
Comparative Genomic Analysis of Lactiplantibacillus plantarum Isolated from Different Niches

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. L. plantarum Strains
2.3. Draft Genome Sequencing of L. plantarum
2.4. Genomic Assembly, Prediction, and Functional Annotation
2.5. Mapping of Homologous Gene Venn Diagram and Phylogenetic Tree Construction
2.6. Average Nucleotide Identity (ANI) Analysis
2.7. Carbohydrate-Active Enzyme Analysis
2.8. Determination of the Carbohydrate Utilization Capacity of L. plantarum
3. Results
3.1. Genome Characteristics of 133 L. plantarum Strains
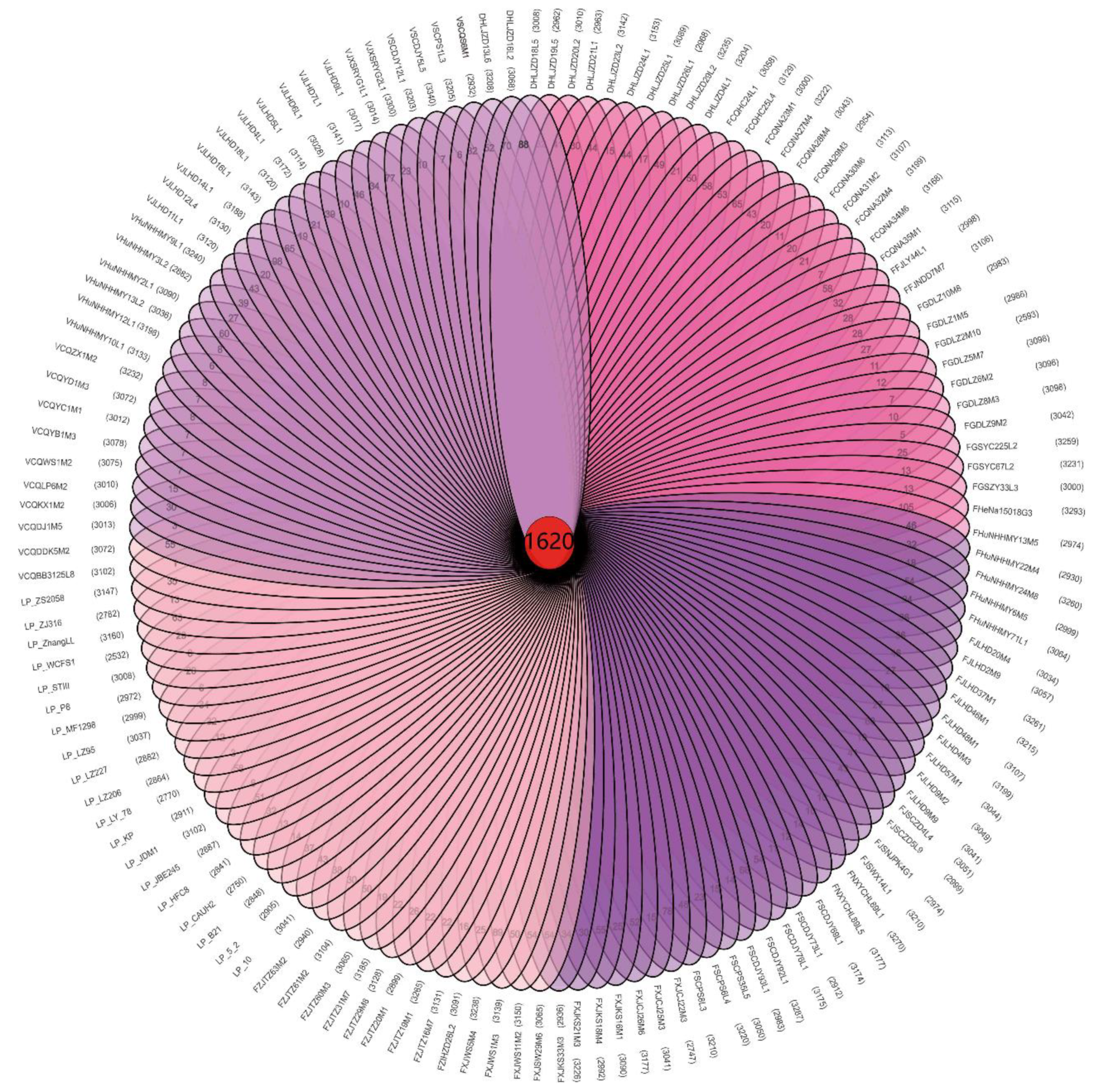
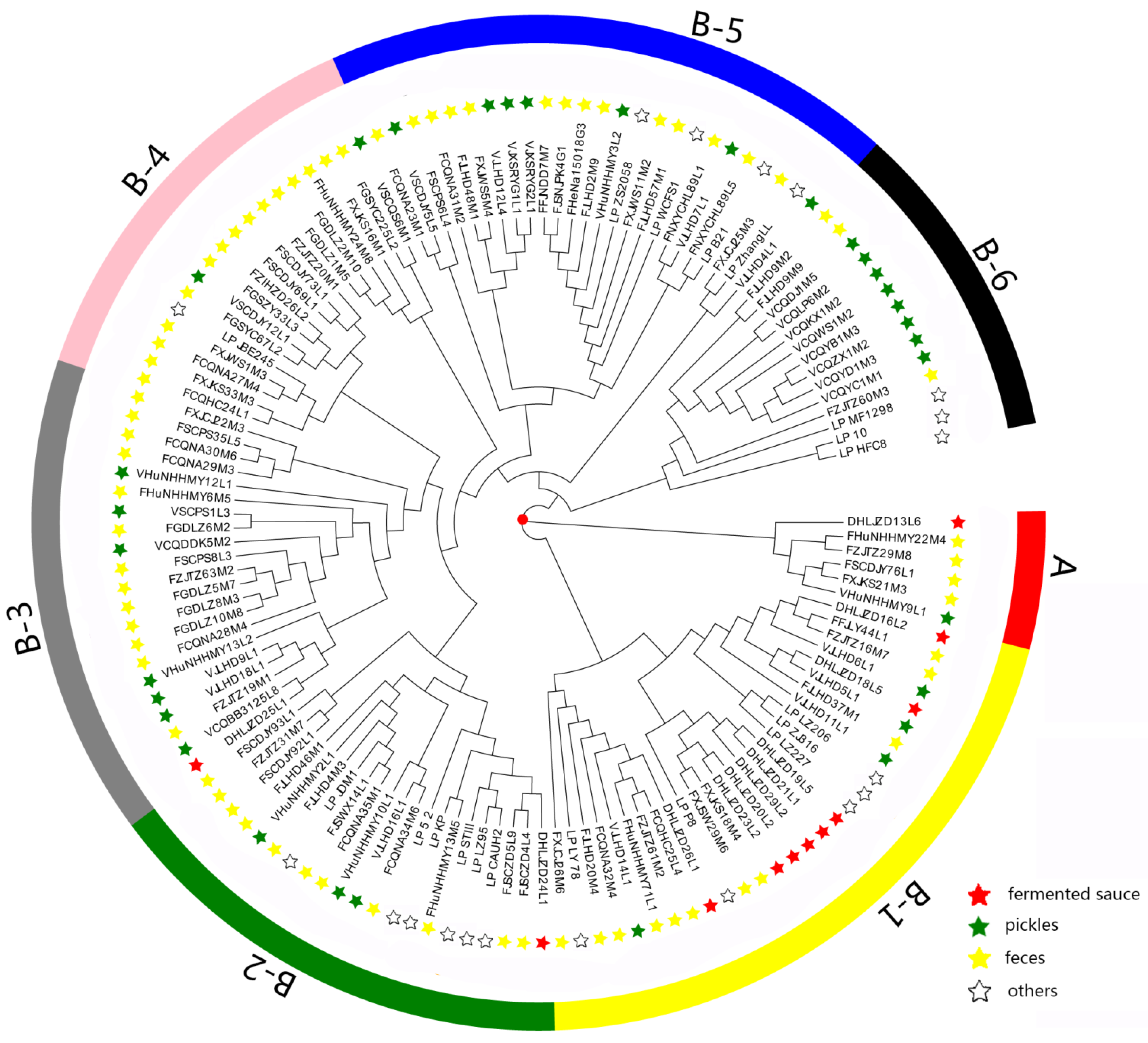
3.2. Analysis of the Phylogenetic Tree with Regard to Homologous Genes
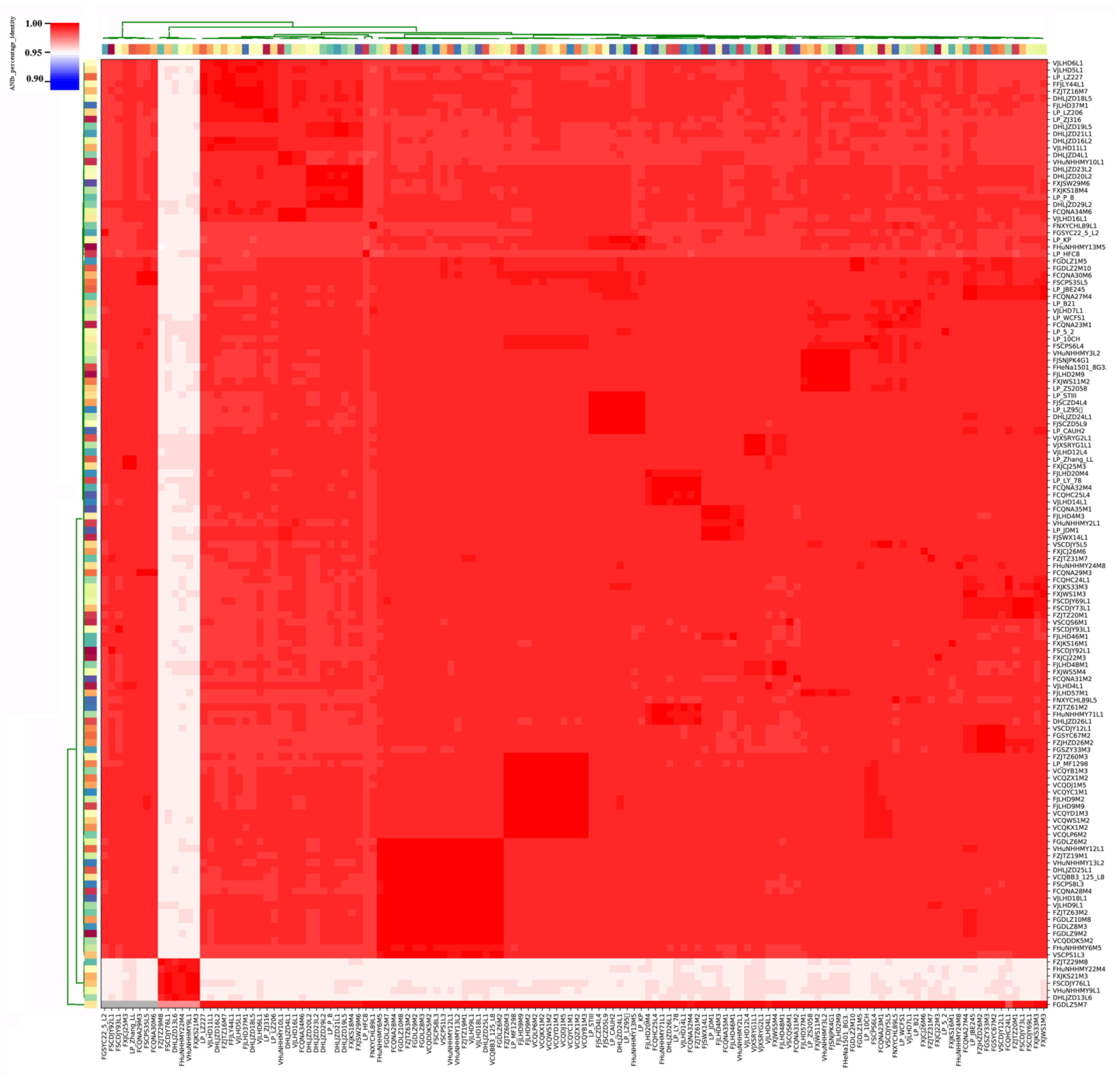
3.3. Average Nucleotide Identity (ANI) Analysis
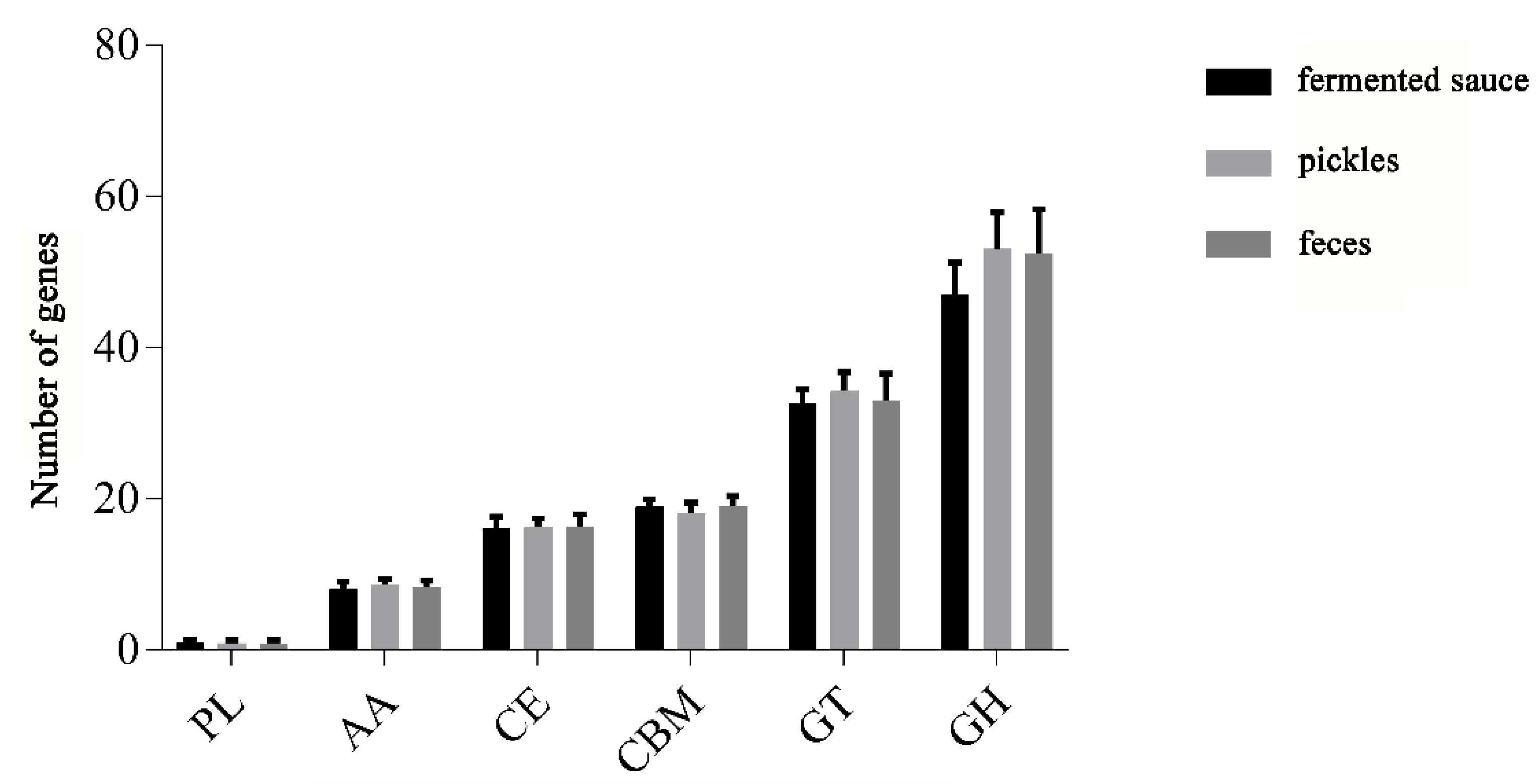
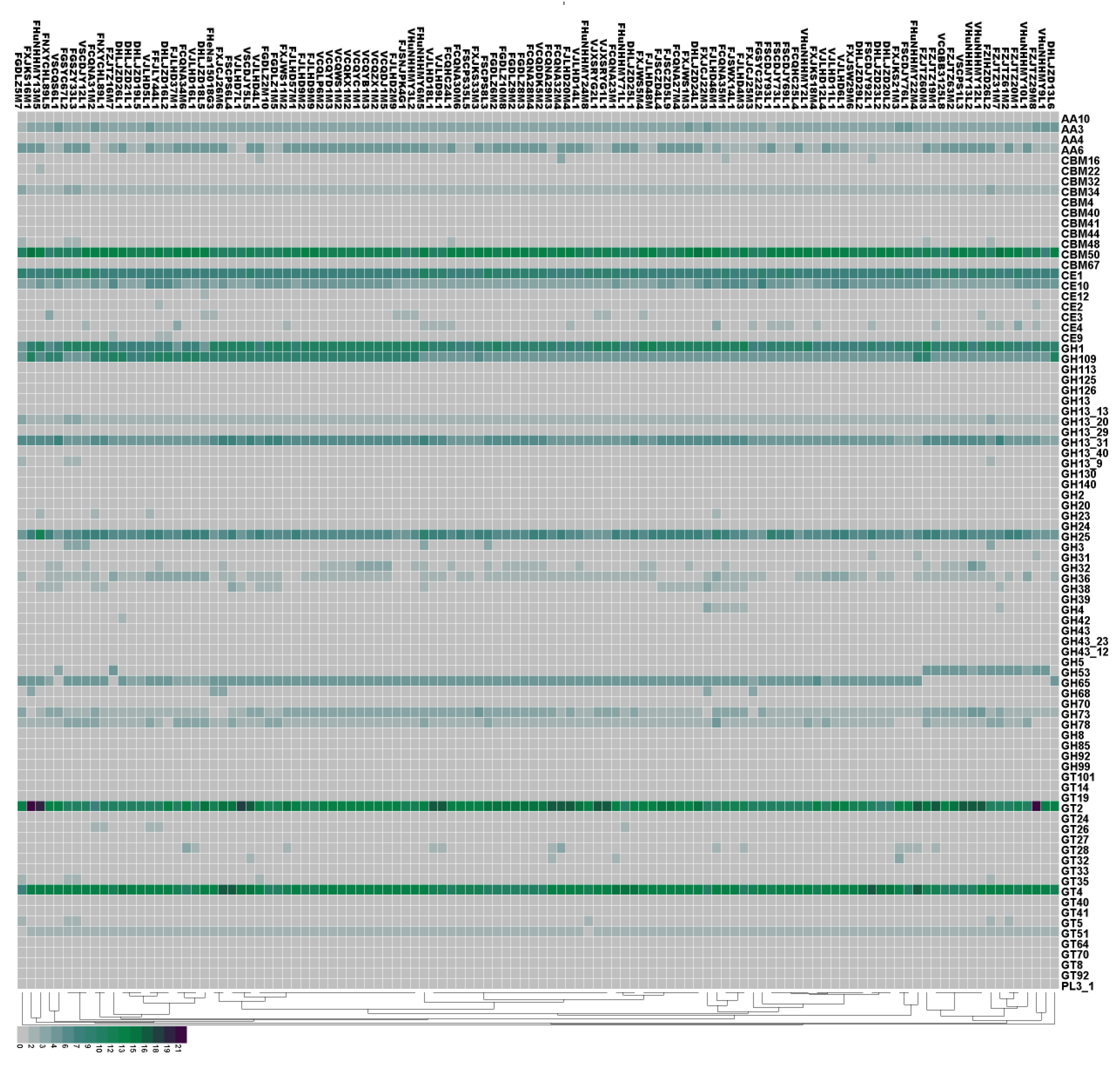
3.4. Analysis of Carbohydrate-Active Enzymes
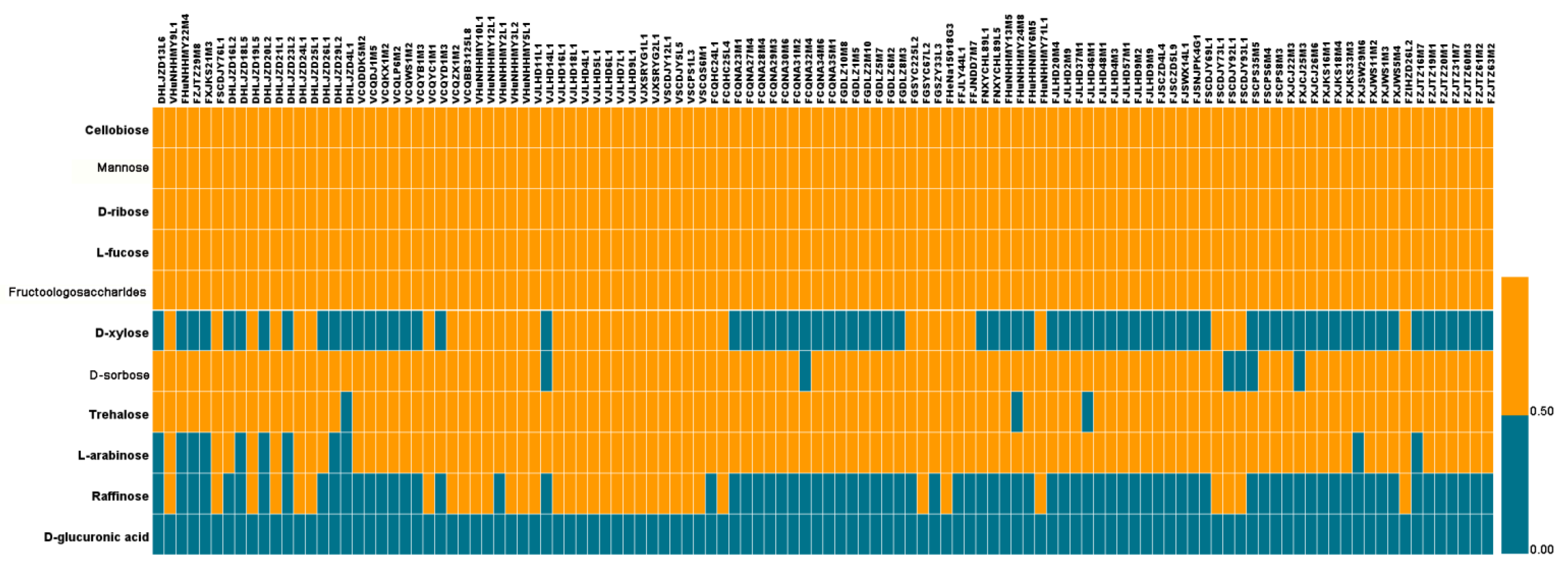
3.5. Analysis of Carbohydrate Use Ability
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Grangette, C.; Nutten, S.; Palumbo, E.; Morath, S.; Hermann, C.; Dewulf, J.; Pot, B.; Hartung, T.; Hols, P.; Mercenier, A. Enhanced anti-inflammatory capacity of a Lactobacillus plantarum mutant synthesizing modified teichoic acids. Proc. Natl. Acad. Sci. USA 2005, 102, 10321–10326. [Google Scholar] [CrossRef] [Green Version]
- Broekaert, I.J.; Walker, W.A. Probiotics and chronic disease. J. Clin. Gastroenterol. 2006, 40, 270–274. [Google Scholar] [CrossRef]
