transforming growth factor-β signaling. J. Biol. Chem. 2001, 276, 17871–17877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lino Cardenas, C.L.; Kessinger, C.W.; Cheng, Y.; MacDonald, C.; MacGillivray, T.; Ghoshhajra, B.; Huleihel, L.; Nuri, S.; Yeri, A.S.; Jaffer, F.A.; et al. An HDAC9-MALAT1-BRG1 complex mediates smooth muscle dysfunction in thoracic aortic aneurysm. Nat. Commun. 2018, 9, 1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boileau, A.; Lindsay, M.E.; Michel, J.B.; Devaux, Y. Epigenetics in Ascending Thoracic Aortic Aneurysm and Dissection. Aorta (Stamford) 2018, 6, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Inamoto, S.; Kwartler, C.S.; Lafont, A.L.; Liang, Y.Y.; Fadulu, V.T.; Duraisamy, S.; Willing, M.; Estrera, A.; Safi, H.; Hannibal, M.C.; et al. TGFBR2 mutations alter smooth muscle cell phenotype and predispose to thoracic aortic aneurysms and dissections. Cardiovasc. Res. 2010, 88, 520–529. [Google Scholar] [CrossRef] [Green Version]
- van der Linde, D.; van de Laar, I.M.; Bertoli-Avella, A.M.; Oldenburg, R.A.; Bekkers, J.A.; Mattace-Raso, F.U.; van den Meiracker, A.H.; Moelker, A.; van Kooten, F.; Frohn-Mulder, I.M.; et al. Aggressive cardiovascular phenotype of aneurysms-osteoarthritis syndrome caused by pathogenic SMAD3 variants. J. Am. Coll. Cardiol. 2012, 60, 397–403. [Google Scholar] [CrossRef]
- van de Laar, I.M.; van der Linde, D.; Oei, E.H.; Bos, P.K.; Bessems, J.H.; Bierma-Zeinstra, S.M.; van Meer, B.L.; Pals, G.; Oldenburg, R.A.; Bekkers, J.A.; et al. Phenotypic spectrum of the SMAD3-related aneurysms-osteoarthritis syndrome. J. Med. Genet. 2012, 49, 47–57. [Google Scholar] [CrossRef]
- Wischmeijer, A.; Van Laer, L.; Tortora, G.; Bolar, N.A.; Van Camp, G.; Fransen, E.; Peeters, N.; di Bartolomeo, R.; Pacini, D.; Gargiulo, G.; et al. Thoracic aortic aneurysm in infancy in aneurysms-osteoarthritis syndrome due to a novel SMAD3 mutation: Further delineation of the phenotype. Am. J. Med. Genet. A 2013, 161A, 1028–1035. [Google Scholar] [CrossRef]
- Schepers, D.; Tortora, G.; Morisaki, H.; MacCarrick, G.; Lindsay, M.; Liang, D.; Mehta, S.G.; Hague, J.; Verhagen, J.; van de Laar, I.; et al. A mutation update on the LDS-associated genes TGFB2/3 and SMAD2/3. Hum. Mutat. 2018, 39, 621–634. [Google Scholar] [CrossRef] [Green Version]
- Zilberberg, L.; Phoon, C.K.; Robertson, I.; Dabovic, B.; Ramirez, F.; Rifkin, D.B. Genetic analysis of the contribution of LTBP-3 to thoracic aneurysm in Marfan syndrome. Proc. Natl. Acad. Sci. USA 2015, 112, 14012–14017. [Google Scholar] [CrossRef] [Green Version]
- Gallo, E.M.; Loch, D.C.; Habashi, J.P.; Calderon, J.F.; Chen, Y.; Bedja, D.; van Erp, C.; Gerber, E.E.; Parker, S.J.; Sauls, K.; et al. Angiotensin II-dependent TGF-β signaling contributes to Loeys-Dietz syndrome vascular pathogenesis. J. Clin. Investig. 2014, 124, 448–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoenhoff, F.S. Increased TGF-β Signaling Precedes Aneurysm Formation in SMAD3 Deficient Mice. EBioMedicine 2016, 12, 26–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Pluijm, I.; van Vliet, N.; von der Thusen, J.H.; Robertus, J.L.; Ridwan, Y.; van Heijningen, P.M.; van Thiel, B.S.; Vermeij, M.; Hoeks, S.E.; Buijs-Offerman, R.; et al. Defective Connective Tissue Remodeling in Smad3 Mice Leads to Accelerated Aneurysmal Growth Through Disturbed Downstream TGF-β Signaling. EBioMedicine 2016, 12, 280–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, B.; Wu, Z.; Phan, S.H. Smad3 mediates transforming growth factor-β-induced α-smooth muscle actin expression. Am. J. Respir. Cell Mol. Biol. 2003, 29, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Li, Q.; Jiao, Y.; Qin, L.; Ali, R.; Zhou, J.; Ferruzzi, J.; Kim, R.W.; Geirsson, A.; Dietz, H.C.; et al. Tgfbr2 disruption in postnatal smooth muscle impairs aortic wall homeostasis. J. Clin. Investig. 2014, 124, 755–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.H.; Wei, H.; Jaffe, M.; Airhart, N.; Du, L.; Angelov, S.N.; Yan, J.; Allen, J.K.; Kang, I.; Wight, T.N.; et al. Postnatal Deletion of the Type II Transforming Growth Factor-β Receptor in Smooth Muscle Cells Causes Severe Aortopathy in Mice. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2647–2656. [Google Scholar] [CrossRef] [Green Version]
- Ye, P.; Chen, W.; Wu, J.; Huang, X.; Li, J.; Wang, S.; Liu, Z.; Wang, G.; Yang, X.; Zhang, P.; et al. GM-CSF contributes to aortic aneurysms resulting from SMAD3 deficiency. J. Clin. Investig. 2013, 123, 2317–2331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindsay, M.E.; Dietz, H.C. Lessons on the pathogenesis of aneurysm from heritable conditions. Nature 2011, 473, 308–316. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Rowitch, D.H.; Soriano, P.; McMahon, A.P.; Sucov, H.M. Fate of the mammalian cardiac neural crest. Development 2000, 127, 1607–1616. [Google Scholar]
- Buckingham, M.; Meilhac, S.; Zaffran, S. Building the mammalian heart from two sources of myocardial cells. Nat. Rev. Genet. 2005, 6, 826–835. [Google Scholar] [CrossRef]
- Sawada, H.; Rateri, D.L.; Moorleghen, J.J.; Majesky, M.W.; Daugherty, A. Smooth Muscle Cells Derived From Second Heart Field and Cardiac Neural Crest Reside in Spatially Distinct Domains in the Media of the Ascending Aorta-Brief Report. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1722–1726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawada, H.; Chen, J.Z.; Wright, B.C.; Sheppard, M.B.; Lu, H.S.; Daugherty, A. Heterogeneity of Aortic Smooth Muscle Cells: A Determinant for Regional Characteristics of Thoracic Aortic Aneurysms? J. Transl. Int. Med. 2018, 6, 93–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, J.; Zhou, D.; Jiang, L.; Qiu, P.; Milewicz, D.M.; Chen, Y.E.; Yang, B. In Vitro Lineage-Specific Differentiation of Vascular Smooth Muscle Cells in Response to SMAD3 Deficiency: Implications for SMAD3-Related Thoracic Aortic Aneurysm. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 1651–1663. [Google Scholar] [CrossRef]
- Schepers, D.; Doyle, A.J.; Oswald, G.; Sparks, E.; Myers, L.; Willems, P.J.; Mansour, S.; Simpson, M.A.; Frysira, H.; Maat-Kievit, A.; et al. The SMAD-binding domain of SKI: A hotspot for de novo mutations causing Shprintzen-Goldberg syndrome. Eur. J. Hum. Genet. 2015, 23, 224–228. [Google Scholar] [CrossRef] [PubMed]
- Gori, I.; George, R.; Purkiss, A.G.; Strohbuecker, S.; Randall, R.A.; Ogrodowicz, R.; Carmignac, V.; Faivre, L.; Joshi, D.; Kjaer, S.; et al. Mutations in SKI in Shprintzen-Goldberg syndrome lead to attenuated TGF-β responses through SKI stabilization. Elife 2021, 10, eLife.63545. [Google Scholar] [CrossRef]
- Meester, J.A.N.; Verstraeten, A.; Alaerts, M.; Schepers, D.; Van Laer, L.; Loeys, B.L. Overlapping but distinct roles for NOTCH receptors in human cardiovascular disease. Clin. Genet. 2019, 95, 85–94. [Google Scholar] [CrossRef]
- Koenig, S.N.; LaHaye, S.; Feller, J.D.; Rowland, P.; Hor, K.N.; Trask, A.J.; Janssen, P.M.; Radtke, F.; Lilly, B.; Garg, V. Notch1 haploinsufficiency causes ascending aortic aneurysms in mice. JCI Insight 2017, 2. [Google Scholar] [CrossRef]
- Kostina, A.; Bjork, H.; Ignatieva, E.; Irtyuga, O.; Uspensky, V.; Semenova, D.; Maleki, S.; Tomilin, A.; Moiseeva, O.; Franco-Cereceda, A.; et al. Notch, BMP and WNT/β-catenin network is impaired in endothelial cells of the patients with thoracic aortic aneurysm. Atheroscler. Suppl. 2018, 35, e6–e13. [Google Scholar] [CrossRef]
- Hans, C.P.; Koenig, S.N.; Huang, N.; Cheng, J.; Beceiro, S.; Guggilam, A.; Kuivaniemi, H.; Partida-Sanchez, S.; Garg, V. Inhibition of Notch1 signaling reduces abdominal aortic aneurysm in mice by attenuating macrophage-mediated inflammation. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 3012–3023. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Koenig, S.N.; Kuivaniemi, H.S.; Garg, V.; Hans, C.P. Pharmacological inhibitor of notch signaling stabilizes the progression of small abdominal aortic aneurysm in a mouse model. J. Am. Heart Assoc. 2014, 3, e001064. [Google Scholar] [CrossRef] [Green Version]
- Michelena, H.I.; Prakash, S.K.; Della Corte, A.; Bissell, M.M.; Anavekar, N.; Mathieu, P.; Bosse, Y.; Limongelli, G.; Bossone, E.; Benson, D.W.; et al. Bicuspid aortic valve: Identifying knowledge gaps and rising to the challenge from the International Bicuspid Aortic Valve Consortium (BAVCon). Circulation 2014, 129, 2691–2704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bravo-Jaimes, K.; Prakash, S.K. Genetics in bicuspid aortic valve disease: Where are we? Prog. Cardiovasc. Dis. 2020, 63, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Kent, K.C.; Crenshaw, M.L.; Goh, D.L.; Dietz, H.C. Genotype-phenotype correlation in patients with bicuspid aortic valve and aneurysm. J. Thorac. Cardiovasc. Surg. 2013, 146, 158–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez, L.; Arheart, K.L.; Colan, S.D.; Stein, N.S.; Lopez-Mitnik, G.; Lin, A.E.; Reller, M.D.; Ventura, R.; Silberbach, M. Turner syndrome is an independent risk factor for aortic dilation in the young. Pediatrics 2008, 121, e1622–e1627. [Google Scholar] [CrossRef] [PubMed]
