
Spectrum of MYO7A Mutations in an Indigenous South African Population Further Elucidates the Nonsyndromic Autosomal Recessive Phenotype of DFNB2 to Include Both Homozygous and Compound Heterozygous Mutations

 , and
, and
Abstract
:1. Introduction
1.1. Background Information
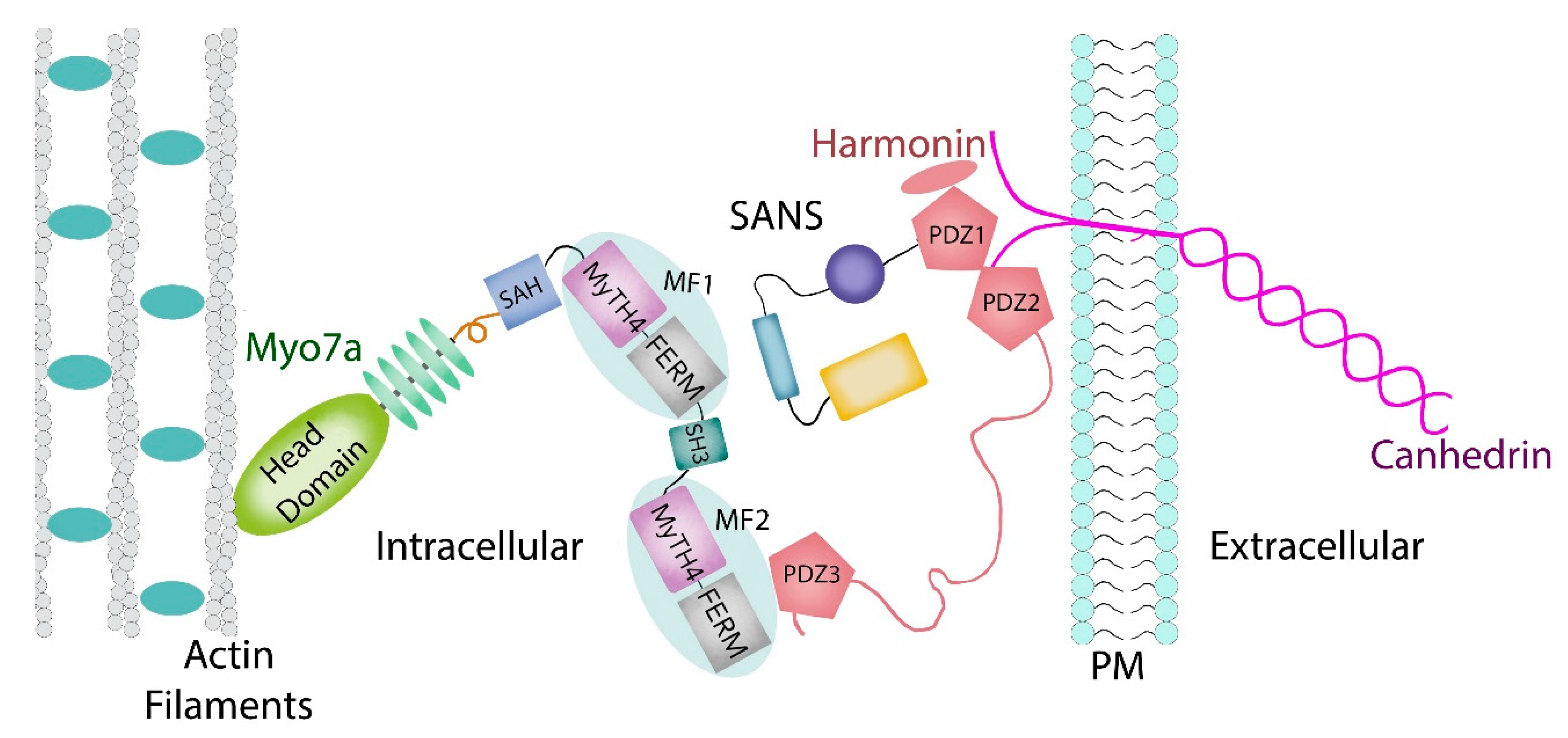
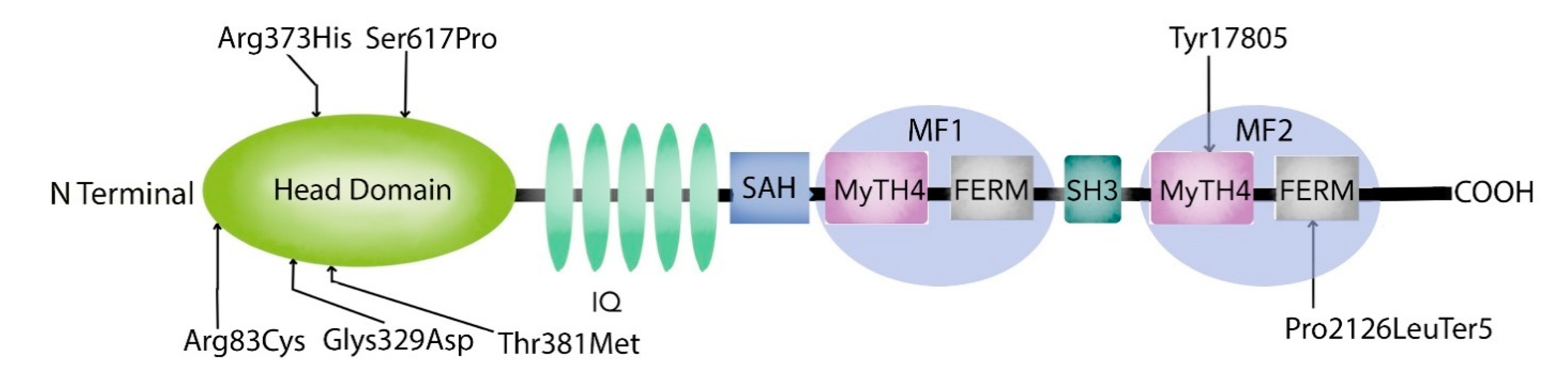
1.2. Myosin VIIA Heavy Chain Structure and Function
1.3. MYO7A Gene Expression and Phenotypes
2. Materials and Methods
2.1. Subjects
2.2. Clinical Evaluation
2.3. Sequencing
2.4. Bioinformatics Analysis
3. Results
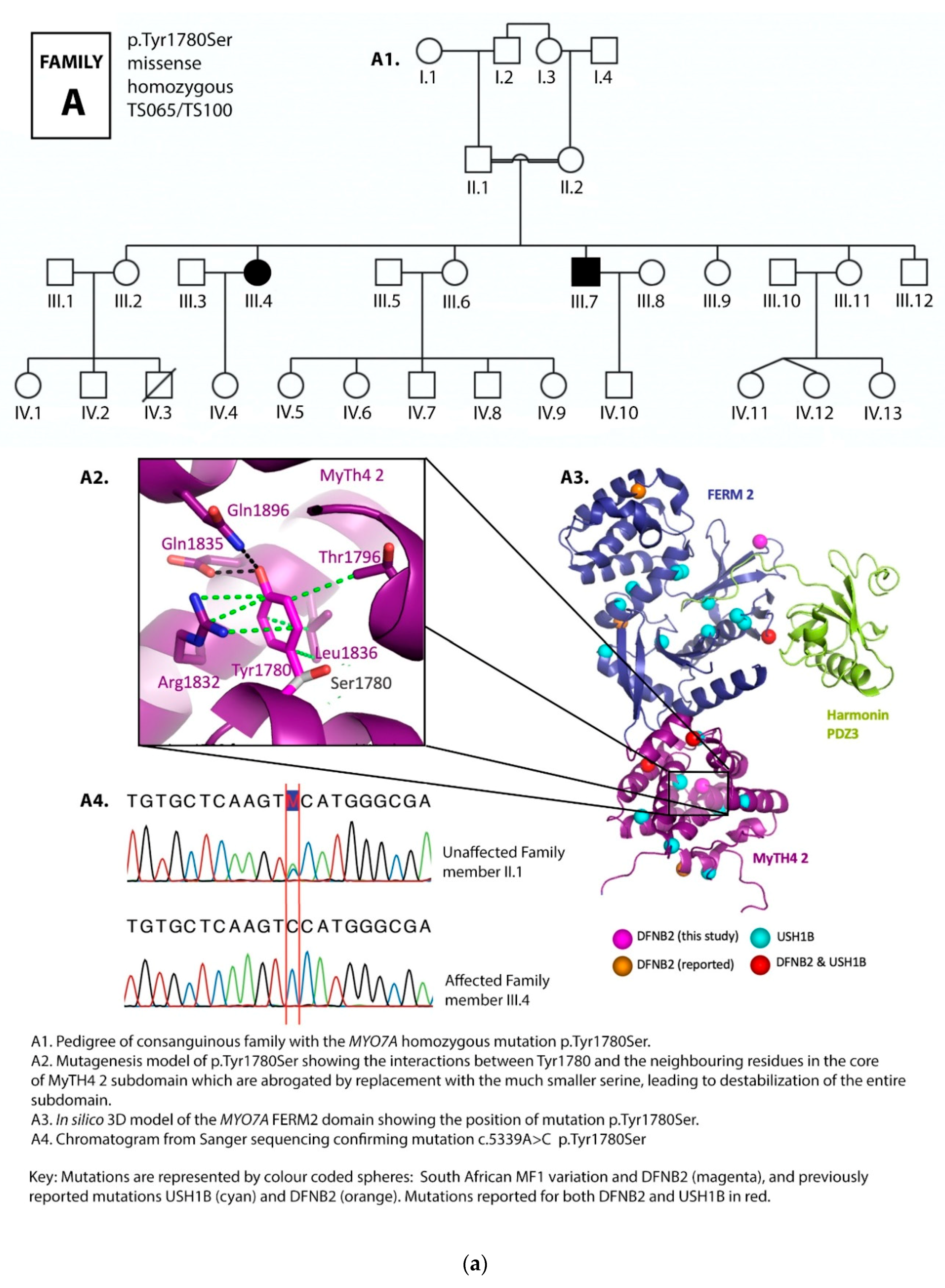
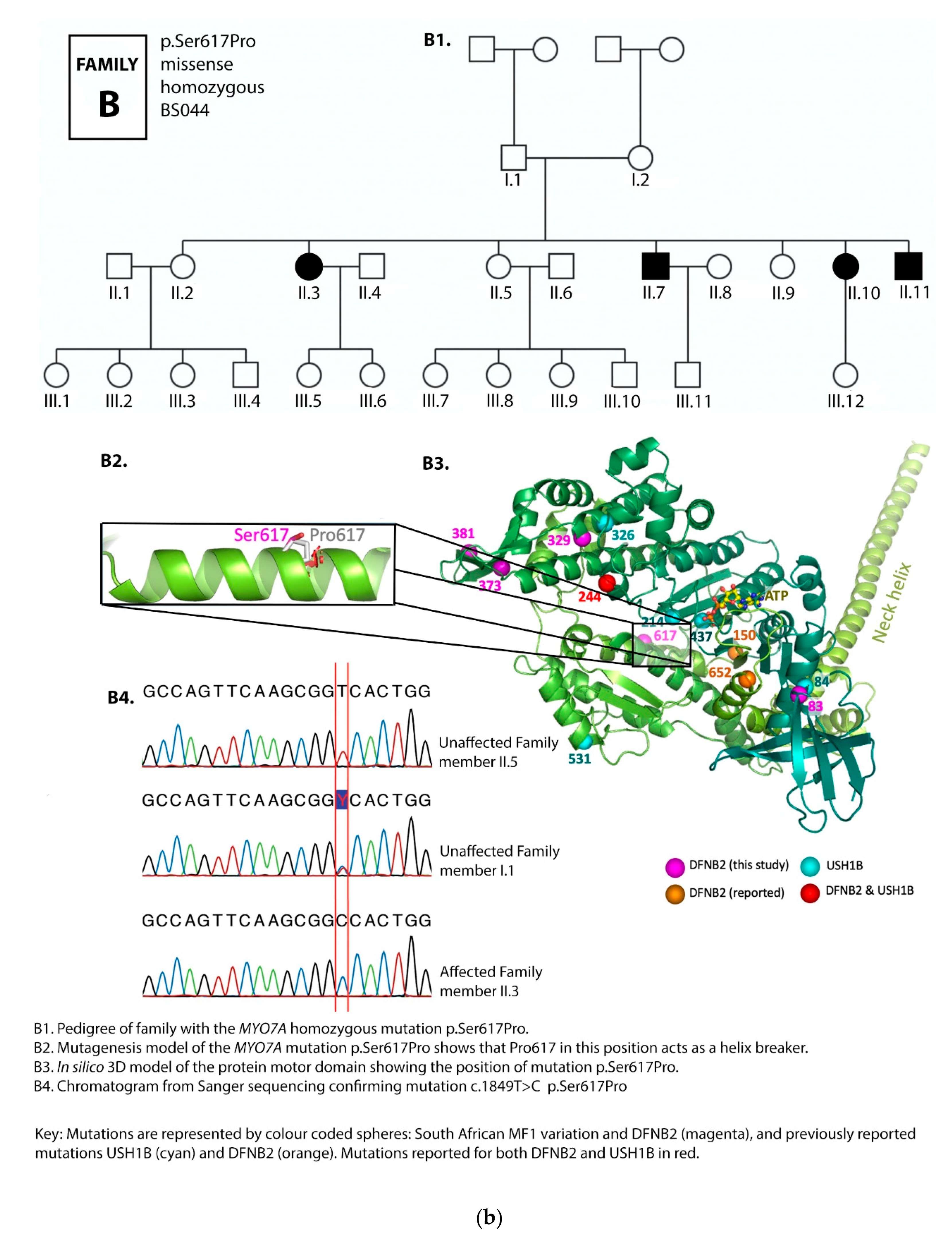
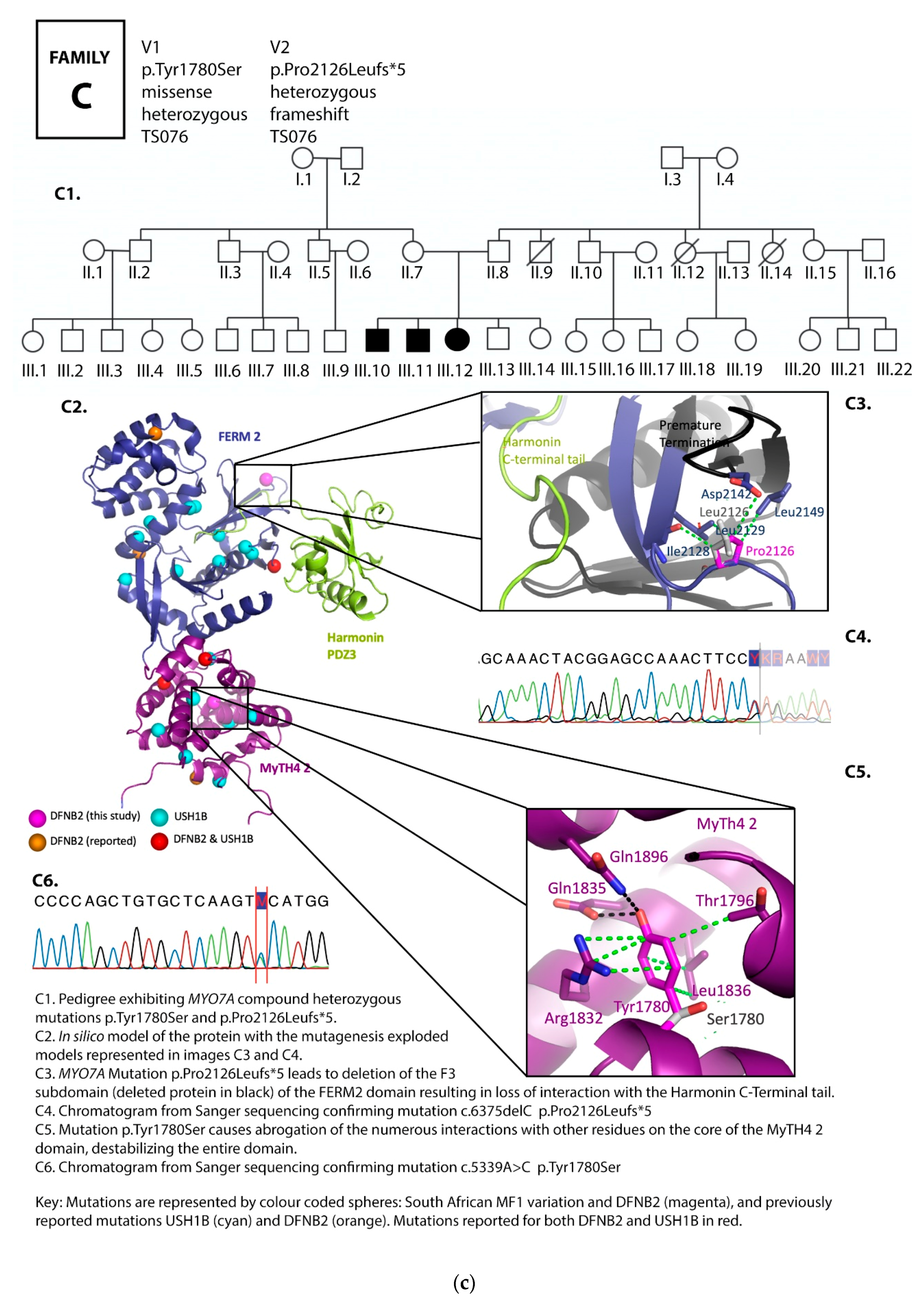
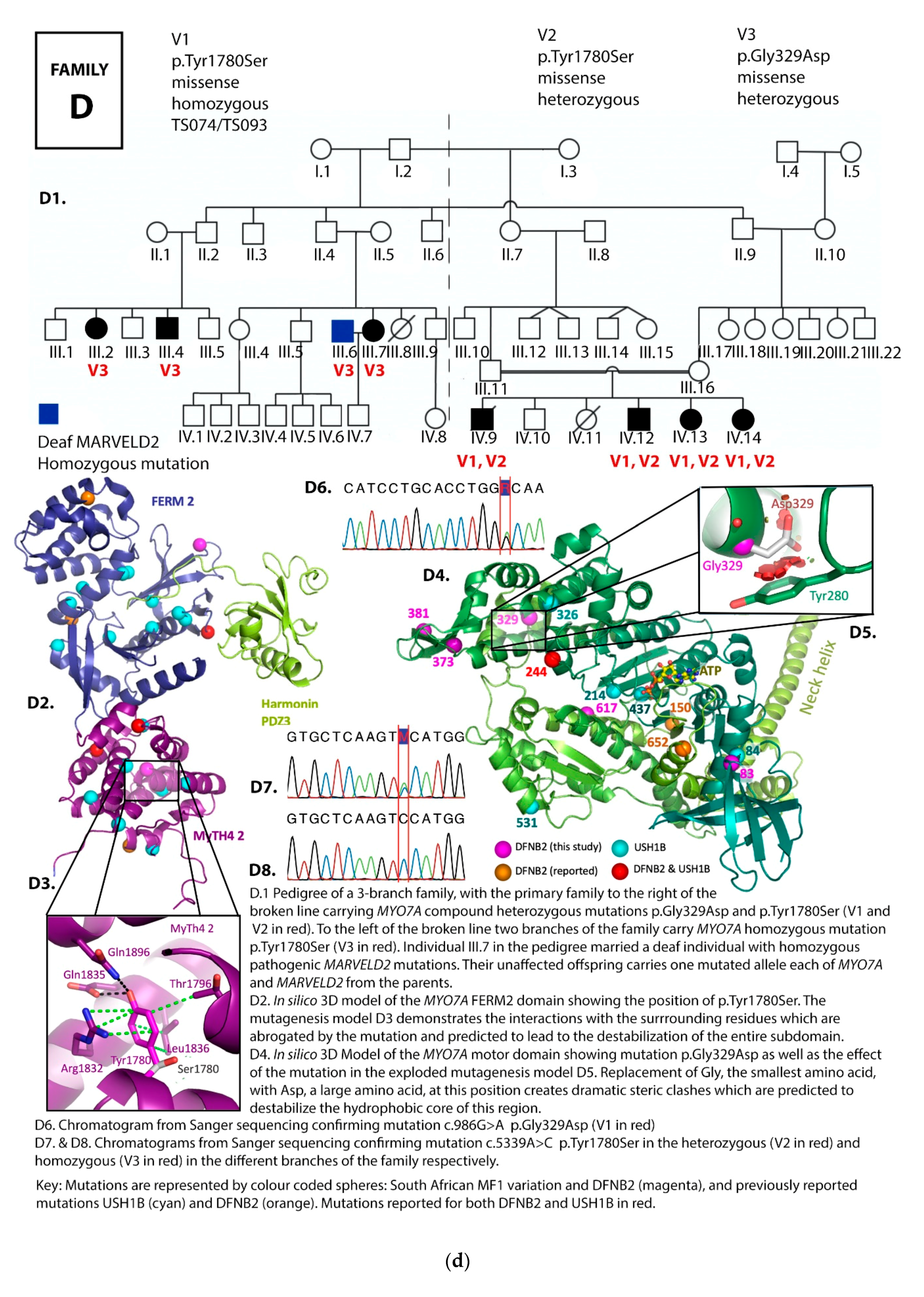
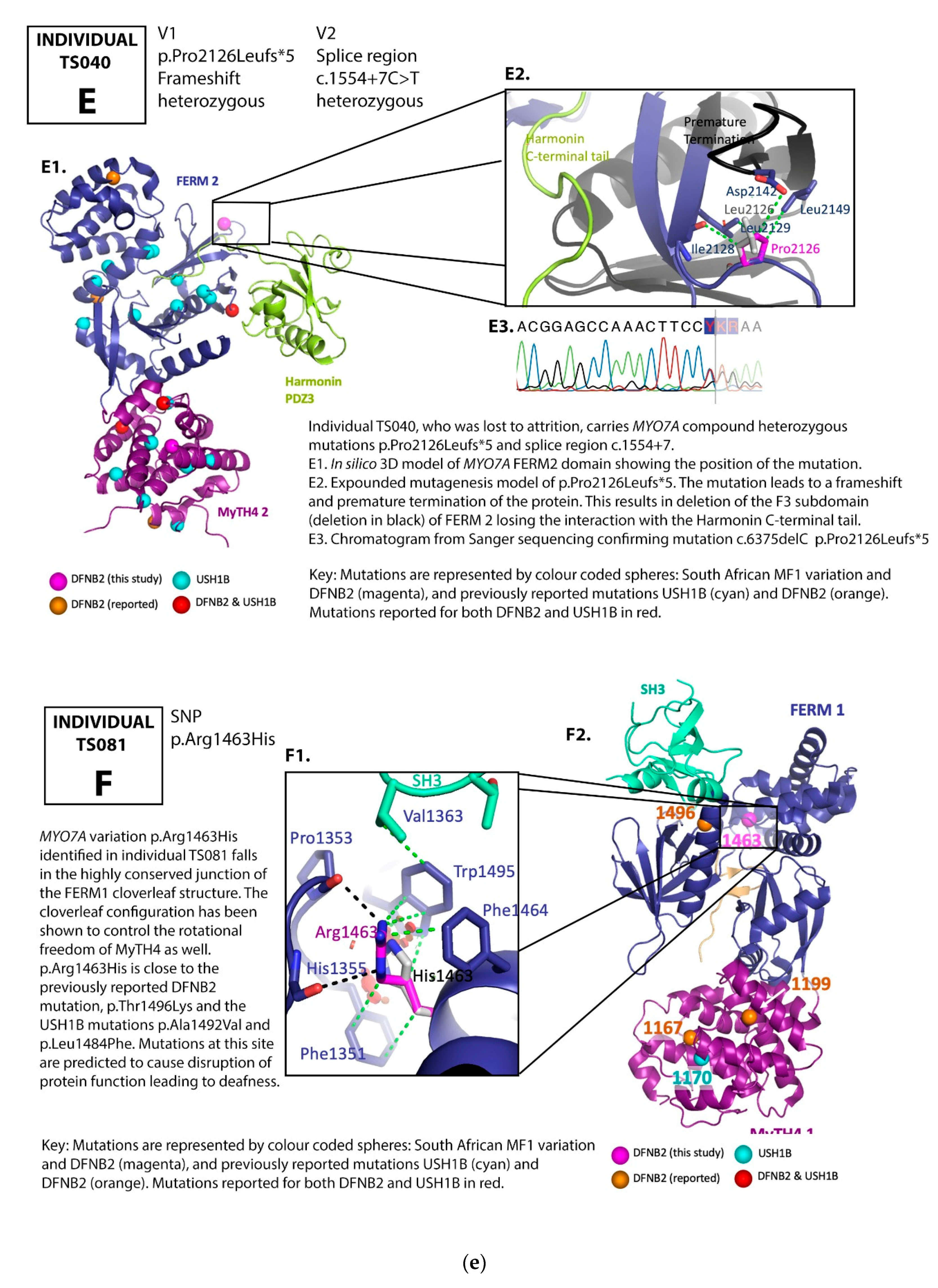
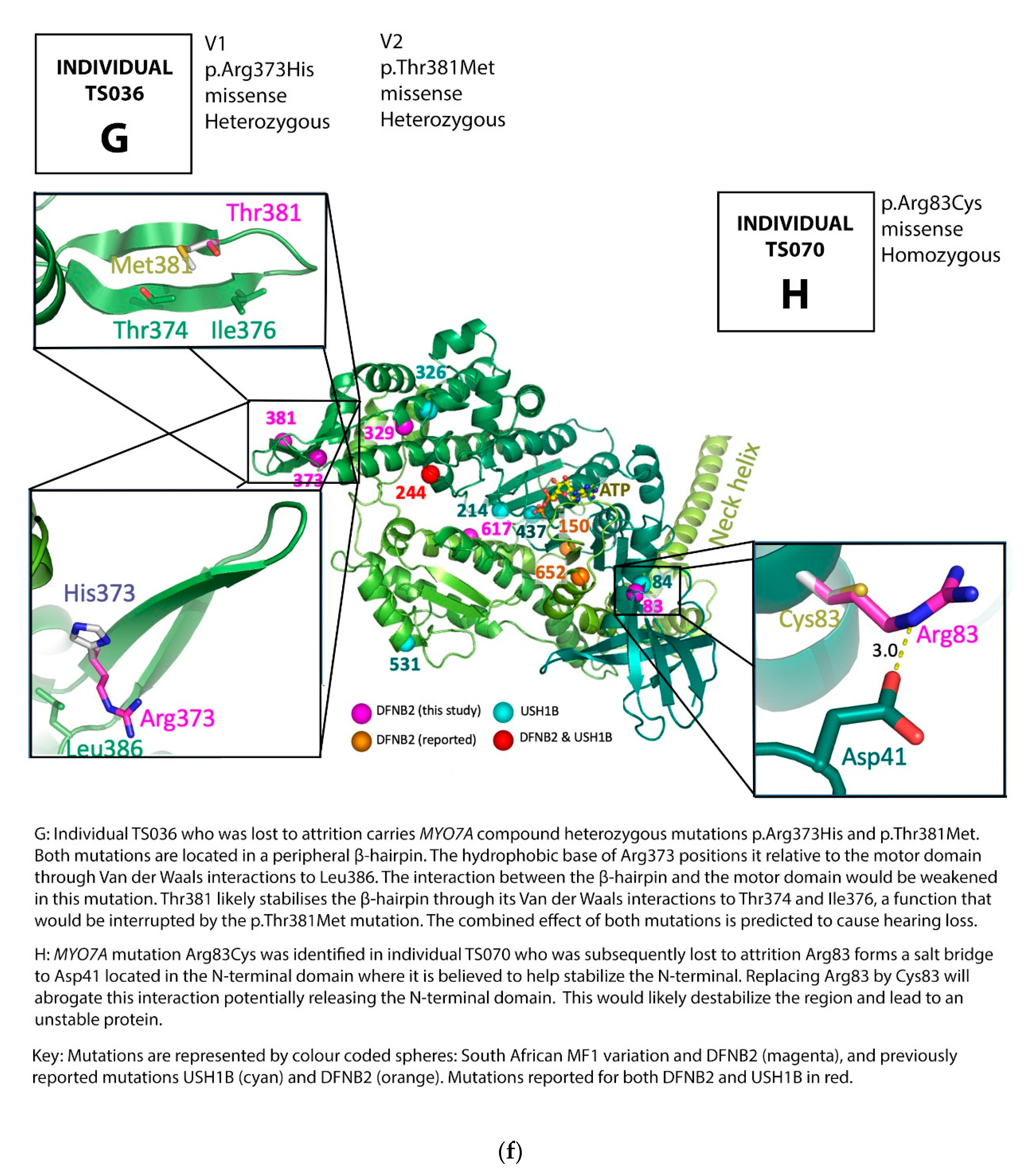
3.1. Family Pedigrees
3.2. MYO7A Mutations
4. Discussion
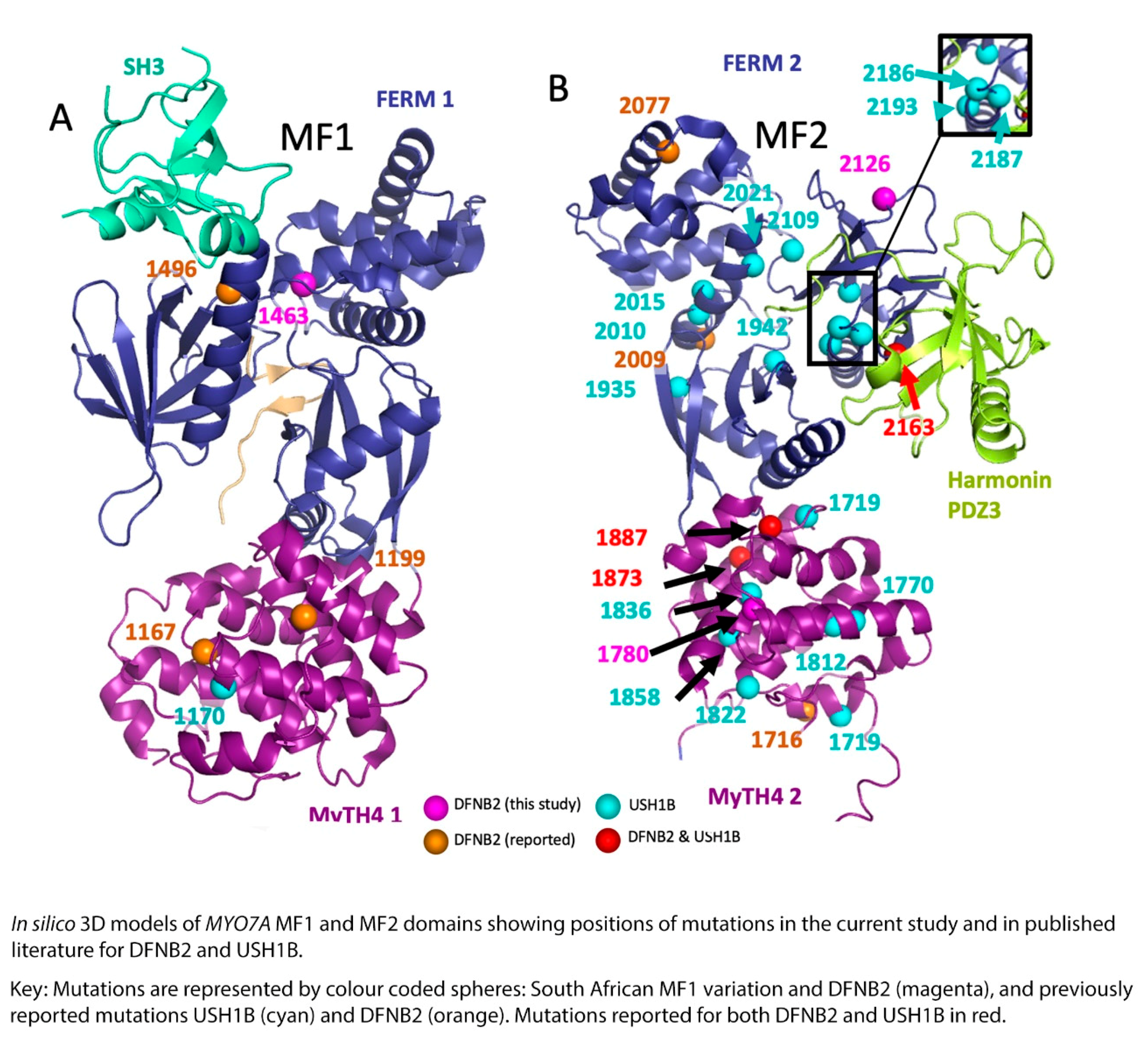
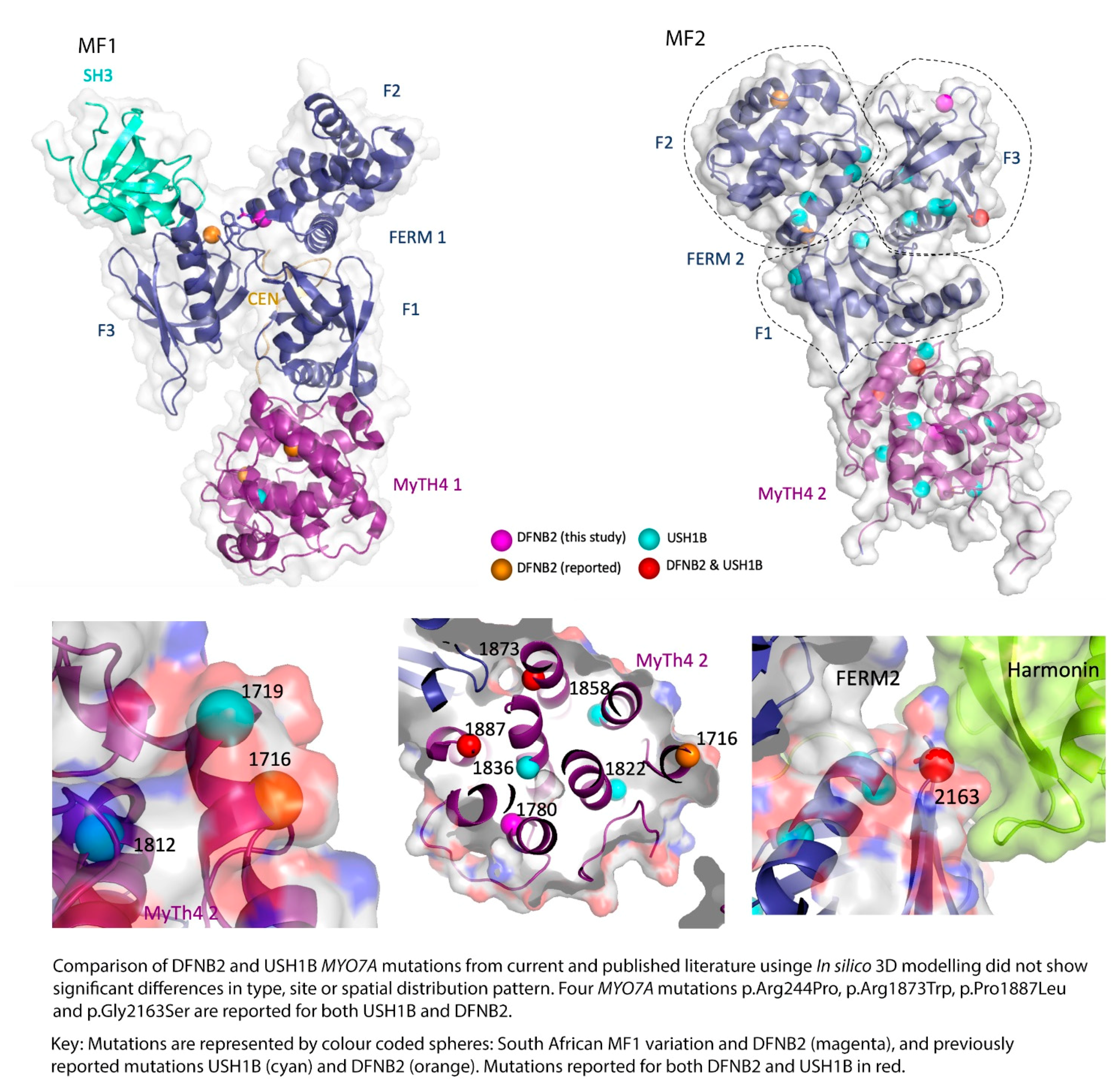
DFNB2 versus Usher 1B Syndrome
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Weil, D.; Blanchard, S.; Kaplan, J.; Guilford, P.; Gibson, F.; Walsh, J.; Mburu, P.; Varela, A.; Levilliers, J.; Weston, M.D.; et al. Defective myosin VIIA gene responsible for Usher syndrome type 1B. Nature 1995, 374, 60–61. [Google Scholar] [CrossRef] [PubMed]
- Lévy, G.; Levi-Acobas, F.; Blanchard, S.; Gerber, S.; Larget-Piet, D.; Chenal, V.; Liu, X.Z.; Newton, V.; Steel, K.P.; Brown, S.D.; et al. Myosin VIIA gene: Heterogeneity of the mutations responsible for Usher syndrome type IB. Hum. Mol. Genet. 1997, 6, 111–116. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.Y.; Hasson, T.; Kelley, P.M.; Schwender, B.J.; Schwartz, M.F.; Ramakrishnan, M.; Kimberling, W.J.; Mooseker, M.S.; Corey, D.P. Molecular cloning and domain structure of human myosin-VIIa, the gene product defective in Usher syndrome 1B. Genomics 1999, 36, 440–448. [Google Scholar] [CrossRef]
- Adato, A.; Vreugde, S.; Joensuu, T.; Avidan, N.; Hamalainen, R.; Belenkiy, O.; Olender, T.; Bonne-Tamir, B.; Ben-Asher, E.; Espinos, C.; et al. USH3A transcripts encode clarin-1, a four-transmembrane-domain protein with a possible role in sensory synapses. Eur. J. Hum. Genet. 2002, 10, 339–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karolyi, I.J.; Probst, F.J.; Beyer, L.; Odeh, H.; Dootz, G.; Cha, K.B.; Martin, D.M.; Avraham, K.B.; Kohrman, D.; Dolan, D.F.; et al. Myo15 function is distinct from Myo6, Myo7a and pirouette genes in development of cochlear stereocilia. Hum Mol Genet. 2003, 12, 2797–2805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corey, D.P.; Hudspeth, A.J. Kinetics of the receptor current in bullfrog saccular hair cells. J. Neurosci. 1983, 3, 962–976. [Google Scholar] [CrossRef] [PubMed]
- Pickles, J.O.; Comis, S.D.; Osborne, M.P. Cross-links between stereocilia in the guinea pig organ of Corti, and their possible relation to sensory transduction. Hear. Res. 1984, 15, 103–112. [Google Scholar] [CrossRef]
- Pickles, J.O.; Brix, J.; Comis, S.D.; Gleich, O.; Köppl, C.; Manley, G.A.; Osborne, M.P. The organization of tip links and stereocilia on hair cells of bird and lizard basilar papillae. Hear. Res. 1989, 41, 31–41. [Google Scholar] [CrossRef] [Green Version]
- Sakaguchi, H.; Tokita, J.; Müller, U.; Kachar, B. Tip links in hair cells: Molecular composition and role in hearing loss. Curr. Opin. Otolaryngol. Head Neck Surg. 2009, 17, 388–393. [Google Scholar] [CrossRef] [Green Version]
- Hudspeth, A.J. Integrating the active process of hair cells with cochlear function. Nat. Rev. Neurosci. 2014, 15, 600–614. [Google Scholar] [CrossRef] [PubMed]
- Pepermans, E.; Petit, C. The tip-link molecular complex of the auditory mechano-electrical transduction machinery. Hear. Res. 2015, 330, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Pan, L.; Zhang, C.; Zhang, M. Large protein assemblies formed by multivalent interactions between cadherin23 and harmonin suggest a stable anchorage structure at the tip link of stereocilia. J. Biol. Chem. 2012, 287, 33460–33471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krendel, M.; Mooseker, M.S. Myosins: Tails (and Heads) of Functional Diversity. Physiology 2005, 20, 239–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woolner, S.; Bement, W.M. Unconventional myosins acting unconventionally. Trends Cell Biol. 2009, 19, 245–252. [Google Scholar] [CrossRef] [Green Version]