- Mayo, B.; Gonzalez, B.; Arca, P.; Suarez, J.E. Cloning and expression of the plasmid-encoded β-d-galactosidase gene from a Lactobacillus-plantarum strain of dairy origin. FEMS Microbiol. Lett. 1994, 122, 145–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kongnum, K.; Hongpattarakere, T. Effect of Lactobacillus plantarum isolated from digestive tract of wild shrimp on growth and survival of white shrimp (Litopenaeus vannamei) challenged with Vibrio harveyi. Fish Shellfish. Immun. 2012, 32, 170–177. [Google Scholar] [CrossRef]
- de Vries, M.C.; Vaughan, E.E.; Kleerebezem, M.; de Vos, W.M. Lactobacillus plantarum- survival, functional and potential probiotic properties in the human intestinal tract. Int. Dairy J. 2006, 16, 1018–1028. [Google Scholar] [CrossRef]
- Bolotin, A.; Wincker, P.; Mauger, S.; Jaillon, O.; Malarme, K.; Weissenbach, J.; Ehrlich, S.D.; Sorokin, A. The complete genome sequence of the lactic acid bacterium Lactococcus lactis ssp lactis IL1403. Genome Res. 2001, 11, 731–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siezen, R.J.; Francke, C.; Renckens, B.; Boekhorst, J.; Wels, M.; Kleerebezem, M.; van Hijum, S.A.F.T. Complete Resequencing and Reannotation of the Lactobacillus plantarum WCFS1 Genome. J. Bacteriol. 2012, 194, 195–196. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.J.; Wang, R.; Gong, F.M.; Liu, X.F.; Zheng, H.J.; Luo, Y.Y.; Li, X.R. Complete genome sequences and comparative genome analysis of Lactobacillus plantarum strain 5-2 isolated from fermented soybean. Genomics 2015, 106, 404–411. [Google Scholar] [CrossRef] [PubMed]
- Axelsson, L.; Rud, I.; Naterstad, K.; Blom, H.; Renckens, B.; Boekhorst, J.; Kleerebezem, M.; van Hijum, S.; Siezen, R.J. Genome Sequence of the Naturally Plasmid-Free Lactobacillus plantarum Strain NC8 (CCUG 61730). J. Bacteriol. 2012, 194, 2391–2392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martino, M.E.; Bayjanov, J.R.; Caffrey, B.E.; Wels, M.; Joncour, P.; Hughes, S.; Gillet, B.; Kleerebezem, M.; van Hijum, S.A.F.T.; Leulier, F. Nomadic lifestyle of Lactobacillus plantarum revealed by comparative genomics of 54 strains isolated from different habitats. Environ. Microbiol. 2016, 18, 4974–4989. [Google Scholar] [CrossRef] [PubMed]