- Pepe, G.; Nistri, S.; Giusti, B.; Sticchi, E.; Attanasio, M.; Porciani, C.; Abbate, R.; Bonow, R.O.; Yacoub, M.; Gensini, G.F. Identification of fibrillin 1 gene mutations in patients with bicuspid aortic valve (BAV) without Marfan syndrome. BMC Med. Genet. 2014, 15, 23. [Google Scholar] [CrossRef] [Green Version]
- Li, R.G.; Xu, Y.J.; Wang, J.; Liu, X.Y.; Yuan, F.; Huang, R.T.; Xue, S.; Li, L.; Liu, H.; Li, Y.J.; et al. GATA4 Loss-of-Function Mutation and the Congenitally Bicuspid Aortic Valve. Am. J. Cardiol. 2018, 121, 469–474. [Google Scholar] [CrossRef]
- Shi, L.M.; Tao, J.W.; Qiu, X.B.; Wang, J.; Yuan, F.; Xu, L.; Liu, H.; Li, R.G.; Xu, Y.J.; Wang, Q.; et al. GATA5 loss-of-function mutations associated with congenital bicuspid aortic valve. Int. J. Mol. Med. 2014, 33, 1219–1226. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.J.; Di, R.M.; Qiao, Q.; Li, X.M.; Huang, R.T.; Xue, S.; Liu, X.Y.; Wang, J.; Yang, Y.Q. GATA6 loss-of-function mutation contributes to congenital bicuspid aortic valve. Gene 2018, 663, 115–120. [Google Scholar] [CrossRef]
- Qu, X.K.; Qiu, X.B.; Yuan, F.; Wang, J.; Zhao, C.M.; Liu, X.Y.; Zhang, X.L.; Li, R.G.; Xu, Y.J.; Hou, X.M.; et al. A novel NKX2.5 loss-of-function mutation associated with congenital bicuspid aortic valve. Am. J. Cardiol. 2014, 114, 1891–1895. [Google Scholar] [CrossRef]
- Gillis, E.; Kumar, A.A.; Luyckx, I.; Preuss, C.; Cannaerts, E.; van de Beek, G.; Wieschendorf, B.; Alaerts, M.; Bolar, N.; Vandeweyer, G.; et al. Corrigendum: Candidate Gene Resequencing in a Large Bicuspid Aortic Valve-Associated Thoracic Aortic Aneurysm Cohort: SMAD6 as an Important Contributor. Front. Physiol. 2017, 8, 730. [Google Scholar] [CrossRef]
- Gillis, E.; Kumar, A.A.; Luyckx, I.; Preuss, C.; Cannaerts, E.; van de Beek, G.; Wieschendorf, B.; Alaerts, M.; Bolar, N.; Vandeweyer, G.; et al. Candidate Gene Resequencing in a Large Bicuspid Aortic Valve-Associated Thoracic Aortic Aneurysm Cohort: SMAD6 as an Important Contributor. Front. Physiol. 2017, 8, 400. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.L.; Glen, E.; Topf, A.; Hall, D.; O’Sullivan, J.J.; Sneddon, L.; Wren, C.; Avery, P.; Lewis, R.J.; ten Dijke, P.; et al. Nonsynonymous variants in the SMAD6 gene predispose to congenital cardiovascular malformation. Hum. Mutat. 2012, 33, 720–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wunnemann, F.; Ta-Shma, A.; Preuss, C.; Leclerc, S.; van Vliet, P.P.; Oneglia, A.; Thibeault, M.; Nordquist, E.; Lincoln, J.; Scharfenberg, F.; et al. Loss of ADAMTS19 causes progressive non-syndromic heart valve disease. Nat. Genet. 2020, 52, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Liu, X.; Wang, L.; Jiang, J.; Sun, Y.; Zhu, Q.; Chen, Z.; He, Y.; Hu, P.; Xu, Q.; et al. Targeted next-generation sequencing identified ADAMTS5 as novel genetic substrate in patients with bicuspid aortic valve. Int. J. Cardiol. 2018, 252, 150–155. [Google Scholar] [CrossRef] [PubMed]
- Luyckx, I.; Kumar, A.A.; Reyniers, E.; Dekeyser, E.; Vanderstraeten, K.; Vandeweyer, G.; Wunnemann, F.; Preuss, C.; Mazzella, J.M.; Goudot, G.; et al. Copy number variation analysis in bicuspid aortic valve-related aortopathy identifies TBX20 as a contributing gene. Eur. J. Hum. Genet. 2019, 27, 1033–1043. [Google Scholar] [CrossRef] [PubMed]
- Gould, R.A.; Aziz, H.; Woods, C.E.; Seman-Senderos, M.A.; Sparks, E.; Preuss, C.; Wunnemann, F.; Bedja, D.; Moats, C.R.; McClymont, S.A.; et al. ROBO4 variants predispose individuals to bicuspid aortic valve and thoracic aortic aneurysm. Nat. Genet. 2019, 51, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Maleki, S.; Poujade, F.A.; Bergman, O.; Gadin, J.R.; Simon, N.; Lang, K.; Franco-Cereceda, A.; Body, S.C.; Bjorck, H.M.; Eriksson, P. Endothelial/Epithelial Mesenchymal Transition in Ascending Aortas of Patients With Bicuspid Aortic Valve. Front. Cardiovasc. Med. 2019, 6, 182. [Google Scholar] [CrossRef]
- Messner, B.; Bernhard, D. Bicuspid aortic valve-associated aortopathy: Where do we stand? J. Mol. Cell Cardiol. 2019, 133, 76–85. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, T.; Hayashi, T.; Tabata, T.; Hirata, K.I. Bicuspid Aortic Valve-Associated Aortic Dilatation- What Is the Mechanism of Bicuspid Aortopathy? Circ. J. 2018, 82, 2470–2471. [Google Scholar] [CrossRef]
- Lo Presti, F.; Guzzardi, D.G.; Bancone, C.; Fedak, P.W.M.; Della Corte, A. The science of BAV aortopathy. Prog. Cardiovasc. Dis. 2020, 63, 465–474. [Google Scholar] [CrossRef]
- Yamashiro, Y.; Yanagisawa, H. Crossing Bridges between Extra- and Intra-Cellular Events in Thoracic Aortic Aneurysms. J. Atheroscler. Thromb. 2018, 25, 99–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedroza, A.J.; Tashima, Y.; Shad, R.; Cheng, P.; Wirka, R.; Churovich, S.; Nakamura, K.; Yokoyama, N.; Cui, J.Z.; Iosef, C.; et al. Single-Cell Transcriptomic Profiling of Vascular Smooth Muscle Cell Phenotype Modulation in Marfan Syndrome Aortic Aneurysm. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2195–2211. [Google Scholar] [CrossRef] [PubMed]
- Yamashiro, Y.; Yanagisawa, H. The molecular mechanism of mechanotransduction in vascular homeostasis and disease. Clin. Sci. 2020, 134, 2399–2418. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, J.; Borkham-Kamphorst, E.; Haas, U.; Van de Leur, E.; Fraga, M.F.; Esteller, M.; Gressner, A.M.; Weiskirchen, R. The expression of CSRP2 encoding the LIM domain protein CRP2 is mediated by TGF-β in smooth muscle and hepatic stellate cells. Biochem. Biophys. Res. Commun. 2006, 345, 1526–1535. [Google Scholar] [CrossRef] [PubMed]
- Mori, M.; Nakagami, H.; Koibuchi, N.; Miura, K.; Takami, Y.; Koriyama, H.; Hayashi, H.; Sabe, H.; Mochizuki, N.; Morishita, R.; et al. Zyxin mediates actin fiber reorganization in epithelial-mesenchymal transition and contributes to endocardial morphogenesis. Mol. Biol. Cell 2009, 20, 3115–3124. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Kulik, M.; Lechleider, R.J. Smad proteins regulate transcriptional induction of the SM22alpha gene by TGF-β. Nucleic Acids Res. 2003, 31, 1302–1310. [Google Scholar] [CrossRef]
- Kihara, T.; Sugimoto, Y.; Shinohara, S.; Takaoka, S.; Miyake, J. Cysteine-rich protein 2 accelerates actin filament cluster formation. PLoS ONE 2017, 12, e0183085. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, L.M.; Jensen, C.C.; Chaturvedi, A.; Yoshigi, M.; Beckerle, M.C. Stretch-induced actin remodeling requires targeting of zyxin to stress fibers and recruitment of actin regulators. Mol. Biol. Cell 2012, 23, 1846–1859. [Google Scholar] [CrossRef]
- Han, X.; Stewart, J.E., Jr.; Bellis, S.L.; Benveniste, E.N.; Ding, Q.; Tachibana, K.; Grammer, J.R.; Gladson, C.L. TGF-beta1 up-regulates paxillin protein expression in malignant astrocytoma cells: Requirement for a fibronectin substrate. Oncogene 2001, 20, 7976–7986. [Google Scholar] [CrossRef] [Green Version]
- Kissin, E.Y.; Lemaire, R.; Korn, J.H.; Lafyatis, R. Transforming growth factor β induces fibroblast fibrillin-1 matrix formation. Arthritis Rheum. 2002, 46, 3000–3009. [Google Scholar] [CrossRef]
- Kahari, V.M.; Olsen, D.R.; Rhudy, R.W.; Carrillo, P.; Chen, Y.Q.; Uitto, J. Transforming growth factor-β up-regulates elastin gene expression in human skin fibroblasts. Evidence for post-transcriptional modulation. Lab. Invest. 1992, 66, 580–588. [Google Scholar] [PubMed]
- Gacheru, S.N.; Thomas, K.M.; Murray, S.A.; Csiszar, K.; Smith-Mungo, L.I.; Kagan, H.M. Transcriptional and post-transcriptional control of lysyl oxidase expression in vascular smooth muscle cells: Effects of TGF-β 1 and serum deprivation. J. Cell Biochem. 1997, 65, 395–407. [Google Scholar] [CrossRef]
- Shanley, C.J.; Gharaee-Kermani, M.; Sarkar, R.; Welling, T.H.; Kriegel, A.; Ford, J.W.; Stanley, J.C.; Phan, S.H. Transforming growth factor-β 1 increases lysyl oxidase enzyme activity and mRNA in rat aortic smooth muscle cells. J. Vasc. Surg. 1997, 25, 446–452. [Google Scholar] [CrossRef] [Green Version]
- Al-Abcha, A.; Saleh, Y.; Mujer, M.; Boumegouas, M.; Herzallah, K.; Charles, L.; Elkhatib, L.; Abdelkarim, O.; Kehdi, M.; Abela, G.S. Meta-analysis Examining the Usefulness of Angiotensin Receptor blockers for the Prevention of Aortic Root Dilation in Patients With the Marfan Syndrome. Am. J. Cardiol. 2020, 128, 101–106. [Google Scholar] [CrossRef]