- Kros, C.J.; Marcotti, W.; van Netten, S.M.; Self, T.J.; Libby, R.T.; Brown, S.D.; Richardson, G.P.; Steel, K.P. Reduced climbing and increased slipping adaptation in cochlear hair cells of mice with Myo7a mutations. Nat. Neurosci. 2002, 5, 41–47. [Google Scholar] [CrossRef]
- Self, T.; Mahony, M.; Fleming, J.; Walsh, J.; Brown, S.D.; Steel, K.P. Shaker-1 mutations reveal roles for myosin VIIA in both development and function of cochlear hair cells. Development 1998, 125, 557–566. [Google Scholar] [PubMed]
- El-Amraoui, A.; Petit, C. Usher I syndrome: Unravelling the mechanisms that underlie the cohesion of the growing hair bundle in inner ear sensory cells. J. Cell Sci. 2005, 118, 4593–4603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanco-Sánchez, B.; Clément, A.; Fierro, J., Jr.; Washbourne, P.; Westerfield, M. Complexes of Usher proteins preassemble at the endoplasmic reticulum and are required for trafficking and ER homeostasis. Dis. Models Mech. 2014, 7, 547–559. [Google Scholar] [CrossRef] [Green Version]
- Riazuddin, S.; Nazli, S.; Ahmed, Z.M.; Yang, Y.; Zulfiqar, F.; Shaikh, R.S.; Zafar, A.U.; Khan, S.N.; Sabar, F.; Javid, F.T.; et al. Mutation spectrum of MYO7A and evaluation of a novel nonsyndromic deafness DFNB2 allele with residual function. Hum. Mutat. 2008, 29, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Crawley, S.W.; Shifrin, D.A., Jr.; Grega-Larson, N.E.; McConnell, R.E.; Benesh, A.E.; Mao, S.; Zheng, Y.; Zheng, Q.Y.; Nam, K.T.; Millis, B.A.; et al. Intestinal Brush Border Assembly Driven by Protocadherin-Based Intermicrovillar Adhesion. Cell 2014, 157, 433–446. [Google Scholar] [CrossRef] [Green Version]
- Crawley, S.W.; Weck, M.L.; Grega-Larson, N.E.; Shifrin, D.A., Jr.; Tyska, M.J. ANKS4B Is Essential for Intermicrovillar Adhesion Complex Formation. Dev. Cell 2016, 36, 190–200. [Google Scholar] [CrossRef] [Green Version]
- Senften, M.; Schwander, M.; Kazmierczak, P.; Lillo, C.; Shin, J.B.; Hasson, T.; Géléoc, G.S.; Gillespie, P.G.; Williams, D.; Holt, J.R.; et al. Physical and functional interaction between protocadherin 15 and myosin VIIa in mechanosensory hair cells. J. Neurosci. 2006, 26, 2060–2071. [Google Scholar] [CrossRef]
- Grati, M.; Kachar, B. Myosin VIIa and sans localization at stereocilia upper tip-link density implicates these Usher syndrome proteins in mechanotransduction. Proc. Natl. Acad. Sci. USA 2011, 108, 11476–11481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belyantseva, I.A.; Boger, E.T.; Friedman, T.B. Myosin XVa localizes to the tips of inner ear sensory cell stereocilia and is essential for staircase formation of the hair bundle. Proc. Natl. Acad. Sci. USA 2003, 100, 13958–13963. [Google Scholar] [CrossRef] [Green Version]
- Indzhykulian, A.A.; Stepanyan, R.; Nelina, A.; Spinelli, K.J.; Ahmed, Z.M.; Belyantseva, I.A.; Friedman, T.B.; Barr-Gillespie, P.G.; Frolenkov, G.I. Molecular Remodeling of Tip Links Underlies Mechanosensory Regeneration in Auditory Hair Cells. PLoS Biol. 2013, 11, 1001583. [Google Scholar] [CrossRef] [Green Version]
- Bahloul, A.; Michel, V.; Hardelin, J.P.; Nouaille, S.; Hoos, S.; Houdusse, A.; England, P.; Petit, C. Cadherin-23, myosin VIIa and harmonin, encoded by Usher syndrome type I genes, form a ternary complex and interact with membrane phospholipids. Hum. Mol. Genet. 2010, 19, 3557–3565. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Pan, L.; Wei, Z.; Zhang, M. Structure of MyTH4-FERM domains in myosin VIIa tail bound to cargo. Science 2011, 331, 757–760. [Google Scholar] [CrossRef] [PubMed]
- Sakai, T.; Jung, H.S.; Sato, O.; Yamada, M.D.; You, D.J.; Ikebe, R.; Ikebe, M. Structure and Regulation of the Movement of Human Myosin VIIA. J. Biol. Chem. 2015, 290, 17587–17598. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; He, Y.; Weck, M.L.; Lu, Q.; Tyska, M.J.; Zhang, M. Structure of Myo7b/USH1C complex suggests a general PDZ domain binding mode by MyTH4-FERM myosins. Proc. Natl. Acad. Sci. USA 2017, 114, 3776–3785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheney, R.E.; Mooseker, M.S. Unconventional myosins. Curr. Opin. Cell Biol. 1992, 4, 27–35. [Google Scholar] [CrossRef]