- Kwak, W.; Kim, K.; Lee, C.; Lee, C.; Kang, J.; Cho, K.; Yoon, S.H.; Kang, D.K.; Kim, H.; Heo, J.; et al. Comparative Analysis of the Complete Genome of Lactobacillus plantarum GB-LP2 and Potential Candidate Genes for Host Immune System Enhancement. J. Microbiol. Biotechnol. 2016, 26, 684–692. [Google Scholar] [CrossRef]
- Torriani, S.; Clementi, F.; Vancanneyt, M.; Hoste, B.; Dellaglio, F.; Kersters, K. Differentiation of Lactobacillus plantarum, L. pentosus and L-paraplantarum species by RAPD-PCR and AFLP. Syst. Appl. Microbiol. 2001, 24, 554–560. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.L.; Quednau, M.; Molin, G.; Ahrne, S. Randomly amplified polymorphic DNA (rapd) for rapid typing of lactobacillus-plantarum strains. Lett. Appl. Microbiol. 1995, 21, 155–159. [Google Scholar] [CrossRef] [PubMed]
- de las Rivas, B.; Marcobal, A.; Munoz, R. Development of a multilocus sequence typing method for analysis of Lactobacillus plantarum strains. Microbiol. Sgm. 2006, 152, 85–93. [Google Scholar] [CrossRef] [Green Version]
- Molenaar, D.; Bringel, F.; Schuren, F.H.; de Vos, W.M.; Siezen, R.J.; Kleerebezem, M. Exploring Lactobacillus plantarum genome diversity by using microarrays. J. Bacteriol. 2005, 187, 6119–6127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siezen, R.J.; Vlieg, J.E.T.V. Genomic diversity and versatility of Lactobacillus plantarum, a natural metabolic engineer. Microb. Cell Factories 2011, 10, S3. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.; Jin, G.D.; Park, J.; You, I.; Kim, E.B. Pan-Genomics of Lactobacillus plantarum Revealed Group-Specific Genomic Profiles without Habitat Association. J. Microbiol. Biotechnol. 2018, 28, 1352–1359. [Google Scholar] [CrossRef]