- Milewicz, D.M.; Ramirez, F. Therapies for Thoracic Aortic Aneurysms and Acute Aortic Dissections. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 126–136. [Google Scholar] [CrossRef] [Green Version]
- Lino Cardenas, C.L.; Kessinger, C.W.; MacDonald, C.; Jassar, A.S.; Isselbacher, E.M.; Jaffer, F.A.; Lindsay, M.E. Inhibition of the methyltranferase EZH2 improves aortic performance in experimental thoracic aortic aneurysm. JCI Insight 2018, 3, e97493. [Google Scholar] [CrossRef] [Green Version]
- Azhar, M.; Schultz Jel, J.; Grupp, I.; Dorn, G.W., 2nd; Meneton, P.; Molin, D.G.; Gittenberger-de Groot, A.C.; Doetschman, T. Transforming growth factor β in cardiovascular development and function. Cytokine Growth Factor Rev. 2003, 14, 391–407. [Google Scholar] [CrossRef] [Green Version]
- Chakrabarti, M.; Al-Sammarraie, N.; Gebere, M.G.; Bhattacharya, A.; Chopra, S.; Johnson, J.; Pena, E.A.; Eberth, J.F.; Poelmann, R.E.; Gittenberger-de Groot, A.C.; et al. Transforming Growth Factor Beta3 is Required for Cardiovascular Development. J. Cardiovasc. Dev. Dis. 2020, 7, 19. [Google Scholar] [CrossRef]
- Kothapalli, D.; Liu, S.L.; Bae, Y.H.; Monslow, J.; Xu, T.; Hawthorne, E.A.; Byfield, F.J.; Castagnino, P.; Rao, S.; Rader, D.J.; et al. Cardiovascular protection by ApoE and ApoE-HDL linked to suppression of ECM gene expression and arterial stiffening. Cell Rep. 2012, 2, 1259–1271. [Google Scholar] [CrossRef] [Green Version]
- Brown, X.Q.; Bartolak-Suki, E.; Williams, C.; Walker, M.L.; Weaver, V.M.; Wong, J.Y. Effect of substrate stiffness and PDGF on the behavior of vascular smooth muscle cells: Implications for atherosclerosis. J. Cell Physiol. 2010, 225, 115–122. [Google Scholar] [CrossRef] [Green Version]
- Xie, S.A.; Zhang, T.; Wang, J.; Zhao, F.; Zhang, Y.P.; Yao, W.J.; Hur, S.S.; Yeh, Y.T.; Pang, W.; Zheng, L.S.; et al. Matrix stiffness determines the phenotype of vascular smooth muscle cell in vitro and in vivo: Role of DNA methyltransferase 1. Biomaterials 2018, 155, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Vorp, D.A.; Schiro, B.J.; Ehrlich, M.P.; Juvonen, T.S.; Ergin, M.A.; Griffith, B.P. Effect of aneurysm on the tensile strength and biomechanical behavior of the ascending thoracic aorta. Ann. Thorac. Surg. 2003, 75, 1210–1214. [Google Scholar] [CrossRef]
- Duprey, A.; Trabelsi, O.; Vola, M.; Favre, J.P.; Avril, S. Biaxial rupture properties of ascending thoracic aortic aneurysms. Acta. Biomater. 2016, 42, 273–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nollen, G.J.; Groenink, M.; Tijssen, J.G.; Van Der Wall, E.E.; Mulder, B.J. Aortic stiffness and diameter predict progressive aortic dilatation in patients with Marfan syndrome. Eur. Heart J. 2004, 25, 1146–1152. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner, D.; Baumgartner, C.; Matyas, G.; Steinmann, B.; Loffler-Ragg, J.; Schermer, E.; Schweigmann, U.; Baldissera, I.; Frischhut, B.; Hess, J.; et al. Diagnostic power of aortic elastic properties in young patients with Marfan syndrome. J. Thorac. Cardiovasc. Surg. 2005, 129, 730–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pees, C.; Michel-Behnke, I. Morphology of the bicuspid aortic valve and elasticity of the adjacent aorta in children. Am. J. Cardiol. 2012, 110, 1354–1360. [Google Scholar] [CrossRef] [PubMed]
- Oulego-Erroz, I.; Alonso-Quintela, P.; Mora-Matilla, M.; Gautreaux Minaya, S.; Lapena-Lopez de Armentia, S. Ascending aorta elasticity in children with isolated bicuspid aortic valve. Int. J. Cardiol. 2013, 168, 1143–1146. [Google Scholar] [CrossRef]
- Teixido-Tura, G.; Redheuil, A.; Rodriguez-Palomares, J.; Gutierrez, L.; Sanchez, V.; Forteza, A.; Lima, J.A.; Garcia-Dorado, D.; Evangelista, A. Aortic biomechanics by magnetic resonance: Early markers of aortic disease in Marfan syndrome regardless of aortic dilatation? Int. J. Cardiol. 2014, 171, 56–61. [Google Scholar] [CrossRef]
- Teixido-Tura, G.; Evangelista, A. Response to Letter by Mkrtchyan regarding article: “Aortic biomechanics by magnetic resonance: Early markers of aortic disease in Marfan syndrome regardless of aortic dilatation?”. Int. J. Cardiol. 2014, 176, 287. [Google Scholar] [CrossRef]
- Mkrtchyan, N.; Fratz, S. Correspondence letter by Mkrtchyan and Fratz regarding article “aortic biomechanics by magnetic resonance: Early markers of aortic disease in Marfan syndrome regardless of aortic dilatation?”. Int. J. Cardiol. 2014, 174, 381. [Google Scholar] [CrossRef]
- Weismann, C.G.; Lombardi, K.C.; Grell, B.S.; Northrup, V.; Sugeng, L. Aortic stiffness and left ventricular diastolic function in children with well-functioning bicuspid aortic valves. Eur. Heart J. Cardiovasc. Imaging 2016, 17, 225–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellini, C.; Korneva, A.; Zilberberg, L.; Ramirez, F.; Rifkin, D.B.; Humphrey, J.D. Differential ascending and descending aortic mechanics parallel aneurysmal propensity in a mouse model of Marfan syndrome. J. Biomech. 2016, 49, 2383–2389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.J.; Galatioto, J.; Rao, S.; Ramirez, F.; Costa, K.D. Losartan Attenuates Degradation of Aorta and Lung Tissue Micromechanics in a Mouse Model of Severe Marfan Syndrome. Ann. Biomed. Eng. 2016, 44, 2994–3006. [Google Scholar] [CrossRef] [Green Version]
- Devos, D.G.; De Groote, K.; Babin, D.; Demulier, L.; Taeymans, Y.; Westenberg, J.J.; Van Bortel, L.; Segers, P.; Achten, E.; De Schepper, J.; et al. Proximal aortic stiffening in Turner patients may be present before dilation can be detected: A segmental functional MRI study. J. Cardiovasc. Magn. Reson. 2017, 19, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellini, C.; Bersi, M.R.; Caulk, A.W.; Ferruzzi, J.; Milewicz, D.M.; Ramirez, F.; Rifkin, D.B.; Tellides, G.; Yanagisawa, H.; Humphrey, J.D. Comparison of 10 murine models reveals a distinct biomechanical phenotype in thoracic aortic aneurysms. J. R Soc. Interface 2017, 14, 20161036. [Google Scholar] [CrossRef] [Green Version]
- Selamet Tierney, E.S.; Levine, J.C.; Sleeper, L.A.; Roman, M.J.; Bradley, T.J.; Colan, S.D.; Chen, S.; Campbell, M.J.; Cohen, M.S.; De Backer, J.; et al. Influence of Aortic Stiffness on Aortic-Root Growth Rate and Outcome in Patients With the Marfan Syndrome. Am. J. Cardiol. 2018, 121, 1094–1101. [Google Scholar] [CrossRef]
- Goudot, G.; Mirault, T.; Bruneval, P.; Soulat, G.; Pernot, M.; Messas, E. Aortic Wall Elastic Properties in Case of Bicuspid Aortic Valve. Front. Physiol. 2019, 10, 299. [Google Scholar] [CrossRef]
- Chen, J.Z.; Sawada, H.; Moorleghen, J.J.; Weiland, M.; Daugherty, A.; Sheppard, M.B. Aortic Strain Correlates with Elastin Fragmentation in Fibrillin-1 Hypomorphic Mice. Circ. Rep. 2019, 1, 199–205. [Google Scholar] [CrossRef] [Green Version]
- Prakash, A.; Adlakha, H.; Rabideau, N.; Hass, C.J.; Morris, S.A.; Geva, T.; Gauvreau, K.; Singh, M.N.; Lacro, R.V. Segmental Aortic Stiffness in Children and Young Adults With Connective Tissue Disorders: Relationships With Age, Aortic Size, Rate of Dilation, and Surgical Root Replacement. Circulation 2015, 132, 595–602. [Google Scholar] [CrossRef]
- Guala, A.; Teixido-Tura, G.; Rodriguez-Palomares, J.; Ruiz-Munoz, A.; Dux-Santoy, L.; Villalva, N.; Granato, C.; Galian, L.; Gutierrez, L.; Gonzalez-Alujas, T.; et al. Proximal aorta longitudinal strain predicts aortic root dilation rate and aortic events in Marfan syndrome. Eur. Heart J. 2019, 40, 2047–2055. [Google Scholar] [CrossRef]
- de Wit, A.; Vis, K.; Jeremy, R.W. Aortic stiffness in heritable aortopathies: Relationship to aneurysm growth rate. Heart Lung Circ. 2013, 22, 3–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellini, C.; Ferruzzi, J.; Roccabianca, S.; Di Martino, E.S.; Humphrey, J.D. A microstructurally motivated model of arterial wall mechanics with mechanobiological implications. Ann. Biomed. Eng. 2014, 42, 488–502. [Google Scholar] [CrossRef] [Green Version]
- Barnett, B.D.; Bird, H.R.; Lalich, J.J.; Strong, F.M. Toxicity of β-amino-propionitrile for turkey poults. Proc. Soc. Exp. Biol. Med. 1957, 94, 67–70. [Google Scholar] [CrossRef] [PubMed]
- Terpin, T.; Roach, M.R. A biophysical and histological analysis of factors that lead to aortic rupture in normal and lathyritic turkeys. Can. J. Physiol. Pharmacol. 1987, 65, 395–400. [Google Scholar] [CrossRef]
