
Clonal Interference and Mutation Bias in Small Bacterial Populations in Droplets


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Strains and Medium
2.2. Experimental Evolution
2.3. Whole Genome Sequencing and Analysis
2.4. Inferring Independent Mutational Events
2.5. Minimum Inhibitory Concentration
2.6. Growth Rate Measurements
2.7. Statistical Analysis
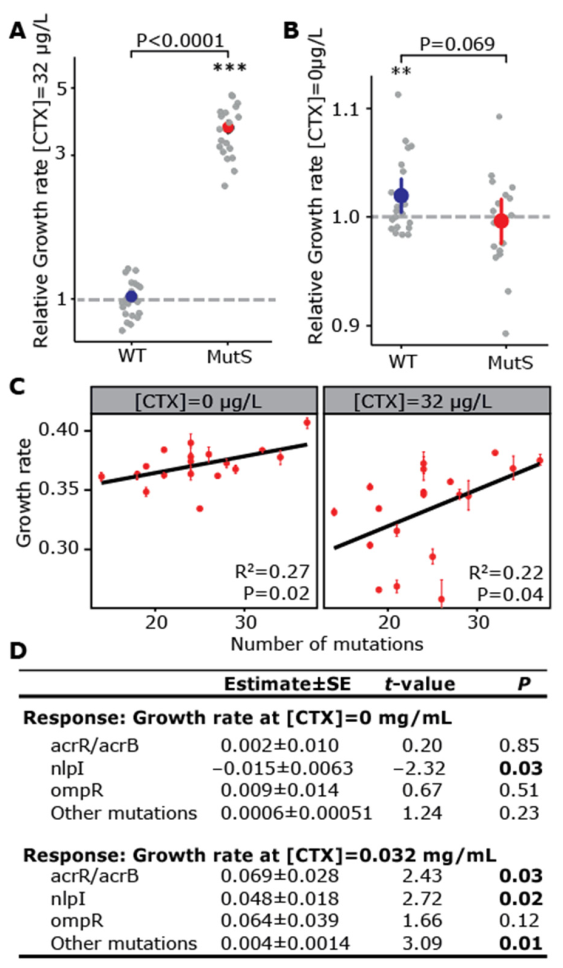
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Burrell, R.A.; McGranahan, N.; Bartek, J.; Swanton, C. The causes and consequences of genetic heterogeneity in cancer evolution. Nat. Cell Biol. 2013, 501, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.; Davies, D. Origins and Evolution of Antibiotic Resistance. Microbiol. Mol. Biol. Rev. 2010, 74, 417–433. [Google Scholar] [CrossRef] [Green Version]
- Blount, Z.D.; Lenski, R.E.; Losos, J.B. Contingency and determinism in evolution: Replaying life’s tape. Science 2018, 362, eaam5979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Visser, J.A.G.; Krug, J. Empirical fitness landscapes and the predictability of evolution. Nat. Rev. Genet. 2014, 15, 480–490. [Google Scholar] [CrossRef]
- Bailey, S.F.; Blanquart, F.; Bataillon, T.; Kassen, R. What drives parallel evolution? BioEssays 2017, 39, e201600176. [Google Scholar] [CrossRef]
- Meyer, J.R.; Dobias, D.T.; Weitz, J.S.; Barrick, J.E.; Quick, R.T.; Lenski, R.E. Repeatability and Contingency in the Evolution of a Key Innovation in Phage Lambda. Science 2012, 335, 428–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lachapelle, J.; Reid, J.; Colegrave, N. Repeatability of adaptation in experimental populations of different sizes. Proc. R. Soc. B Boil. Sci. 2015, 282, 20143033. [Google Scholar] [CrossRef] [Green Version]
- Szendro, I.G.; Franke, J.; De Visser, J.A.G.M.; Krug, J. Predictability of evolution depends nonmonotonically on population size. Proc. Natl. Acad. Sci. USA 2012, 110, 571–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez, K.; Bertram, J.; Masel, J. Mutation Bias Can Shape Adaptation in Large Asexual Populations Experiencing Clonal Interference; Cold Spring Harbor Laboratory: Cold Spring Harbor, NY, USA, 2020. [Google Scholar]
- Schenk, M.F.; Zwart, M.P.; Hwang, S.; Ruelens, P.; Severing, E.; Krug, J.; De Visser, J.A.G.M. Population size mediates the contribution of high-rate and large-benefit mutations to parallel evolution. bioRxiv 2021, 429281. [Google Scholar]
- Tenaillon, O.; Barrick, J.E.; Ribeck, N.; Deatherage, D.E.; Blanchard, J.L.; Dasgupta, A.; Wu, G.C.; Wielgoss, S.; Cruveiller, S.; Médigue, C.; et al. Tempo and mode of genome evolution in a 50,000-generation experiment. Nat. Cell Biol. 2016, 536, 165–170. [Google Scholar] [CrossRef] [Green Version]
- Tenaillon, O.; Rodríguez-Verdugo, A.; Gaut, R.L.; McDonald, P.; Bennett, A.F.; Long, A.D.; Gaut, B.S. The Molecular Diversity of Adaptive Convergence. Science 2012, 335, 457–461. [Google Scholar] [CrossRef] [Green Version]