- Todorov, P.T.; Hardisty, R.E.; Brown, S.D. Myosin VIIA is specifically associated with calmodulin and microtubule-associated protein-2B (MAP-2B). Biochem. J. 2001, 354, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Udovichenko, I.P.; Gibbs, D.; Williams, D.S. Actin-based motor properties of native myosin VIIa. J. Cell Sci. 2002, 115, 445–450. [Google Scholar]
- Inoue, A.; Ikebe, M. Characterization of the Motor Activity of Mammalian Myosin VIIA. J. Biol. Chem. 2003, 278, 5478–5487. [Google Scholar] [CrossRef] [Green Version]
- Adato, A.; Michel, V.; Kikkawa, Y.; Reiners, J.; Alagramam, K.N.; Weil, D.; Yonekawa, H.; Wolfrum, U.; El-Amraoui, A.; Petit, C. Interactions in the network of Usher syndrome type 1 proteins. Hum. Mol. Genet. 2005, 14, 347–356. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Berg, J.S.; Li, Z.; Wang, Y.; Lang, P.; Sousa, A.D.; Bhaskar, A.; Cheney, R.E.; Stromblad, S. MyosinX provides a motor-based link between integrins and the cytoskeleton. Nat. Cell Biol. 2004, 6, 523–531. [Google Scholar] [CrossRef]
- Liu, L.; Srikakulam, R.; Winkelmann, J. Unc45 activates Hsp90-dependent folding of the myosin motor domain. J. Biol. Chem. 2008, 283, 13185–13193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boëda, B.; El-Amraoui, A.; Bahloul, A.; Goodyear, R.; Daviet, L.; Blanchard, S.; Perfettini, I.; Fath, K.R.; Shorte, S.; Reiners, J.; et al. Myosin VIIa, harmonin and cadherin 23, three Usher I gene products that cooperate to shape the sensory hair cell bundle. EMBO J. 2002, 21, 6689–6699. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.J.; Wang, C.Z.; Dai, P.G.; Xie, Y.; Song, N.N.; Liu, Y.; Du, Q.S.; Mei, L.; Ding, Y.Q.; Xiong, W.C. Myosin X regulates netrin receptors and functions in axonal path-finding. Nat. Cell Biol. 2007, 9, 184–192. [Google Scholar] [CrossRef]
- Yang, Y.; Baboolal, T.G.; Siththanandan, V.; Chen, M.; Walker, M.L.; Knight, P.J.; Sellers, J.R. A FERM domain autoregulates Drosophila myosin 7a activity. Proc. Natl. Acad. Sci. USA 2009, 106, 4189–4194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pi, X.; Ren, R.; Kelley, R.; Zhang, C.; Moser, M.; Bohil, A.B.; Divito, M.; Cheney, R.E.; Patterson, C. Sequential roles for myosin-X in BMP6-dependent filopodial extension, migration, and activation of BMP receptors. J. Cell Biol. 2007, 179, 1569–1582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umeki, N.; Jung, H.S.; Sakai, T.; Sato, O.; Ikebe, R.; Ikebe, M. Phospholipid-dependent regulation of the motor activity of myosin X. Nat. Struct. Mol. Biol. 2011, 18, 783–788. [Google Scholar] [CrossRef]
- Küssel-Andermann, P.; El-Amraoui, A.; Safieddine, S.; Hardelin, J.P.; Nouaille, S.; Camonis, J.; Petit, C. Unconventional myosin VIIA is a novel A-kinase-anchoring protein. J. Biol. Chem. 2000, 275, 29654–29659. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.Z.; Hope, C.; Walsh, J.; Newton, V.; Ke, X.M.; Liang, C.Y.; Xu, L.R.; Zhou, J.M.; Trump, D.; Steel, K.P.; et al. Mutations in the myosin VIIA gene cause a wide phenotypic spectrum, including atypical Usher syndrome. Am. J. Hum. Genet. 1998, 63, 909–912. [Google Scholar] [CrossRef] [Green Version]
- Gibbs, D.; Kitamoto, J.; Williams, D.S. Abnormal phagocytosis by retinal pigmented epithelium that lacks myosin VIIa, the Usher syndrome 1B protein. Proc. Natl. Acad. Sci. USA 2003, 100, 6481–6486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Udovichenko, I.P.; Brown, S.D.; Steel, K.P.; Williams, D.S. Myosin VIIa participates in opsin transport through the photoreceptor cilium. J. Neurosci. 1999, 19, 6267–6274. [Google Scholar] [CrossRef]
- Wolfrum, U.; Schmitt, A. Rhodopsin transport in the membrane of the connecting cilium of mammalian photoreceptor cells. Cell Motil. Cytoskelet. 2000, 46, 95–107. [Google Scholar] [CrossRef]