- Li, J.S.; Li, H.Y.; Wang, L.; Zhang, L.; Jing, Z.P. Comparison of β-aminopropionitrile-induced aortic dissection model in rats by different administration and dosage. Vascular 2013, 21, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.C.; Lee, M.T.; Chen, Y.S.; Lee, S.H.; Chen, Y.S.; Chen, S.C.; Chang, S.C. Risk of Aortic Dissection and Aortic Aneurysm in Patients Taking Oral Fluoroquinolone. JAMA Intern. Med. 2015, 175, 1839–1847. [Google Scholar] [CrossRef] [PubMed]
- LeMaire, S.A.; Zhang, L.; Luo, W.; Ren, P.; Azares, A.R.; Wang, Y.; Zhang, C.; Coselli, J.S.; Shen, Y.H. Effect of Ciprofloxacin on Susceptibility to Aortic Dissection and Rupture in Mice. JAMA Surg. 2018, 153, e181804. [Google Scholar] [CrossRef]
- Noman, A.T.; Qazi, A.H.; Alqasrawi, M.; Ayinde, H.; Tleyjeh, I.M.; Lindower, P.; Bin Abdulhak, A.A. Fluoroquinolones and the risk of aortopathy: A systematic review and meta-analysis. Int. J. Cardiol. 2019, 274, 299–302. [Google Scholar] [CrossRef]
- Guzzardi, D.G.; Teng, G.; Kang, S.; Geeraert, P.J.; Pattar, S.S.; Svystonyuk, D.A.; Belke, D.D.; Fedak, P.W.M. Induction of human aortic myofibroblast-mediated extracellular matrix dysregulation: A potential mechanism of fluoroquinolone-associated aortopathy. J. Thorac. Cardiovasc. Surg. 2019, 157, 109–119. [Google Scholar] [CrossRef]
- Peyton, S.R.; Putnam, A.J. Extracellular matrix rigidity governs smooth muscle cell motility in a biphasic fashion. J. Cell Physiol. 2005, 204, 198–209. [Google Scholar] [CrossRef]
- Lindeman, J.H.; Ashcroft, B.A.; Beenakker, J.W.; van Es, M.; Koekkoek, N.B.; Prins, F.A.; Tielemans, J.F.; Abdul-Hussien, H.; Bank, R.A.; Oosterkamp, T.H. Distinct defects in collagen microarchitecture underlie vessel-wall failure in advanced abdominal aneurysms and aneurysms in Marfan syndrome. Proc. Natl. Acad. Sci. USA 2010, 107, 862–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sazonova, O.V.; Lee, K.L.; Isenberg, B.C.; Rich, C.B.; Nugent, M.A.; Wong, J.Y. Cell-cell interactions mediate the response of vascular smooth muscle cells to substrate stiffness. Biophys. J. 2011, 101, 622–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaterji, S.; Kim, P.; Choe, S.H.; Tsui, J.H.; Lam, C.H.; Ho, D.S.; Baker, A.B.; Kim, D.H. Synergistic effects of matrix nanotopography and stiffness on vascular smooth muscle cell function. Tissue Eng. Part. A 2014, 20, 2115–2126. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, B.; Zhou, J.; Li, P.; Thomas, S.; Kaartinen, V.; Sucov, H.M. Absence of TGFbeta signaling in embryonic vascular smooth muscle leads to reduced lysyl oxidase expression, impaired elastogenesis, and aneurysm. Genesis 2009, 47, 115–121. [Google Scholar] [CrossRef]
- Choudhary, B.; Ito, Y.; Makita, T.; Sasaki, T.; Chai, Y.; Sucov, H.M. Cardiovascular malformations with normal smooth muscle differentiation in neural crest-specific type II TGFbeta receptor (Tgfbr2) mutant mice. Dev. Biol. 2006, 289, 420–429. [Google Scholar] [CrossRef] [Green Version]
- Wei, H.; Hu, J.H.; Angelov, S.N.; Fox, K.; Yan, J.; Enstrom, R.; Smith, A.; Dichek, D.A. Aortopathy in a Mouse Model of Marfan Syndrome Is Not Mediated by Altered Transforming Growth Factor β Signaling. J. Am. Heart Assoc. 2017, 6, e004968. [Google Scholar] [CrossRef]
- Qiu, P.; Ritchie, R.P.; Fu, Z.; Cao, D.; Cumming, J.; Miano, J.M.; Wang, D.Z.; Li, H.J.; Li, L. Myocardin enhances Smad3-mediated transforming growth factor-beta1 signaling in a CArG box-independent manner: Smad-binding element is an important cis element for SM22alpha transcription in vivo. Circ. Res. 2005, 97, 983–991. [Google Scholar] [CrossRef] [Green Version]
- Kwak, H.J.; Park, M.J.; Cho, H.; Park, C.M.; Moon, S.I.; Lee, H.C.; Park, I.C.; Kim, M.S.; Rhee, C.H.; Hong, S.I. Transforming growth factor-beta1 induces tissue inhibitor of metalloproteinase-1 expression via activation of extracellular signal-regulated kinase and Sp1 in human fibrosarcoma cells. Mol. Cancer Res. 2006, 4, 209–220. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.H.; Zheng, B.; Gu, C.; Fu, J.R.; Wen, J.K. TGF-beta1 downregulates AT1 receptor expression via PKC-delta-mediated Sp1 dissociation from KLF4 and Smad-mediated PPAR-γ association with KLF4. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1015–1023. [Google Scholar] [CrossRef] [Green Version]
- Gomez, D.; Kessler, K.; Borges, L.F.; Richard, B.; Touat, Z.; Ollivier, V.; Mansilla, S.; Bouton, M.C.; Alkoder, S.; Nataf, P.; et al. Smad2-dependent protease nexin-1 overexpression differentiates chronic aneurysms from acute dissections of human ascending aorta. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2222–2232. [Google Scholar] [CrossRef] [Green Version]
- Ferruzzi, J.; Murtada, S.I.; Li, G.; Jiao, Y.; Uman, S.; Ting, M.Y.; Tellides, G.; Humphrey, J.D. Pharmacologically Improved Contractility Protects Against Aortic Dissection in Mice With Disrupted Transforming Growth Factor-β Signaling Despite Compromised Extracellular Matrix Properties. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 919–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ignotz, R.A.; Massague, J. Transforming growth factor-β stimulates the expression of fibronectin and collagen and their incorporation into the extracellular matrix. J. Biol. Chem. 1986, 261, 4337–4345. [Google Scholar] [CrossRef]
- Lindahl, G.E.; Chambers, R.C.; Papakrivopoulou, J.; Dawson, S.J.; Jacobsen, M.C.; Bishop, J.E.; Laurent, G.J. Activation of fibroblast procollagen α 1(I) transcription by mechanical strain is transforming growth factor-β-dependent and involves increased binding of CCAAT-binding factor (CBF/NF-Y) at the proximal promoter. J. Biol. Chem. 2002, 277, 6153–6161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goel, S.A.; Guo, L.W.; Shi, X.D.; Kundi, R.; Sovinski, G.; Seedial, S.; Liu, B.; Kent, K.C. Preferential secretion of collagen type 3 versus type 1 from adventitial fibroblasts stimulated by TGF-β/Smad3-treated medial smooth muscle cells. Cell Signal. 2013, 25, 955–960. [Google Scholar] [CrossRef] [Green Version]
- Hong, H.H.; Trackman, P.C. Cytokine regulation of gingival fibroblast lysyl oxidase, collagen, and elastin. J. Periodontol. 2002, 73, 145–152. [Google Scholar] [CrossRef]
- Schonherr, E.; Jarvelainen, H.T.; Sandell, L.J.; Wight, T.N. Effects of platelet-derived growth factor and transforming growth factor-β 1 on the synthesis of a large versican-like chondroitin sulfate proteoglycan by arterial smooth muscle cells. J. Biol. Chem. 1991, 266, 17640–17647. [Google Scholar] [CrossRef]
- Wight, T.N. Arterial remodeling in vascular disease: A key role for hyaluronan and versican. Front. Biosci 2008, 13, 4933–4937. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.N.; Burch, M.L.; Tannock, L.R.; Evanko, S.; Osman, N.; Little, P.J. Transforming growth factor-β regulation of proteoglycan synthesis in vascular smooth muscle: Contribution to lipid binding and accelerated atherosclerosis in diabetes. J. Diabetes 2010, 2, 233–242. [Google Scholar] [CrossRef] [Green Version]
- Rostam, M.A.; Kamato, D.; Piva, T.J.; Zheng, W.; Little, P.J.; Osman, N. The role of specific Smad linker region phosphorylation in TGF-β mediated expression of glycosaminoglycan synthesizing enzymes in vascular smooth muscle. Cell Signal. 2016, 28, 956–966. [Google Scholar] [CrossRef]
- Kim, E.S.; Kim, M.S.; Moon, A. TGF-β-induced upregulation of MMP-2 and MMP-9 depends on p38 MAPK, but not ERK signaling in MCF10A human breast epithelial cells. Int. J. Oncol. 2004, 25, 1375–1382. [Google Scholar] [CrossRef]
- Safina, A.; Vandette, E.; Bakin, A.V. ALK5 promotes tumor angiogenesis by upregulating matrix metalloproteinase-9 in tumor cells. Oncogene 2007, 26, 2407–2422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinpitaksakul, S.N.; Pimkhaokham, A.; Sanchavanakit, N.; Pavasant, P. TGF-beta1 induced MMP-9 expression in HNSCC cell lines via Smad/MLCK pathway. Biochem. Biophys. Res. Commun. 2008, 371, 713–718. [Google Scholar] [CrossRef] [PubMed]
- Cucoranu, I.; Clempus, R.; Dikalova, A.; Phelan, P.J.; Ariyan, S.; Dikalov, S.; Sorescu, D. NAD(P)H oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ. Res. 2005, 97, 900–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samarakoon, R.; Overstreet, J.M.; Higgins, P.J. TGF-β signaling in tissue fibrosis: Redox controls, target genes and therapeutic opportunities. Cell Signal. 2013, 25, 264–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thallas-Bonke, V.; Jandeleit-Dahm, K.A.; Cooper, M.E. Nox-4 and progressive kidney disease. Curr. Opin. Nephrol. Hypertens. 2015, 24, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Altayo, F.; Meirelles, T.; Crosas-Molist, E.; Sorolla, M.A.; Del Blanco, D.G.; Lopez-Luque, J.; Mas-Stachurska, A.; Siegert, A.M.; Bonorino, F.; Barbera, L.; et al. Redox stress in Marfan syndrome: Dissecting the role of the NADPH oxidase NOX4 in aortic aneurysm. Free Radic. Biol. Med. 2018, 118, 44–58. [Google Scholar] [CrossRef] [PubMed]