- Siezen, R.J.; Tzeneva, V.A.; Castioni, A.; Wels, M.; Phan, H.T.K.; Rademaker, J.L.W.; Starrenburg, M.J.C.; Kleerebezem, M.; Molenaar, D.; Vlieg, J.E.T.V. Phenotypic and genomic diversity of Lactobacillus plantarum strains isolated from various environmental niches. Environ. Microbiol. 2010, 12, 758–773. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.Z.; Chen, Y.X.; Pan, Q.; Liu, Y.J.; et al. Erratum: SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. GigaScience 2015, 4, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.Z.; Chen, Y.X.; Pan, Q.; Liu, Y.J.; et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. Gigascience 2012, 1, 18. [Google Scholar] [CrossRef] [PubMed]
- Borodovsky, M.; Mills, R.; Besemer, J.; Lomsadze, A. Prokaryotic Gene Prediction Using GeneMark and GeneMark.hmm. Curr. Protoc. Bioinform. 2003, 1, 4.5.1–4.5.16. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wu, J.; Yang, J.; Sun, S.; Xiao, J.; Yu, J. PGAP: Pan-genomes analysis pipeline. Bioinformatics 2012, 28, 416–418. [Google Scholar] [CrossRef] [Green Version]
- Skarp-de Haan, C.P.A.; Culebro, A.; Schott, T.; Revez, J.; Schweda, E.K.H.; Hanninen, M.L.; Rossi, M. Comparative genomics of unintrogressed Campylobacter coli clades 2 and 3. BMC Genom. 2014, 15, 129. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [Green Version]
- Wickstead, B.; Gull, K.; Richards, T.A. Patterns of kinesin evolution reveal a complex ancestral eukaryote with a multifunctional cytoskeleton. BMC Evol. Biol. 2010, 10, 110. [Google Scholar] [CrossRef] [Green Version]