- El-Amraoui, A.; Sahly, I.; Picaud, S.; Sahel, J.; Abitbol, M.; Petit, C. Human Usher 1B/mouse shaker-1: The retinal phenotype discrepancy explained by the presence/absence of myosin VIIA in the photoreceptor cells. Hum. Mol. Genet. 1996, 5, 1171–1178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guilford, P.; Ben Arab, S.; Blanchard, S.; Levilliers, J.; Weissenbach, J.; Belkahia, A.; Petit, C. A non-syndrome form of neurosensory, recessive deafness maps to the pericentromeric region of chromosome 13q. Nat. Genet. 1994, 6, 24–28. [Google Scholar] [CrossRef] [PubMed]
- Weil, D.; Küssel, P.; Blanchard, S.; Lévy, G.; Levi-Acobas, F.; Drira, M.; Ayadi, H.; Petit, C. The autosomal recessive isolated deafness, DFNB2, and the Usher 1B syndrome are allelic defects of the myosin-VIIA gene. Nat. Genet. 1997, 16, 191–193. [Google Scholar] [CrossRef]
- Liu, X.Z.; Walsh, J.; Mburu, P.; Kendrick-Jones, J.; Cope, M.J.; Steel, K.P.; Brown, S.D. Mutations in the myosin VIIA gene cause non-syndromic recessive deafness. Nat. Genet. 1997, 16, 188–190. [Google Scholar] [CrossRef]
- Zina, Z.B.; Masmoudi, S.; Ayadi, H.; Chaker, F.; Ghorbel, A.M.; Drira, M.; Petit, C. From DFNB2 to Usher syndrome: Variable expressivity of the same disease. Am. J. Med Genet. 2001, 101, 181–183. [Google Scholar] [CrossRef] [PubMed]
- Astuto, L.M.; Bork, J.M.; Weston, M.D.; Askew, J.W.; Fields, R.R.; Orten, D.J.; Kimberling, W.J. CDH23 mutation and phenotype heterogeneity: A profile of 107 diverse families with Usher syndrome and nonsyndromic deafness. Am. J. Hum. Genet. 2002, 71, 262–275. [Google Scholar] [CrossRef] [Green Version]
- Hildebrand, M.S.; Thorne, N.P.; Bromhead, C.J.; Kahrizi, K.; Webster, J.A.; Fattahi, Z.; Bataejad, M.; Kimberling, W.J.; Stephan, D.; Najmabadi, H.; et al. Specific isoforms of drosophila shroom define spatial requirements for the induction of apical constriction. Clin. Genet. 2010, 77, 563–571. [Google Scholar] [CrossRef] [Green Version]
- Brownstein, Z.; Abu-Rayyan, A.; Karfunkel-Doron, D.; Sirigu, S.; Davidov, B.; Shohat, M.; Frydman, M.; Houdusse, A.; Kanaan, M.; Avraham, K.B. Novel myosin mutations for hereditary hearing loss revealed by targeted genomic capture and massively parallel sequencing. Eur. J. Hum. Genet. 2014, 22, 768–775. [Google Scholar] [CrossRef] [Green Version]
- Chen, A.; Wayne, S.; Bell, A.; Ramesh, A.; Srisailapathy, C.R.; Scott, D.A.; Sheffield, V.C.; Van Hauwe, P.; Zbar, R.I.; Ashley, J.; et al. New gene for autosomal recessive non-syndromic hearing loss maps to either chromosome 3q or 19p. Am. J. Med Genet. 1997, 71, 467–471. [Google Scholar] [CrossRef]
- Sun, Y.; Chen, J.; Sun, H.; Cheng, J.; Li, J.; Lu, Y.; Lu, Y.; Jin, Z.; Zhu, Y.; Ouyang, X.; et al. Novel missense mutations in MYO7A underlying postlingual high- or low-frequency non-syndromic hearing impairment in two large families from China. J. Hum. Genet. 2011, 56, 64–70. [Google Scholar] [CrossRef] [Green Version]
- Ben-Salem, S.; Rehm, H.L.; Willems, P.J.; Tamimi, Z.A.; Ayadi, H.; Ali, B.R.; Al-Gazali, L. Analysis of two Arab families reveals additional support for a DFNB2 nonsyndromic phenotype of MYO7A. Mol. Biol. Rep. 2014, 41, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Asgharzade, S.; Reiisi, S.; Tabatabaiefar, M.A.; Chaleshtori, M.H. Screening of Myo7A Mutations in Iranian Patients with Autosomal Recessive Hearing Loss from West of Iran. Iran. J. Public Health 2017, 46, 76–82. [Google Scholar]
- Sadeghi, A.; Sanati, M.; Alasti, F.; Hashemzadeh Chaleshtori, M.; Mahmoudian, S.; Ataei, M. Contribution of GJB2 Mutations and Four Common DFNB Loci in Autosomal Recessive Non-Syndromic Hearing Impairment in Markazi and Qom Provinces of Iran. Iran. J. Biotechnol. 2009, 7, 108–111. [Google Scholar]
- Talbi, S.; Bonnet, C.; Riahi, Z.; Boudjenah, F.; Dahmani, M.; Hardelin, J.P.; Wong Jun Tai, F.; Louha, M.; Ammar-Khodja, F.; Petit, C. Genetic heterogeneity of congenital hearing impairment in Algerians from the Ghardaïa province. Int. J. Pediatr. Otorhinolaryngol. 2018, 112, 1–5. [Google Scholar] [CrossRef]