- Angelov, S.N.; Hu, J.H.; Wei, H.; Airhart, N.; Shi, M.; Dichek, D.A. TGF-β (Transforming Growth Factor-β) Signaling Protects the Thoracic and Abdominal Aorta From Angiotensin II-Induced Pathology by Distinct Mechanisms. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 2102–2113. [Google Scholar] [CrossRef] [Green Version]
- Cook, J.R.; Clayton, N.P.; Carta, L.; Galatioto, J.; Chiu, E.; Smaldone, S.; Nelson, C.A.; Cheng, S.H.; Wentworth, B.M.; Ramirez, F. Dimorphic effects of transforming growth factor-β signaling during aortic aneurysm progression in mice suggest a combinatorial therapy for Marfan syndrome. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 911–917. [Google Scholar] [CrossRef] [Green Version]
- Huang, K.; Wang, Y.; Siu, K.L.; Zhang, Y.; Cai, H. Targeting feed-forward signaling of TGFbeta/NOX4/DHFR/eNOS uncoupling/TGFbeta axis with anti-TGFbeta and folic acid attenuates formation of aortic aneurysms: Novel mechanisms and therapeutics. Redox Biol. 2020, 38, 101757. [Google Scholar] [CrossRef]
- Holm, T.M.; Habashi, J.P.; Doyle, J.J.; Bedja, D.; Chen, Y.; van Erp, C.; Lindsay, M.E.; Kim, D.; Schoenhoff, F.; Cohn, R.D.; et al. Noncanonical TGFbeta signaling contributes to aortic aneurysm progression in Marfan syndrome mice. Science 2011, 332, 358. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.H.; LeMaire, S.A.; Webb, N.R.; Cassis, L.A.; Daugherty, A.; Lu, H.S. Aortic Aneurysms and Dissections Series: Part II: Dynamic Signaling Responses in Aortic Aneurysms and Dissections. Arterioscler. Thromb. Vasc. Biol. 2020, 40, e78–e86. [Google Scholar] [CrossRef] [PubMed]
- Moltzer, E.; te Riet, L.; Swagemakers, S.M.; van Heijningen, P.M.; Vermeij, M.; van Veghel, R.; Bouhuizen, A.M.; van Esch, J.H.; Lankhorst, S.; Ramnath, N.W.; et al. Impaired vascular contractility and aortic wall degeneration in fibulin-4 deficient mice: Effect of angiotensin II type 1 (AT1) receptor blockade. PLoS ONE 2011, 6, e23411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, W.; Meisinger, T.; Knispel, R.; Worth, J.M.; Baxter, B.T. MMP-2 regulates Erk1/2 phosphorylation and aortic dilatation in Marfan syndrome. Circ. Res. 2012, 110, e92–e101. [Google Scholar] [CrossRef] [Green Version]
- Kuang, S.Q.; Geng, L.; Prakash, S.K.; Cao, J.M.; Guo, S.; Villamizar, C.; Kwartler, C.S.; Peters, A.M.; Brasier, A.R.; Milewicz, D.M. Aortic remodeling after transverse aortic constriction in mice is attenuated with AT1 receptor blockade. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2172–2179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galatioto, J.; Caescu, C.I.; Hansen, J.; Cook, J.R.; Miramontes, I.; Iyengar, R.; Ramirez, F. Cell Type-Specific Contributions of the Angiotensin II Type 1a Receptor to Aorta Homeostasis and Aneurysmal Disease-Brief Report. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 588–591. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.Z.; Sawada, H.; Moorleghen, J.J.; Franklin, M.K.; Howatt, D.A.; Sheppard, M.B.; Mullick, A.E.; Lu, H.S.; Daugherty, A. Inhibition of Angiotensin II Dependent AT1a Receptor Stimulation Attenuates Thoracic Aortic Pathology in Fibrillin-1C1041G/+Mice. bioRxiv 2020. [Google Scholar]
- Habashi, J.P.; Doyle, J.J.; Holm, T.M.; Aziz, H.; Schoenhoff, F.; Bedja, D.; Chen, Y.; Modiri, A.N.; Judge, D.P.; Dietz, H.C. Angiotensin II type 2 receptor signaling attenuates aortic aneurysm in mice through ERK antagonism. Science 2011, 332, 361–365. [Google Scholar] [CrossRef]
- Papakonstantinou, E.; Roth, M.; Kokkas, B.; Papadopoulos, C.; Karakiulakis, G. Losartan inhibits the angiotensin II-induced modifications on fibrinolysis and matrix deposition by primary human vascular smooth muscle cells. J. Cardiovasc. Pharmacol. 2001, 38, 715–728. [Google Scholar] [CrossRef] [Green Version]
- Muino-Mosquera, L.; De Nobele, S.; Devos, D.; Campens, L.; De Paepe, A.; De Backer, J. Efficacy of losartan as add-on therapy to prevent aortic growth and ventricular dysfunction in patients with Marfan syndrome: A randomized, double-blind clinical trial. Acta Cardiol. 2017, 72, 616–624. [Google Scholar] [CrossRef]
- Chiu, H.H.; Wu, M.H.; Wang, J.K.; Lu, C.W.; Chiu, S.N.; Chen, C.A.; Lin, M.T.; Hu, F.C. Losartan added to β-blockade therapy for aortic root dilation in Marfan syndrome: A randomized, open-label pilot study. Mayo Clin. Proc. 2013, 88, 271–276. [Google Scholar] [CrossRef]
- Teixido-Tura, G.; Forteza, A.; Rodriguez-Palomares, J.; Gonzalez Mirelis, J.; Gutierrez, L.; Sanchez, V.; Ibanez, B.; Garcia-Dorado, D.; Evangelista, A. Losartan Versus Atenolol for Prevention of Aortic Dilation in Patients With Marfan Syndrome. J. Am. Coll Cardiol 2018, 72, 1613–1618. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Yamani, N.; Al-Naimat, S.; Khurshid, A.; Usman, M.S. Role of losartan in prevention of aortic dilatation in Marfan syndrome: A systematic review and meta-analysis. Eur. J. Prev. Cardiol 2020, 27, 1447–1450. [Google Scholar] [CrossRef] [PubMed]
- Jondeau, G.; Milleron, O.; Boileau, C. Marfan sartan saga, episode X. Eur. Heart J. 2020. [Google Scholar] [CrossRef] [PubMed]
- Lacro, R.V.; Dietz, H.C.; Sleeper, L.A.; Yetman, A.T.; Bradley, T.J.; Colan, S.D.; Pearson, G.D.; Selamet Tierney, E.S.; Levine, J.C.; Atz, A.M.; et al. Atenolol versus losartan in children and young adults with Marfan’s syndrome. N. Engl. J. Med. 2014, 371, 2061–2071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milleron, O.; Arnoult, F.; Ropers, J.; Aegerter, P.; Detaint, D.; Delorme, G.; Attias, D.; Tubach, F.; Dupuis-Girod, S.; Plauchu, H.; et al. Marfan Sartan: A randomized, double-blind, placebo-controlled trial. Eur. Heart J. 2015, 36, 2160–2166. [Google Scholar] [CrossRef] [Green Version]
- Forteza, A.; Evangelista, A.; Sanchez, V.; Teixido-Tura, G.; Sanz, P.; Gutierrez, L.; Gracia, T.; Centeno, J.; Rodriguez-Palomares, J.; Rufilanchas, J.J.; et al. Efficacy of losartan vs. atenolol for the prevention of aortic dilation in Marfan syndrome: A randomized clinical trial. Eur. Heart J. 2016, 37, 978–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franken, R.; den Hartog, A.W.; Radonic, T.; Micha, D.; Maugeri, A.; van Dijk, F.S.; Meijers-Heijboer, H.E.; Timmermans, J.; Scholte, A.J.; van den Berg, M.P.; et al. Beneficial Outcome of Losartan Therapy Depends on Type of FBN1 Mutation in Marfan Syndrome. Circ. Cardiovasc. Genet. 2015, 8, 383–388. [Google Scholar] [CrossRef] [Green Version]
- Groenink, M.; den Hartog, A.W.; Franken, R.; Radonic, T.; de Waard, V.; Timmermans, J.; Scholte, A.J.; van den Berg, M.P.; Spijkerboer, A.M.; Marquering, H.A.; et al. Losartan reduces aortic dilatation rate in adults with Marfan syndrome: A randomized controlled trial. Eur. Heart J. 2013, 34, 3491–3500. [Google Scholar] [CrossRef] [Green Version]
- Milewicz, D.M.; Prakash, S.K.; Ramirez, F. Therapeutics Targeting Drivers of Thoracic Aortic Aneurysms and Acute Aortic Dissections: Insights from Predisposing Genes and Mouse Models. Annu. Rev. Med. 2017, 68, 51–67. [Google Scholar] [CrossRef] [Green Version]
- Ford, C.M.; Li, S.; Pickering, J.G. Angiotensin II stimulates collagen synthesis in human vascular smooth muscle cells. Involvement of the AT(1) receptor, transforming growth factor-β, and tyrosine phosphorylation. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 1843–1851. [Google Scholar] [CrossRef] [Green Version]
- Che, Z.Q.; Gao, P.J.; Shen, W.L.; Fan, C.L.; Liu, J.J.; Zhu, D.L. Angiotensin II-stimulated collagen synthesis in aortic adventitial fibroblasts is mediated by connective tissue growth factor. Hypertens. Res. 2008, 31, 1233–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milewicz, D.M.; Guo, D.C.; Tran-Fadulu, V.; Lafont, A.L.; Papke, C.L.; Inamoto, S.; Kwartler, C.S.; Pannu, H. Genetic basis of thoracic aortic aneurysms and dissections: Focus on smooth muscle cell contractile dysfunction. Annu. Rev. Genomics Hum. Genet. 2008, 9, 283–302. [Google Scholar] [CrossRef] [PubMed]
- Muino-Mosquera, L.; De Backer, J. Angiotensin-II receptor blockade in Marfan syndrome. Lancet 2019, 394, 2206–2207. [Google Scholar] [CrossRef] [Green Version]
- Abraham, H.M.; White, C.M.; White, W.B. The comparative efficacy and safety of the angiotensin receptor blockers in the management of hypertension and other cardiovascular diseases. Drug Saf. 2015, 38, 33–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nair, A.B.; Jacob, S. A simple practice guide for dose conversion between animals and human. J. Basic Clin. Pharm. 2016, 7, 27–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullen, M.; Jin, X.Y.; Child, A.; Stuart, A.G.; Dodd, M.; Aragon-Martin, J.A.; Gaze, D.; Kiotsekoglou, A.; Yuan, L.; Hu, J.; et al. Irbesartan in Marfan syndrome (AIMS): A double-blind, placebo-controlled randomised trial. Lancet 2019, 394, 2263–2270. [Google Scholar] [CrossRef] [Green Version]