- Richter, M.; Rossello-Mora, R. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. USA 2009, 106, 19126–19131. [Google Scholar] [CrossRef] [Green Version]
- Yoon, S.H.; Ha, S.M.; Lim, J.; Kwon, S.; Chun, J. A large-scale evaluation of algorithms to calculate average nucleotide identity. Antonie Leeuw Int. J. G. 2017, 110, 1281–1286. [Google Scholar] [CrossRef]
- Mistry, J.; Finn, R.D.; Eddy, S.R.; Bateman, A.; Punta, M. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 2013, 41, e121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, Y.; Mao, X.; Yang, J.; Chen, X.; Mao, F.; Xu, Y. dbCAN: A web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2012, 40, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Lombard, V.; Ramulu, H.G.; Drula, E.; Coutinho, P.M.; Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014, 42, D490–D495. [Google Scholar] [CrossRef] [Green Version]
- Rodin, S.N.; Parkhomchuk, D.V. Position-associated GC asymmetry of gene duplicates. J. Mol. Evol. 2004, 59, 372–384. [Google Scholar] [CrossRef]
- Jorgensen, R. Altered gene expression in plants due to trans interactions between homologous genes. Trends Biotechnol. 1990, 8, 340–344. [Google Scholar] [CrossRef]
- Kant, R.; Rintahaka, J.; Yu, X.; Sigvart-Mattila, P.; Paulin, L.; Mecklin, J.P.; Saarela, M.; Palva, A.; von Ossowski, I. A Comparative Pan-Genome Perspective of Niche-Adaptable Cell-Surface Protein Phenotypes in Lactobacillus rhamnosus. PLoS ONE 2014, 9, e102762. [Google Scholar] [CrossRef]