- Bakhchane, A.; Charif, M.; Bousfiha, A.; Boulouiz, R.; Nahili, H.; Rouba, H.; Charoute, H.; Lenaers, G.; Barakat, A. Novel compound heterozygous MYO7A mutations in Moroccan families with autosomal recessive non-syndromic hearing loss. PLoS ONE 2017, 12, e0176516. [Google Scholar] [CrossRef]
- Goebel, J. The Ten-Minute Examination of the Dizzy Patient. Semin. Neurol. 2002, 21, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Tekin, D.; Yan, D.; Bademci, G.; Feng, Y.; Guo, S.; Foster, J., 2nd; Blanton, S.; Tekin, M.; Liu, X. A next-generation sequencing gene panel (MiamiOtoGenes) for comprehensive analysis of deafness genes. Hear Res. 2016, 333, 179–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with burrows-wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [Green Version]
- Garrison, E.; Marth, G. Haplotype-based variant detection from short-read sequencing. arXiv 2012, arXiv:1207.3907. [Google Scholar]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. ACMG Laboratory Quality Assurance Committee (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. Off. J. Am. Coll. Med Genet. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Oza, A.M.; DiStefano, M.T.; Hemphill, S.E.; Cushman, B.J.; Grant, A.R.; Siegert, R.K.; Shen, J.; Chapin, A.; Boczek, N.J.; Schimmenti, L.A.; et al. ClinGen Hearing Loss Clinical Domain Working Group Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss. Hum. Mutat. 2018, 39, 1593–1613. [Google Scholar] [CrossRef]
- Nord, A.S.; Lee, M.; King, M.; Walsh, T. Accurate and exact CNV identification from targeted high-throughput sequence data. BMC Genom. 2011, 12, 184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabahuma, R.I.; Ouyang, X.; Du, L.L.; Yan, D.; Hutchin, T.; Ramsay, M.; Penn, C.; Liu, X.Z. Absence of GJB2 gene mutations, the GJB6 deletion (GJB6-D13S1830) and four common mitochondrial mutations in nonsyndromic genetic hearing loss in a South African population. Int. J. Pediatr. Otorhinolaryngol. 2011, 75, 611–617. [Google Scholar] [CrossRef] [Green Version]
- Yan, D.; Tekin, D.; Bademci, G.; Foster, J., 2nd; Cengiz, F.B.; Kannan-Sundhari, A.; Guo, S.; Mittal, R.; Zou, B.; Grati, M.; et al. Spectrum of DNA variants for non-syndromic deafness in a large cohort from multiple continents. Hum. Genet. 2017, 135, 953–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ammar-Khodja, F.; Faugère, V.; Baux, D.; Giannesini, C.; Léonard, S.; Makrelouf, M.; Malek, R.; Djennaoui, D.; Zenati, A.; Claustres, M.; et al. Molecular screening of deafness in Algeria: High genetic heterogeneity involving DFNB1 and the Usher loci, DFNB2/USH1B, DFNB12/USH1D and DFNB23/USH1F. Eur. J. Med Genet. 2009, 52, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Adato, A.; Weil, D.; Kalinski, H.; Pel-Or, Y.; Ayadi, H.; Petit, C.; Korostishevsky, M.; Bonne-Tamir, B. Mutation profile of all 49 exons of the human myosin VIIA gene, and haplotype analysis, in Usher 1B families from diverse origins. Am. J. Hum. Genet. 1997, 61, 813–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaijo, T.; Aller, E.; Beneyto, M.; Najera, C.; Graziano, C.; Turchetti, D.; Seri, M.; Ayuso, C.; Baiget, M.; Moreno, F.; et al. MYO7A mutation screening in Usher syndrome type I patients from diverse origins. J. Med. Genet. 2007, 44, e71. [Google Scholar] [CrossRef] [Green Version]
- Le Guédard-Méreuze, S.; Vaché, C.; Baux, D.; Faugère, V.; Larrieu, L.; Abadie, C.; Janecke, A.; Claustres, M.; Roux, A.F. Tuffery-Giraud, S. Ex vivo splicing assays of mutations at noncanonical positions of splice sites in USHER genes. Hum. Mutat. 2010, 31, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Cuevas, J.M.; Espinós, C.; Millan, J.M.; Sanchez, F.; Trujillo, M.J.; Ayuso, C.; Beneyto, M.; Najera, C. Identification of three novel mutations in the MYO7A gene. Hum. Mutat. 1999, 14, 181. [Google Scholar] [CrossRef]
- Duzkale, H.; Shen, J.; McLaughlin, H.; Alfares, A.; Kelly, M.A.; Pugh, T.J.; Funke, B.H.; Rehm, H.L.; Lebo, M.S. A systematic approach to assessing the clinical significance of genetic variants. Clin. Genet. 2013, 84, 453–463. [Google Scholar] [CrossRef]
- Janecke, A.R.; Meins, M.; Sadeghi, M.; Grundmann, K.; Apfelstedt-Sylla, E.; Zrenner, E.; Rosenberg, T.; Gal, A. Twelve novel myosin VIIA mutations in 34 patients with Usher syndrome type I: Confirmation of genetic heterogeneity. Hum. Mutat. 1999, 13, 133–140. [Google Scholar] [CrossRef]