- Bergh, B.V.D.; Swings, T.; Fauvart, M.; Michiels, J. Experimental Design, Population Dynamics, and Diversity in Microbial Experimental Evolution. Microbiol. Mol. Biol. Rev. 2018, 82. [Google Scholar] [CrossRef] [Green Version]
- Kawecki, T.J.; Lenski, R.E.; Ebert, D.; Hollis, B.; Olivieri, I.; Whitlock, M.C. Experimental evolution. Trends Ecol. Evol. 2012, 27, 547–560. [Google Scholar] [CrossRef] [Green Version]
- Abel, S.; Wiesch, P.A.Z.; Davis, B.M.; Waldor, M.K. Analysis of Bottlenecks in Experimental Models of Infection. PLoS Pathog. 2015, 11, e1004823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Handel, A.; Bennett, M.R. Surviving the Bottleneck: Transmission Mutants and the Evolution of Microbial Populations. Genetics 2008, 180, 2193–2200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garoff, L.; Pietsch, F.; Huseby, D.L.; Lilja, T.; Brandis, G.; Hughes, D. Population Bottlenecks Strongly Influence the Evolutionary Trajectory to Fluoroquinolone Resistance in Escherichia coli. Mol. Biol. Evol. 2020, 37, 1637–1646. [Google Scholar] [CrossRef]
- Wein, T.; Dagan, T. The Effect of Population Bottleneck Size and Selective Regime on Genetic Diversity and Evolvability in Bacteria. Genome Biol. Evol. 2019, 11, 3283–3290. [Google Scholar] [CrossRef]
- Wahl, L.M.; Gerrish, P.J.; Saika-Voivod, I. Evaluating the impact of population bottlenecks in experimental evolution. Genetics 2002, 162, 961–971. [Google Scholar]
- Boitard, L.; Cottinet, D.; Bremond, N.; Baudry, J.; Bibette, J. Growing microbes in millifluidic droplets. Eng. Life Sci. 2015, 15, 318–326. [Google Scholar] [CrossRef]
- Siegel, E.C.; Wain, S.L.; Meltzer, S.F.; Binion, M.L.; Steinberg, J.L. Mutator mutations in Escherichia coli induced by the insertionof phage Mu and the transposable resistance elements Tn5 and Tn10. Mutat. Res. Mol. Mech. Mutagen. 1982, 93, 25–33. [Google Scholar] [CrossRef]
- De Visser, J.A.G.M.; Zeyl, C.W.; Gerrish, P.J.; Blanchard, J.L.; Lenski, R.E. Diminishing Returns from Mutation Supply Rate in Asexual Populations. Science 1999, 283, 404–406. [Google Scholar]
- Deatherage, D.E.; Barrick, J.E. Identification of mutations in laboratory-evolved microbes from next-generation se-quencing data using breseq. In Engineering and Analyzing Multicellular Systems; Springer: Berlin/Heidelberg, Germany, 2014; pp. 165–188. [Google Scholar]
- Schliep, K.P. phangorn: Phylogenetic analysis in R. Bioinformatics 2010, 27, 592–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Foundation for Statistical Computing. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013. [Google Scholar]
- Hartig, F. DHARMa: Residual Diagnostics for Hierarchical (Multi-Level/Mixed) Regression Models. R package Version 0.1. 2017. Available online: http://cran.nexr.com/web/packages/DHARMa/vignettes/DHARMa.html (accessed on 29 January 2021).