- Zhang, S.; Song, W.; Yu, M.; Lin, X. Comparative genomics analysis of five Psychrobacter strains isolated from world-wide habitats reveal high intra-genus variations. Extremophiles 2017, 21, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Zhang, C.; Yang, Q.; Guo, Z.; Yang, B.; Lu, W.; Li, D.Y.; Tian, F.W.; Liu, X.M.; Zhang, H.; et al. Selection of Taste Markers Related to Lactic Acid Bacteria Microflora Metabolism for Chinese Traditional Paocai: A Gas Chromatography-Mass Spectrometry -Based Metabolomics Approach. J. Agric. Food Chem. 2016, 64, 2415–2422. [Google Scholar] [CrossRef]
- Ciufo, S.; Kannan, S.; Sharma, S.; Badretdin, A.; Clark, K.; Turner, S.; Brover, S.; Schoch, C.L.; Kimchi, A.; DiCuccio, M. Using average nucleotide identity to improve taxonomic assignments in prokaryotic genomes at the NCBI. Int. J. Syst. Evol. Microbiol. 2018, 68, 2386–2392. [Google Scholar] [CrossRef]
- Duan, C.J.; Xian, L.; Zhao, G.C.; Feng, Y.; Pang, H.; Bai, X.L.; Tang, J.L.; Ma, Q.S.; Feng, J.X. Isolation and partial characterization of novel genes encoding acidic cellulases from metagenomes of buffalo rumens. J. Appl. Microbiol. 2009, 107, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Zdunczyk, Z.; Jankowski, J.; Juskiewicz, J.; Lecewicz, A.; Slominski, B. Application of soybean meal, soy protein concentrate and isolate differing in α-galactosides content to low- and high-fibre diets in growing turkeys. J. Anim. Physiol. Anim. Nutr. 2010, 94, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Douillard, F.P.; Ribbera, A.; Kant, R.; Pietila, T.E.; Jarvinen, H.M.; Messing, M.; Randazzo, C.L.; Paulin, L.; Laine, P.; Ritari, J.; et al. Comparative Genomic and Functional Analysis of 100 Lactobacillus rhamnosus Strains and Their Comparison with Strain GG. PLoS Genet. 2013, 9, e1003683. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Han, G.G.; Kim, E.B.; Choi, Y.J. Comparative genomics of Lactobacillus salivarius strains focusing on their host adaptation. Microbiol. Res. 2017, 205, 48–58. [Google Scholar] [CrossRef]