- Vaché, C.; Besnard, T.; Blanchet, C.; Baux, D.; Larrieu, L.; Faugère, V.; Mondain, M.; Hamel, C.; Malcolm, S.; Claustres, M.; et al. Nasal epithelial cells are a reliable source to study splicing variants in Usher syndrome. Hum. Mutat. 2010, 31, 734–741. [Google Scholar] [CrossRef] [Green Version]
- Nájera, C.; Beneyto, M.; Blanca, J.; Aller, E.; Fontcuberta, A.; Millán, J.M.; Ayuso, C. Mutations in myosin VIIA (MYO7A) and usherin (USH2A) in Spanish patients with Usher syndrome types I and II, respectively. Hum. Mutat. 2002, 20, 76–77. [Google Scholar] [CrossRef]
- Jaijo, T.; Aller, E.; Oltra, S.; Beneyto, M.; Nájera, C.; Ayuso, C.; Baiget, M.; Carballo, M.; Antiñolo, G.; Valverde, D.; et al. Mutation profile of the MYO7A gene in Spanish patients with Usher syndrome type I. Hum. Mutat. 2006, 27, 290–301. [Google Scholar] [CrossRef] [PubMed]
- Roux, A.F.; Faugère, V.; Vaché, C.; Baux, D.; Besnard, T.; Léonard, S.; Blanchet, C.; Hamel, C.; Mondain, M.; Gilbert-Dussardier, B.; et al. Four-year follow-up of diagnostic service in USH1 patients. Investig. Ophthalmol. Vis. Sci. 2011, 52, 4063–4071. [Google Scholar] [CrossRef]
- Bharadwaj, A.K.; Kasztejna, J.P.; Huq, S.; Berson, E.L.; Dryja, T.P. Evaluation of the myosin VIIA gene and visual function in patients with Usher syndrome type I. Exp. Eye Res. 2000, 71, 173–181. [Google Scholar] [CrossRef]
- Cremers, F.P.; Kimberling, W.J.; Külm, M.; de Brouwer, A.P.; van Wijk, E.; te Brinke, H.; Cremers, C.W.; Hoefsloot, L.H.; Banfi, S.; Simonelli, F.; et al. Development of a genotyping microarray for Usher syndrome. J. Med. Genet. 2007, 44, 153–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roux, A.F.; Faugère, V.; Le Guédard, S.; Pallares-Ruiz, N.; Vielle, A.; Chambert, S.; Marlin, S.; Hamel, C.; Gilbert, B.; Malcolm, S.; et al. French Usher Syndrome Collaboration. Survey of the frequency of USH1 gene mutations in a cohort of Usher patients shows the importance of cadherin 23 and protocadherin 15 genes and establishes a detection rate of above 90%. J. Med. Genet. 2006, 43, 763–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouyang, X.M.; Yan, D.; Du, L.L.; Hejtmancik, J.F.; Jacobson, S.G.; Nance, W.E.; Li, A.R.; Angeli, S.; Kaiser, M.; Newton, V.; et al. Characterization of Usher syndrome type I gene mutations in an Usher syndrome patient population. Hum. Genet. 2005, 116, 292–299. [Google Scholar] [CrossRef]
- Duman, D.; Sirmaci, A.; Cengiz, F.B.; Ozdag, H.; Tekin, M. Screening of 38 genes identifies mutations in 62% of families with nonsyndromic deafness in Turkey. Genet. Test. Mol. Biomarks 2011, 15, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Dong, C.; Wang, Q.; Zhong, Z.; Qi, Y.; Ke, X.; Liu, Y. Targeted Next-Generation Sequencing Successfully Detects Causative Genes in Chinese Patients with Hereditary Hearing Loss. Genet. Test. Mol. Biomarkers 2016, 20, 660–665. [Google Scholar] [CrossRef]
- Bonnet, C.; Grati, M.; Marlin, S.; Levilliers, J.; Hardelin, J.P.; Parodi, M.; Niasme-Grare, M.; Zelenika, D.; Délépine, M.; Feldmann, D.; et al. Complete exon sequencing of all known Usher syndrome genes greatly improves molecular diagnosis. Orphanet J. Rare Dis. 2011, 6, 21. [Google Scholar] [CrossRef] [Green Version]
- Kiehart, D.P.; Franke, J.D.; Chee, M.K.; Montague, R.A.; Chen, T.L.; Roote, J.; Ashburner, M. Drosophila crinkled, mutations of which disrupt morphogenesis and cause lethality, encodes fly myosin VIIA. Genetics 2004, 168, 1337–1352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearson, M.A.; Reczek, D.; Bretscher, A.; Karplus, P.A. Structure of the ERM protein moesin reveals the FERM domain fold masked by an extended actin binding tail domain. Cell 2000, 101, 259–270. [Google Scholar] [CrossRef] [Green Version]
- Smith, D.A.; Geeves, M.A. Cooperative regulation of myosin-actin interactions by a continuous flexible chain II: Actin-tropomyosin-troponin and regulation by calcium. Biophys. J. 2003, 84, 3168–3180. [Google Scholar] [CrossRef] [Green Version]