- Whelton, P.K.; Carey, R.M.; Aronow, W.S.; Casey, D.E., Jr.; Collins, K.J.; Dennison Himmelfarb, C.; DePalma, S.M.; Gidding, S.; Jamerson, K.A.; Jones, D.W.; et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J. Am. Coll. Cardiol. 2018, 71, 2199–2269. [Google Scholar]
- van Andel, M.M.; Indrakusuma, R.; Jalalzadeh, H.; Balm, R.; Timmermans, J.; Scholte, A.J.; van den Berg, M.P.; Zwinderman, A.H.; Mulder, B.J.M.; de Waard, V.; et al. Long-term clinical outcomes of losartan in patients with Marfan syndrome: Follow-up of the multicentre randomized controlled COMPARE trial. Eur. Heart J. 2020, 41, 4181–4187. [Google Scholar] [CrossRef]
- Jana, S.; Hu, M.; Shen, M.; Kassiri, Z. Extracellular matrix, regional heterogeneity of the aorta, and aortic aneurysm. Exp. Mol. Med. 2019, 51, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Ko, Y.S.; Coppen, S.R.; Dupont, E.; Rothery, S.; Severs, N.J. Regional differentiation of desmin, connexin43, and connexin45 expression patterns in rat aortic smooth muscle. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 355–364. [Google Scholar] [CrossRef] [Green Version]
- Absi, T.S.; Sundt, T.M., 3rd; Tung, W.S.; Moon, M.; Lee, J.K.; Damiano, R.R., Jr.; Thompson, R.W. Altered patterns of gene expression distinguishing ascending aortic aneurysms from abdominal aortic aneurysms: Complementary DNA expression profiling in the molecular characterization of aortic disease. J. Thorac. Cardiovasc. Surg. 2003, 126, 344–357. [Google Scholar] [CrossRef] [Green Version]
- Sinha, I.; Bethi, S.; Cronin, P.; Williams, D.M.; Roelofs, K.; Ailawadi, G.; Henke, P.K.; Eagleton, M.J.; Deeb, G.M.; Patel, H.J.; et al. A biologic basis for asymmetric growth in descending thoracic aortic aneurysms: A role for matrix metalloproteinase 9 and 2. J. Vasc. Surg. 2006, 43, 342–348. [Google Scholar] [CrossRef] [Green Version]
- Majesky, M.W. Choosing Smads: Smooth muscle origin-specific transforming growth factor-β signaling. Circ. Res. 2013, 113, 946–948. [Google Scholar] [CrossRef] [Green Version]
- Majesky, M.W. Developmental basis of vascular smooth muscle diversity. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1248–1258. [Google Scholar] [CrossRef] [Green Version]
- Trigueros-Motos, L.; Gonzalez-Granado, J.M.; Cheung, C.; Fernandez, P.; Sanchez-Cabo, F.; Dopazo, A.; Sinha, S.; Andres, V. Embryological-origin-dependent differences in homeobox expression in adult aorta: Role in regional phenotypic variability and regulation of NF-kappaB activity. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1248–1256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfaltzgraff, E.R.; Bader, D.M. Heterogeneity in vascular smooth muscle cell embryonic origin in relation to adult structure, physiology, and disease. Dev. Dyn. 2015, 244, 410–416. [Google Scholar] [CrossRef] [Green Version]
- Le Lievre, C.S.; Le Douarin, N.M. Mesenchymal derivatives of the neural crest: Analysis of chimaeric quail and chick embryos. J. Embryol. Exp. Morphol. 1975, 34, 125–154. [Google Scholar]
- Christ, B.; Huang, R.; Scaal, M. Formation and differentiation of the avian sclerotome. Anat. Embryol. (Berl.) 2004, 208, 333–350. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.G.; Buckingham, M.E.; Moorman, A.F. Heart fields and cardiac morphogenesis. Cold Spring Harb. Perspect. Med. 2014, 4, a015750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waldo, K.L.; Hutson, M.R.; Ward, C.C.; Zdanowicz, M.; Stadt, H.A.; Kumiski, D.; Abu-Issa, R.; Kirby, M.L. Secondary heart field contributes myocardium and smooth muscle to the arterial pole of the developing heart. Dev. Biol. 2005, 281, 78–90. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, D. Making or breaking the heart: From lineage determination to morphogenesis. Cell 2006, 126, 1037–1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pouget, C.; Gautier, R.; Teillet, M.A.; Jaffredo, T. Somite-derived cells replace ventral aortic hemangioblasts and provide aortic smooth muscle cells of the trunk. Development 2006, 133, 1013–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wasteson, P.; Johansson, B.R.; Jukkola, T.; Breuer, S.; Akyurek, L.M.; Partanen, J.; Lindahl, P. Developmental origin of smooth muscle cells in the descending aorta in mice. Development 2008, 135, 1823–1832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gadson, P.F., Jr.; Rossignol, C.; McCoy, J.; Rosenquist, T.H. Expression of elastin, smooth muscle α-actin, and c-jun as a function of the embryonic lineage of vascular smooth muscle cells. In Vitro Cell Dev. Biol. Anim. 1993, 29A, 773–781. [Google Scholar] [CrossRef]
- Topouzis, S.; Catravas, J.D.; Ryan, J.W.; Rosenquist, T.H. Influence of vascular smooth muscle heterogeneity on angiotensin converting enzyme activity in chicken embryonic aorta and in endothelial cells in culture. Circ. Res. 1992, 71, 923–931. [Google Scholar] [CrossRef] [Green Version]
- Topouzis, S.; Majesky, M.W. Smooth muscle lineage diversity in the chick embryo. Two types of aortic smooth muscle cell differ in growth and receptor-mediated transcriptional responses to transforming growth factor-β. Dev. Biol. 1996, 178, 430–445. [Google Scholar] [CrossRef]
- Wrenn, R.W.; Raeuber, C.L.; Herman, L.E.; Walton, W.J.; Rosenquist, T.H. Transforming growth factor-β: Signal transduction via protein kinase C in cultured embryonic vascular smooth muscle cells. In Vitro Cell Dev. Biol. 1993, 29A, 73–78. [Google Scholar] [CrossRef]
- Thieszen, S.L.; Dalton, M.; Gadson, P.F.; Patterson, E.; Rosenquist, T.H. Embryonic lineage of vascular smooth muscle cells determines responses to collagen matrices and integrin receptor expression. Exp. Cell Res. 1996, 227, 135–145. [Google Scholar] [CrossRef]
- Gadson, P.F., Jr.; Dalton, M.L.; Patterson, E.; Svoboda, D.D.; Hutchinson, L.; Schram, D.; Rosenquist, T.H. Differential response of mesoderm- and neural crest-derived smooth muscle to TGF-beta1: Regulation of c-myb and alpha1 (I) procollagen genes. Exp. Cell Res. 1997, 230, 169–180. [Google Scholar] [CrossRef]
- Gittenberger-de Groot, A.C.; DeRuiter, M.C.; Bergwerff, M.; Poelmann, R.E. Smooth muscle cell origin and its relation to heterogeneity in development and disease. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 1589–1594. [Google Scholar] [CrossRef] [Green Version]
- Aird, W.C. Endothelial cell heterogeneity. Cold Spring Harb. Perspect. Med. 2012, 2, a006429. [Google Scholar] [CrossRef] [PubMed]
- Ishii, Y.; Langberg, J.; Rosborough, K.; Mikawa, T. Endothelial cell lineages of the heart. Cell Tissue Res. 2009, 335, 67–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalluri, A.S.; Vellarikkal, S.K.; Edelman, E.R.; Nguyen, L.; Subramanian, A.; Ellinor, P.T.; Regev, A.; Kathiresan, S.; Gupta, R.M. Single-Cell Analysis of the Normal Mouse Aorta Reveals Functionally Distinct Endothelial Cell Populations. Circulation 2019, 140, 147–163. [Google Scholar] [CrossRef] [PubMed]
- Warkala, M.; Chen, D.; Ramirez, A.; Jubran, A.; Schonning, M.J.; Wang, X.; Zhao, H.; Astrof, S. Cell—ECM Interactions Play Multiple Essential Roles in Aortic Arch Development. Circ. Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Cheung, C.; Bernardo, A.S.; Trotter, M.W.; Pedersen, R.A.; Sinha, S. Generation of human vascular smooth muscle subtypes provides insight into embryological origin-dependent disease susceptibility. Nat. Biotechnol. 2012, 30, 165–173. [Google Scholar] [CrossRef] [Green Version]
- Sinha, S.; Iyer, D.; Granata, A. Embryonic origins of human vascular smooth muscle cells: Implications for in vitro modeling and clinical application. Cell Mol. Life Sci. 2014, 71, 2271–2288. [Google Scholar] [CrossRef] [Green Version]
- Granata, A.; Bernard, W.G.; Zhao, N.; McCafferty, J.; Lilly, B.; Sinha, S. Temporal and embryonic lineage-dependent regulation of human vascular SMC development by NOTCH3. Stem Cells Dev. 2015, 24, 846–856. [Google Scholar] [CrossRef] [Green Version]
- Bargehr, J.; Low, L.; Cheung, C.; Bernard, W.G.; Iyer, D.; Bennett, M.R.; Gambardella, L.; Sinha, S. Embryological Origin of Human Smooth Muscle Cells Influences Their Ability to Support Endothelial Network Formation. Stem Cells Transl. Med. 2016, 5, 946–959. [Google Scholar] [CrossRef] [Green Version]