- Yu, J.; Zhao, J.; Song, Y.; Zhang, J.; Yu, Z.; Zhang, H.; Sun, Z.H. Comparative Genomics of the Herbivore Gut Symbiont Lactobacillus reuteri Reveals Genetic Diversity and Lifestyle Adaptation. Front. Microbiol. 2018, 9, 1151. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, C.; Ai, L.; Zhou, F.; Zhou, Z.; Wang, L.; Zhang, H.; Chen, W.; Guo, B.H. Complete Genome Sequence of the Probiotic Lactobacillus plantarum ST-III. J. Bacteriol. 2011, 193, 313–314. [Google Scholar] [CrossRef] [Green Version]
- Miller, K.A.; Phillips, R.S.; Mrazek, J.; Hoover, T.R. Salmonella Utilizes D-Glucosaminate via a Mannose Family Phosphotransferase System Permease and Associated Enzymes. J. Bacteriol. 2013, 195, 4057–4066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibson, G.R.; Hutkins, R.; Sanders, M.E.; Prescott, S.L.; Reimer, R.A.; Salminen, S.J.; Scott, K.; Stanton, C.; Swanson, K.S.; Cani, P.D.; et al. The International Scientific Association for Probiotics and Prebiotics (ISAPP) consensus statement on the definition and scope of prebiotics. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 491–502. [Google Scholar] [CrossRef] [Green Version]
- Jo, S.; Yoon, J.; Lee, S.M.; Um, Y.; Han, S.O.; Woo, H.M. Modular pathway engineering of Corynebacterium glutamicum to improve xylose utilization and succinate production. J. Biotechnol. 2017, 258, 69–78. [Google Scholar] [CrossRef]
- Jeffrey, G.A.; Huang, D.B. The hydrogen bonding in the crystal structure of raffinose pentahydrate. Carbohyd. Res. 1990, 206, 173–182. [Google Scholar] [CrossRef]
- Mao, B.; Li, D.; Zhao, J.; Liu, X.; Gu, Z.; Chen, Y.Q.; Zhang, H.; Chen, W. In Vitro Fermentation of Lactulose by Human Gut Bacteria. J. Agric. Food Chem. 2014, 62, 10970–10977. [Google Scholar] [CrossRef]