- Hamada, K.; Shimizu, T.; Yonemura, S.; Tsukita, S.; Tsukita, S.; Hakoshima, T. Structural basis of adhesion-molecule recognition by ERM proteins revealed by the crystal structure of the radixin-ICAM-2 complex. EMBO J. 2003, 22, 502–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, I.M.; Planelles-Herrero, V.J.; Sourigues, Y.; Moussaoui, D.; Sirkia, H.; Kikuti, C.; Stroebel, D.; Titus, M.A.; Houdusse, A. Myosin 7 and its adaptors link cadherins to actin. Nat. Commun. 2017, 8, 15864. [Google Scholar] [CrossRef] [PubMed]
- Vastinsalo, H.; Isosomppi, J.; Aittakorpi, A.; Sankila, E.M. Two Finnish USH1B patients with three novel mutations in myosin VIIA. Mol. Vis. 2006, 12, 1093–1097. [Google Scholar]
- Rudman, J.R.; Kabahuma, R.I.; Bressler, S.E.; Feng, Y.; Blanton, S.H.; Yan, D.; Liu, X.Z. The genetic basis of deafness in populations of African descent. J. Genet. Genom. 2017, 44, 285–294. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| cDNA | HGVS Protein Notation | Sequence_Ontology | GRCh38.p12 | Family IDs | Cosegregation in Family | Novelty ClinVar | ClinVar Allele ID (Accession) | ACMG Criteria with HL-EP Specifications |
|---|---|---|---|---|---|---|---|---|
| c.1849T>C | p.Ser617Pro | missense | chr11:77172799 | Family B BS044 | Yes | ClinVar Uncertain significance (26 August 2019) | 431763 (VCV000438172) | Pathogenic PP1_Strong PM3_Very Strong |
| c.986G>A | p.Gly329Asp | missense | chr11:77158413 | Family D1 TS074/TS093 | Yes | Novel | Not reported | Pathogenic PP1_Strong, PM3_Very Strong |
| c.5339A>C | p.Tyr1780Ser | missense | chr11:77204088 | Family A TS065/TS100 | Yes | ClinVar Uncertain significance (10 Febuary 2017) | 511947 (VCV000521209) | Pathogenic PP1_Strong, PM3_Very Strong PS3_Supporting |
| Family D2 | Yes | |||||||
| Family D3 | Yes | |||||||
| Family D1 TS074/TS093 | Yes | |||||||
| Family C TS076 | Yes | |||||||
| c.6375delC | p.Pro2126Leufs*5 | frameshift | chr11:77,212,972 | Family C TS076 | Yes | Novel | Not reported | Pathogenic PVS1_Strong PP1_Moderate PM3_Very Strong PS3_Supporting |
| TS040 | N/A Family not available | |||||||
| c.4388G>A | p.Arg1463His | missense | chr11:77197545 | TS081 | N/A Family not available | Novel | Not reported | Likely Pathogenic PM2 |
| c.1118G>A | p.Arg373His | missense | chr11:77160200 | TS036 | N/A Family not available | Novel | Not reported | Likely Pathogenic PM3_Supporting |
| c.1142C>T | p.Thr381Met | missense | chr11:77160224 | TS036 | N/A Family not available | ClinVar Uncertain significance (30 Junuary 2017) | 546742 (VCV000552693) | Likely Pathogenic PM3_Supporting |
| c.1554+7C>T | splice region | chr11:77162337 | TS040 | N/A Family not available | ClinVar Conflicting interpretations pathogenicity (31 December 2019) | 178242 (VCV000178480) | Likely Pathogenic PS1, PM3_Moderate | |
| c.247C>A | p.Arg83Cys | missense | chr11:77147912 | TS070 | N/A Family not available | ClinVar Uncertain significance (30 August 2018) | 552016 VCV000560896 | Likely Pathogenic PP1, PM3_Supporting. |
| Race/Population Group | Number of DFNB2 Families in Cohort | Citation | Summary of Families | cDNA | Protein Change |
|---|---|---|---|---|---|
| Indigenous sub-Saharan South Africans | 9/89 | Current study | Nine families identified with either homozygous or compound heterozygous mutations in MYO7A leading to DFNB2 | c.247C>A | p.Arg83Cys |
| c.986G>A | p.Gly329Asp | ||||
| c.1118G>A | p.Arg373His | ||||
| c.1142C>T | p.Thr381Met | ||||
| c.1849T>C | p.Ser617Pro | ||||
| c.5339A>C | p.Tyr1780Ser | ||||
| c.6375delC | p.Pro2126Leufs*5 | ||||
| Iranian | 2/30 | Asgharzade et al., 2017 | Two out of 30 deaf families displayed linkage to and were cofirmed DFNB2 | c.6487G>A | p.G2163S |
| c.448 C>T | p.Arg150X | ||||
| Moroccan | 2/61 | Bakhchane A et al., 2017 | Compound heterozygous mutations | c.6025delG | p.Ala2009Profs*32 |
| c.6229T>A | p.Trp2077Arg | ||||
| c.3500T>A | p.Leu1167His | ||||
| c.5617C>T | p.Arg1873Trp | ||||
| c.4487C>A | p.Thr1496Lys | ||||
| Iraqi | 1/1 | Ben-Salem et al., 2014 | Homozygous mutations | c.1952_1953insAG | p.Cys652Glyfs*11 |
| Palestinians | 1/1 | Ben-Salem et al., 2014 | Homozygous mutations | c.5660C>T | p.Pro1887Leu |