- Pos, K.M. Drug transport mechanism of the AcrB efflux pump. Biochim. Biophys. Acta Proteins Proteom. 2009, 1794, 782–793. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Gu, R.; Su, C.-C.; Routh, M.D.; Harris, K.C.; Jewell, E.S.; McDermott, G.; Yu, E.W. Crystal Structure of the Transcriptional Regulator AcrR from Escherichia coli. J. Mol. Biol. 2007, 374, 591–603. [Google Scholar] [CrossRef]
- Schwechheimer, C.; Rodriguez, D.L.; Kuehn, M.J. NlpI-mediated modulation of outer membrane vesicle production through peptidoglycan dynamics in Escherichia coli. MicrobiologyOpen 2015, 4, 375–389. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.W.; Park, S.B.; Im, S.P.; Lee, J.S.; Jung, J.W.; Gong, T.W.; Lazarte, J.M.S.; Kim, J.; Seo, J.-S.; Kim, J.-H.; et al. Outer membrane vesicles from beta-lactam-resistant Escherichia coli enable the survival of be-ta-lactam-susceptible E. coli in the presence of beta-lactam antibiotics. Sci. Rep. 2018, 8, 5402. [Google Scholar] [CrossRef] [Green Version]
- Schwechheimer, C.; Kuehn, M.J. Outer-membrane vesicles from Gram-negative bacteria: Biogenesis and functions. Nat. Rev. Genet. 2015, 13, 605–619. [Google Scholar] [CrossRef] [Green Version]
- Shimada, T.; Takada, H.; Yamamoto, K.; Ishihama, A. Expanded roles of two-component response regulator OmpR inEscherichia coli: Genomic SELEX search for novel regulation targets. Genes Cells 2015, 20, 915–931. [Google Scholar] [CrossRef]
- Seo, S.; Gao, Y.; Kim, D.; Szubin, R.; Yang, J.; Cho, B.-K.; Palsson, B.O. Revealing genome-scale transcriptional regulatory landscape of OmpR highlights its expanded regulatory roles under osmotic stress in Escherichia coli K-12 MG1655. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Woods, R.; Schneider, D.; Winkworth, C.L.; Riley, M.A.; Lenski, R.E. Tests of parallel molecular evolution in a long-term experiment with Escherichia coli. Proc. Natl. Acad. Sci. USA 2006, 103, 9107–9112. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruelens, P.; de Visser, J.A.G.M. Clonal Interference and Mutation Bias in Small Bacterial Populations in Droplets. Genes 2021, 12, 223. https://doi.org/10.3390/genes12020223
Ruelens P, de Visser JAGM. Clonal Interference and Mutation Bias in Small Bacterial Populations in Droplets. Genes. 2021; 12(2):223. https://doi.org/10.3390/genes12020223
Chicago/Turabian StyleRuelens, Philip, and J. Arjan G. M. de Visser. 2021. "Clonal Interference and Mutation Bias in Small Bacterial Populations in Droplets" Genes 12, no. 2: 223. https://doi.org/10.3390/genes12020223
APA StyleRuelens, P., & de Visser, J. A. G. M. (2021). Clonal Interference and Mutation Bias in Small Bacterial Populations in Droplets. Genes, 12(2), 223. https://doi.org/10.3390/genes12020223