- Mao, B.; Li, D.; Zhao, J.; Liu, X.; Gu, Z.; Chen, Y.Q.; Zhang, H.; Chen, W. In vitro fermentation of fructooligosaccharides with human gut bacteria. Food Funct. 2015, 6, 947–954. [Google Scholar] [CrossRef]
- Mao, B.; Tang, H.; Gu, J.; Li, D.; Cui, S.; Zhao, J.; Zhang, H.; Chen, W. In vitro fermentation of raffinose by the human gut bacteria. Food Funct. 2018, 9, 5824–5831. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Source of Isolation | Number of Strains | Genomic Size (Mb) | Number of Genes | GC Content (%) |
|---|---|---|---|---|
| Fermented sauce | 11 | 3.14–3.45 | 3049~3416 | 45.43–46.33 |
| Pickles | 32 | 3.23–3.50 | 2931–3464 | 45.16–46.55 |
| Feces | 71 | 2.94–3.90 | 2767–3487 | 45.25–46.29 |
| Others * | 19 | 2.95–3.69 | 2860–3650 | 44.08–44.90 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mao, B.; Yin, R.; Li, X.; Cui, S.; Zhang, H.; Zhao, J.; Chen, W. Comparative Genomic Analysis of Lactiplantibacillus plantarum Isolated from Different Niches. Genes 2021, 12, 241. https://doi.org/10.3390/genes12020241
Mao B, Yin R, Li X, Cui S, Zhang H, Zhao J, Chen W. Comparative Genomic Analysis of Lactiplantibacillus plantarum Isolated from Different Niches. Genes. 2021; 12(2):241. https://doi.org/10.3390/genes12020241
Chicago/Turabian StyleMao, Bingyong, Ruimin Yin, Xiaoshu Li, Shumao Cui, Hao Zhang, Jianxin Zhao, and Wei Chen. 2021. "Comparative Genomic Analysis of Lactiplantibacillus plantarum Isolated from Different Niches" Genes 12, no. 2: 241. https://doi.org/10.3390/genes12020241
APA StyleMao, B., Yin, R., Li, X., Cui, S., Zhang, H., Zhao, J., & Chen, W. (2021). Comparative Genomic Analysis of Lactiplantibacillus plantarum Isolated from Different Niches. Genes, 12(2), 241. https://doi.org/10.3390/genes12020241