- Jiao, J.; Xiong, W.; Wang, L.; Yang, J.; Qiu, P.; Hirai, H.; Shao, L.; Milewicz, D.; Chen, Y.E.; Yang, B. Differentiation defect in neural crest-derived smooth muscle cells in patients with aortopathy associated with bicuspid aortic valves. EBioMedicine 2016, 10, 282–290. [Google Scholar] [CrossRef] [Green Version]
- Granata, A.; Serrano, F.; Bernard, W.G.; McNamara, M.; Low, L.; Sastry, P.; Sinha, S. An iPSC-derived vascular model of Marfan syndrome identifies key mediators of smooth muscle cell death. Nat. Genet. 2017, 49, 97–109. [Google Scholar] [CrossRef] [Green Version]
- Grewal, N.; Gittenberger-de Groot, A.C.; Poelmann, R.E.; Klautz, R.J.; Lindeman, J.H.; Goumans, M.J.; Palmen, M.; Mohamed, S.A.; Sievers, H.H.; Bogers, A.J.; et al. Ascending aorta dilation in association with bicuspid aortic valve: A maturation defect of the aortic wall. J. Thorac. Cardiovasc. Surg. 2014, 148, 1583–1590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crosas-Molist, E.; Meirelles, T.; Lopez-Luque, J.; Serra-Peinado, C.; Selva, J.; Caja, L.; Gorbenko Del Blanco, D.; Uriarte, J.J.; Bertran, E.; Mendizabal, Y.; et al. Vascular smooth muscle cell phenotypic changes in patients with Marfan syndrome. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 960–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Zhao, Z.; Hou, L.; Xiao, Y.; Qin, F.; Yan, J.; Zhou, J.; Jing, Z. The regulatory role of smooth muscle 22 on the proliferation of aortic smooth muscle cells participates in the development of aortic dissection. J. Vasc. Surg. 2017, 66, 875–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Yu, C.; Chang, Q.; Luo, X.; Qiu, J.; Liu, S. Comparison of gene expression profiles in aortic dissection and normal human aortic tissues. Biomed. Rep. 2016, 5, 421–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Ren, P.; Dawson, A.; Vasquez, H.G.; Ageedi, W.; Zhang, C.; Luo, W.; Chen, R.; Li, Y.; Kim, S.; et al. Single-Cell Transcriptome Analysis Reveals Dynamic Cell Populations and Differential Gene Expression Patterns in Control and Aneurysmal Human Aortic Tissue. Circulation 2020, 142, 1374–1388. [Google Scholar] [CrossRef]
- Avril, S.; Bersi, M.R.; Bellini, C.; Genovese, K.; Humphrey, J.D. Regional identification of mechanical properties in arteries. Comput Methods Biomech. Biomed. Engin. 2015, 18 Suppl 1, 1874–1875. [Google Scholar] [CrossRef] [Green Version]
- Halloran, B.G.; Davis, V.A.; McManus, B.M.; Lynch, T.G.; Baxter, B.T. Localization of aortic disease is associated with intrinsic differences in aortic structure. J. Surg. Res. 1995, 59, 17–22. [Google Scholar] [CrossRef]
- Bell, V.; Mitchell, G.F. Influence of vascular function and pulsatile hemodynamics on cardiac function. Curr. Hypertens. Rep. 2015, 17, 580. [Google Scholar] [CrossRef]
- Beller, C.J.; Labrosse, M.R.; Thubrikar, M.J.; Robicsek, F. Role of aortic root motion in the pathogenesis of aortic dissection. Circulation 2004, 109, 763–769. [Google Scholar] [CrossRef] [Green Version]
- Roccabianca, S.; Figueroa, C.A.; Tellides, G.; Humphrey, J.D. Quantification of regional differences in aortic stiffness in the aging human. J. Mech. Behav. Biomed. Mater. 2014, 29, 618–634. [Google Scholar] [CrossRef] [Green Version]
- Bell, V.; Mitchell, W.A.; Sigurethsson, S.; Westenberg, J.J.; Gotal, J.D.; Torjesen, A.A.; Aspelund, T.; Launer, L.J.; de Roos, A.; Gudnason, V.; et al. Longitudinal and circumferential strain of the proximal aorta. J. Am. Heart Assoc. 2014, 3, e001536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bersi, M.R.; Bellini, C.; Humphrey, J.D.; Avril, S. Local variations in material and structural properties characterize murine thoracic aortic aneurysm mechanics. Biomech. Model. Mechanobiol 2019, 18, 203–218. [Google Scholar] [CrossRef]
- Yu, X.; Turcotte, R.; Seta, F.; Zhang, Y. Micromechanics of elastic lamellae: Unravelling the role of structural inhomogeneity in multi-scale arterial mechanics. J. R Soc. Interface 2018, 15, 20180492. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Wang, Y.; Zhang, Y. Transmural variation in elastin fiber orientation distribution in the arterial wall. J. Mech Behav Biomed. Mater. 2018, 77, 745–753. [Google Scholar] [CrossRef] [PubMed]
- Michel, J.B.; Thaunat, O.; Houard, X.; Meilhac, O.; Caligiuri, G.; Nicoletti, A. Topological determinants and consequences of adventitial responses to arterial wall injury. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1259–1268. [Google Scholar] [CrossRef] [Green Version]



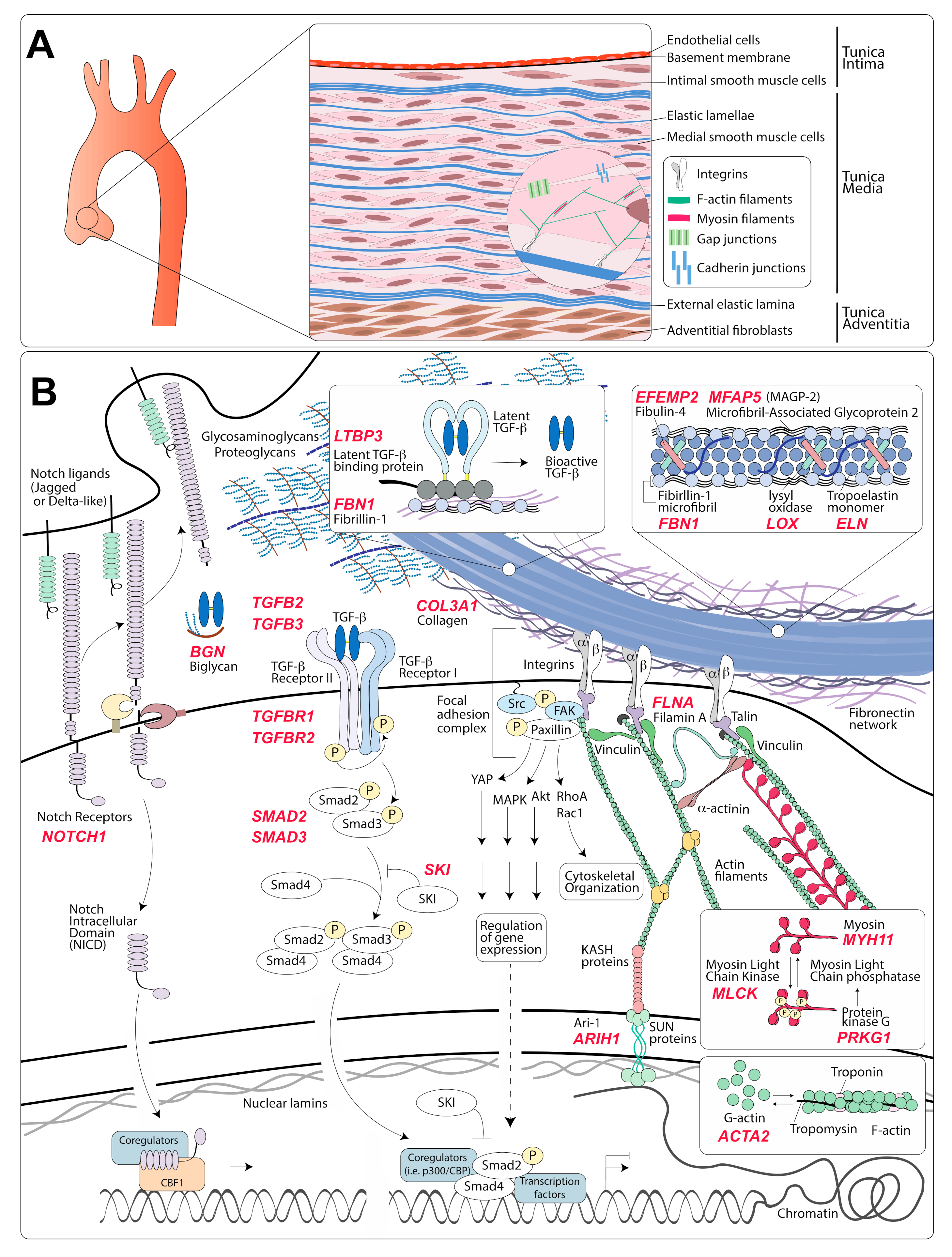{kind=link}
{kind=link}
| Genes Coding for Components of the Extracellular Matrix | ||||
|---|---|---|---|---|
| Gene | Inheritance | Protein Name | Primary Effect of TAA-Causing Variants | Disease (Phenotype MIM Number) |
| FBN1 [188] | AD | Fibrillin-1 | Fibrillin-1 is an extracellular matrix glycoprotein that serves as the structural component of microfibrils. TAA-causing variants are predicted to impair protein synthesis, secretion, or incorporation of mutant fibrillin in the microfibrillar architecture. | Marfan Syndrome (154700) |
| EFEMP2 [189,190] | AR | EGF-containing fibulin-like extracellular matrix protein 2 (Fibulin-4) | Fibulin-4 is necessary for elastic fiber formation. The known variants are predicted to result in defective maturation of elastic fibers in consequence of reduced cross-linking. | Cutis laxa type 1B (614437) |
| COL3A1 [191,192,193,194] | AD | Collagen α-1(III) chain | The alpha1 chains of type III collagen are a component of a fibrillar collagen that is found in the vascular system, often in association with type I collagen. The known variants are predicted to result in defective assembly. These mutations tend to cause dissection without preceding dilatation. | Vascular Ehlers–Danlos Syndrome (130050) |
| BGN [195] | X-linked | Biglycan | Biglycan is a secreted proteoglycan that interacts with other components of the ECM, including collagen type I/II/III/VI, elastin, microfibrils, and TGF-β. Although it is known that TAA-causing variants disrupt protein function, the exact effect on ECM deposition and/or TGF-β activity is not clear. | Meester–Loeys syndrome (300989) |
| MFAP5 [196] | AD | Microfibrillar-associated protein 5 | Microfibrillar-associated protein 5 is a component of microfibrils. The known variants are predicted to disrupt protein function, although the exact effect on ECM deposition or activity is not clear. | -- |
| LOX [197,198] | AD | Lysyl oxidase | Lysyl oxidase is an enzyme required for cross-linking and maturation of collagen and elastic fibers. TAA-causing variants are predicted to result in reduced enzymatic activity and thus reduced cross-linking. | Familial thoracic aortic aneurysm 10 (617168) |
| ELN [199] | AD | Tropoelastin | Tropoelastin self-assembles into polymer networks of elastin via a process that includes fibulin-4/5 complexes, lysyl oxidase, and other matrix proteins. It is the core component of elastic lamellae. A 25-bp deletion in exon 30 of ELN was found to be associated with aortic dilatation and rupture in some patients [199]. | Cutis laxa (123700) |
| FBN2 [200,201] | AD | Fibrillin-2 | Fibrillin-2 is a component of microfibrils and may be involved in elastic fiber assembly. Most loss-of-function mutations cause congenital contractural arachnodactyly; evidence for a causal role in TAA is limited. | Congenital contractural arachnodactyly (121050) |
| Genes coding for proteins involved in transduction of biochemical signals | ||||
| Gene | Inheritance | Protein name | Primary effect of TAA-causing variants | Disease (PhenotypeMIM number) |
| TGFBR1 [202] | AD | TGF-β receptor type I | TGF-β receptor type I is one of the two components of the TGF-β Receptor heterodimer. Upon binding to TGF-β, it dimerizes with, and is phosphorylated by TGF-β Receptor II. Thus activated, it phosphorylates and activates SMAD2 and SMAD3 proteins. TAA-causing mutations are predicted to result in decreased kinase activity and thus reduced levels of SMAD phosphorylation. | Loeys–Dietz syndrome type 1 (609192) |
| TGFBR2 [202] | AD | TGF-β receptor type II | TGF-β receptor type II is one of the two components of the TGF-β receptor heterodimer. Upon binding to TGF-β, it phosphorylates and activates TGF-β receptor I, which in turns phosphorylates and activates SMAD proteins. TAA-causing mutations are predicted to result in decreased kinase activity and thus reduced levels of SMAD phosphorylation. | Loeys–Dietz syndrome type 2 (610168) |
| SMAD3 [203] | AD | Mothers against decapentaplegic drosophila homolog 3 (SMAD3) | SMAD3 is one of the major signal transduction molecules activated by TGF-β receptors through phosphorylation. TAA-causing mutations are predicted to result in either decreased protein levels or decreased SMAD3-dependent transcriptional activity. | Loeys–Dietz syndrome type 3 (613795) |
| SMAD2 [204] | AD | Mothers against decapentaplegic drosophila homolog 2 (SMAD2) | SMAD2 is one of the major signal transduction molecules activated by TGF-β receptors though phosphorylation. TAA-causing mutations are predicted to result in decreased protein levels or decreased SMAD2-dependent transcriptional activity. | Loeys–Dietz syndrome type 6 |
| TGFB2 [205,206] | AD | Transforming growth factor β-2 proprotein(TGF-β2) | TGF-β2 is one of the three TGF-β ligands that bind and activate signaling by TGF-β receptors. TAA-causing mutations are predicted to result in decreased protein levels or decreased binding to TGF-β receptors. | Loeys–Dietz syndrome type 4 (614816) |
| TGFB3 [207] | AD | Transforming growth factor β-3 proprotein(TGF-β3) | TGF-β3 is one of the three TGF-β ligands that bind and activate signaling by TGF-β receptors. TAA-causing mutations are predicted to result in decreased protein levels or decreased binding to TGF-β receptors. | Loeys–Dietz syndrome type 5 (615582) |
| SMAD4 [208,209] | AD | Mothers against decapentaplegic drosophila homolog 4 (SMAD4) | SMAD4 binds to phosphorylated SMAD2 and SMAD3 to facilitate translocation to the nucleus and transcriptional regulation downstream of TGF-β receptors. Variants associated with TAA are predicted to reduce SMAD4 activity and thus decrease TGF-β signaling output. | Juvenile polyposis/hereditary hemorrhagic telangiectasia syndrome (175050) |
| LTBP3 [210] | AR | Latent transforming growth factor β binding protein 3 (LTBP-3) | LTBP-3 belongs to a family of proteins that regulate TGF-β activity by enabling its secretion and incorporation of its latent form into the ECM. They also participate in its conversion from latent to active form. Homozygous loss-of-function mutations associated with TAA are predicted to both decrease LTBP-3 levels in fibrillin-1–containing microfibrils and also decrease the overall secretion of TGF-β. | Dental anomalies and short stature syndrome (601216) |
| SKI [211,212] | AD | SKI protooncogene | SKI encodes a transcriptional repressor of TGF-β signaling. Mutations associated with TAA are predicted to disrupt the binding of SKI to SMAD proteins and other transcriptional co-regulators, resulting in a loss of inhibitory activity on the TGF-β signaling pathway. | Shprintzen–Goldberg syndrome (182212) |
| NOTCH1 [213,214,215,216] | AD | Neurogenic locus notch homolog protein 1 (Notch 1) | Notch1 is one of the four receptors that are activated by binding to one of the membrane-bound Notch ligands (Delta-like 1, 3, and 4, and Jagged 1 and 2). In humans, loss-of-function mutations in Notch1 cause aortic valve disease and, in some limited cases, TAA. | Aortic valve disease (with or without thoracic aortic aneurysm) (109730) |
| Genes coding for proteins involved in transduction of mechanical signals | ||||
| Gene | Inheritance | Protein name | Primary effect of TAA-causing variants | Disease (PhenotypeMIM number) |
| ACTA2 [217] | AD | Smooth muscle actin α 2 (α-SMA) | α-SMA is a smooth muscle-specific form of actin that, as other actins, exists in two states: the globular monomeric G-actin and the structural filament F-actin. It is a major constituent of the cell contractile apparatus. TAA-associated mutations are predicted to result in structurally altered actin monomers and less stable actin filaments. | Familial thoracic aortic aneurysm 6 (611788) |
| MYH11 [62,218] | AD | Myosin-11 or Smooth muscle myosin heavy chain (SM-MHC) | SM-MHC is a subunit of the hexameric myosin protein complex, which consists of two heavy chain subunits and two pairs of non-identical light chain subunits. It functions as a major contractile protein, converting chemical energy into mechanical energy through the hydrolysis of ATP. TAA-associated mutations are predicted to impair the ability of the mutant myosin to polymerize into thick filaments and form a quaternary structure. Haploinsufficiency for MYH11 does not appear to cause aneurysm. | Familial thoracic aortic aneurysm 4 (132900) |
| MYLK [219] | AD | Myosin light chain kinase (MLCK) | MLCK is a calcium/calmodulin-dependent kinase that phosphorylates myosin regulatory light chains to facilitate myosin interaction with actin filaments and thus produce contractile activity. TAA-associated mutations are predicted to either cause haploinsufficiency or impair kinase activity. | Familial thoracic aortic aneurysm 7 (613780) |
| PRKG1 [220] | AD | cGMP-dependent protein kinase 1 (PKG-1) | PKG-1 is a cGMP-activated kinase that promotes the relaxation of VSMCs. It activates the phosphatase that dephosphorylates myosin regulatory light chains. One recurring gain-of-function variant in the PRKG1 gene results in the constitutive activation and TAA. | Familial thoracic aortic aneurysm 8 (615436) |
| FLNA [221,222] | X-linked | Filamin-A | Filamin-A is an actin-binding protein that links membrane glycoproteins, including integrins, to actin filaments. It also serves as a scaffold and integrator for a wide range of cytoplasmic signaling proteins [223]. Loss-of-function mutations are associated with a broad range of congenital malformations and with increased risk of TAA [224]. | Periventricular nodular heterotopia type 1 (300049) |
| ARIH1 [225] | AD | E3 ubiquitin–protein ligase ARIH1 (Ari-1) | The Ari-1 protein is an E3-ubiquitin ligase that controls the degradation of SUN2, which is a component of the LINC (Linker of Nucleoskeleton and Cytoskeleton) complex. The LINC complex is involved in the coupling of mechanical signals to nuclear regulation, including chromatin and transcriptional regulation [111]. The ARI-1 variants associated with aneurysm are predicted to interfere with LINC complex function, although the mechanism remains unclear. | |
| Unclear function or mechanism | ||||
| Gene | Inheritance | Protein name | Primary effect of TAA-causing variants | Disease (phenotypeMIM number) |
| SLC2A10 [226] | AR | Solute carrier family 2, facilitated glucose transporter member 10 (GLUT10) | GLUT10 is a member of the class III facilitative glucose transporter family. Loss-of-function mutations in this transporter have been associated with aneurysms of large and medium-sized arteries. The pathogenic mechanisms remain unclear, although effects on mitochondrial function, TGF-β signaling, and synthesis of ECM glycoproteins have been proposed [226,227,228]. | Arterial tortuosity syndrome (208050) |
| HCN4 [229,230] | AD | Potassium/sodium hyperpolarization-activated cyclic nucleotide-gated channel 4 (HCN4) | HCN4 is a member of the hyperpolarization-activated cyclic nucleotide-gated potassium channels. Loss-of-function mutations in this gene have been linked to sick sinus syndrome 2, which is characterized by atrial fibrillation with bradyarrhythmia as well as increased risk of aneurysm. The pathogenic mechanism remains unclear. | Sick Sinus Syndrome 2 (163800) |
| MAT2A [231] | AD | S-adenosylmethionine synthase isoform type-2 (METK2), also known as methionine adenosyltransferase II α (MAT-IIα) | MAT-IIα catalyzes synthesis of S-adenosylmethionine from methionine and ATP. Mutations that predispose individuals to TAA are predicted to reduce or disrupt the activity of the enzyme. It remains unclear how reduced levels of S-adenosylmethionine cause disease. | |
| FOXE3 [232] | AD | Forkhead box protein E3 (FOXE3) | FOXE3 is a transcription factor of the forkhead family of transcription factors. Mutations associated with TAA are predicted to disrupt the forkhead domain and result in defective transcriptional regulation. | Aortic aneurysm, familial thoracic 11 (617349) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Creamer, T.J.; Bramel, E.E.; MacFarlane, E.G. Insights on the Pathogenesis of Aneurysm through the Study of Hereditary Aortopathies. Genes 2021, 12, 183. https://doi.org/10.3390/genes12020183
Creamer TJ, Bramel EE, MacFarlane EG. Insights on the Pathogenesis of Aneurysm through the Study of Hereditary Aortopathies. Genes. 2021; 12(2):183. https://doi.org/10.3390/genes12020183
Chicago/Turabian StyleCreamer, Tyler J., Emily E. Bramel, and Elena Gallo MacFarlane. 2021. "Insights on the Pathogenesis of Aneurysm through the Study of Hereditary Aortopathies" Genes 12, no. 2: 183. https://doi.org/10.3390/genes12020183
APA StyleCreamer, T. J., Bramel, E. E., & MacFarlane, E. G. (2021). Insights on the Pathogenesis of Aneurysm through the Study of Hereditary Aortopathies. Genes, 12(2), 183. https://doi.org/10.3390/genes12020183