| Pakistani | 1/24 | Riazuddin et al., 2008 | Segregated nonsyndromic hearing loss due to a novel three-nucleotide deletion in an exon of MYO7A encoding a region of the tail domain | c.5142_5144del | p.Glu1716del |
| Chinese | 2/2 | Liu et al., 1997 | Homozygous and compound heterozygous mutations | c.133-2A>G | Splice region p.Arg244Pro |
| c.731G>C | p.V1199insT | ||||
| Jewish | 2/248 | Brownstein et al., 2014 | Compound heterozygous and homozygous mutations | c.29T4C | p.Val10Ala |
| c.1969C4T | p.Arg657Trp | ||||
| c.620A4G | p.Asn207Ser | ||||
| Arab Palestinian | 3/611 | Brownstein et al., 2014 | Homozygous mutations (including splice site mutations) | c.4153-2A4G | Splice region |
| c.6211C4T | p.Gln2071* | ||||
| c.G6487A | p.Gly2163Ser |
| Phenotype | Nucleotide Change | Protein Change | Citation | Ref |
|---|---|---|---|---|
| DFNB2 | c.29T>C | p.Val10Ala | Brownstein et al., 2014 | [54] |
| DFNB2 | c.247C>A | p.Arg83Cys | Current study | |
| DFNB2 | c.133-2A>G | Splice acceptor variant | Liu et al., 1997 | [50] |
| DFNB2 | c.448C>T | p.Arg150Ter | Asgharzade et al., 2017 | [58] |
| DFNB2 | c.620>4G | p.Asn207Ser | Brownstein et al. 2014 | [54] |
| DFNB2 | c.731G>C | p.Arg244Pro | Liu et al., 1997 | [50] |
| DFNB2 | c.986G>A | p.Gly329Asp | Current study | |
| DFNB2 | c.1118G>A | p.Arg373His | Current study | |
| DFNB2 | c.1142C>T | p.Thr381Met | Current study | |
| DFNB2 | c.1554+7C>T | Splice region | Current study | |
| DFNB2 | c.1849T>C | p.Ser617Pro | Current study | |
| DFNB2 | c.1969C>T | p.Arg657Trp | Brownstein et al., 2014 | [54] |
| DFNB2 | c.1952_1953insAG | p.Cys652Glyfs*11 | Ben-Salem et al., 2014 | [57] |
| DFNB2 | c.3500T>A | p.Leu1167His | Bakhchane A et al., 2017 | [61] |
| DFNB2 | c.4153-2A>G | Splice acceptor variant | Brownstein et al., 2014 | [54] |
| DFNB2 | c.4388G>A | p.Arg1463His | Current study | |
| DFNB2 | c.4487C>A | p.Thr1496Lys | Bakhchane A et al., 2017 | [61] |
| DFNB2 | c.5146_5148delGAG | p.Glu1716del | Riazuddin et al., 2008 | [19] |
| DFNB2 | c.5339A>C | p.Tyr1780Ser | Current study | |
| DFNB2 | c.5617C>T | p.Arg1873Trp | Bakhchane A et al., 2017 | [61] |
| DFNB2 | c.5660C>T | p.Pro1887Leu | Ben-Salem et al., 2014 | [54] |
| DFNB2 | c.6025delG | p.Ala2009Profs*32 | Bakhchane A et al., 2017 | [61] |
| DFNB2 | c.6211C>T | p.Gln2071* | Brownstein et al., 2014 | [54] |
| DFNB2 | c.6229T>A | p.Trp2077Arg | Bakhchane A et al., 2017 | [61] |
| DFNB2 | c.6375delC | p.Pro2126Leufs*5 | Current study | |
| DFNB2 | c.G6487A | p.Gly2163Ser | Ammar-Khodja et al., 2009 | [71] |
| USH1B | c.252C4G | p.Asn84Lys | Riazuddin et al., 2008 | [19] |
| USH1B | c.398_399insC | p.His133fs*139 | Riazuddin et al., 2008 | [19] |
| USH1B | c.495delG | p.Glu166fs*170 | Riazuddin et al., 2008 | [19] |
| USH1B | c.640G>A | p.Gly214Arg | Adato et al., 1997 | [72] |
| USH1B | c.731G>A | p.Arg244Pro | Liu et al., 1997 | [50] |
| USH1B | c.471-1G>A | Splice acceptor variation | Riazuddin et al., 2008 | [19] |
| USH1B | c.977T>A | p.Leu326Gln | Riazuddin et al., 2008 | [19] |
| USH1B | c.1309G4A | p.Asp437Asn | Riazuddin et al., 2008 | [19] |
| USH1B | c.1591C4T | p.Gln531X | Riazuddin et al., 2008 | [19] |
| USH1B | c.1935+1G>A | Splice region | Riazuddin et al., 2008 | [19] |
| USH1B | c.2476G>A | p.Ala826Thr | Adato et al., 1997 | [72] |
| USH1B | c.2914C4T | p.Arg972X | Riazuddin et al., 2008 | [19] |
| USH1B | c.3135_3136insC | p.Leu1046fs*1054 | Riazuddin et al., 2008 | [19] |
| USH1B | c.3134T>C | p.Ile1045Thr | Jaijo et al., 2007 | [73] |
| USH1B | c.3502C>T | p.Arg1168Pro/Trp | Le Guédard-Méreuze et al., 2010 | [74] |
| USH1B | c.3508G>A | p.Glu1170Lys | Cuevas et al., 1999 | [75] |
| USH1B | c.3631delT | p.Tyr1211fs*1231 | Riazuddin et al., 2008 | [19] |
| USH1B | c.3652G>A | p.Gly1218Arg | Duzkale et al., 2013 | [76] |
| USH1B | c.3719G>A | p.Arg1240Gln | Janecke et al., 1999 | [77] |
| c.3718C>T | p.Arg1240Trp | Vaché et al., 2010 | [78] | |
| USH1B | c.3979G>A | p.Glu1327Lys | Nájera et al., 2002 | [79] |
| USH1B | c.4029G>C | p. Arg1343Ser | Janecke et al., 1999 | [77] |
| USH1B | c.4450C>T | p.Leu1484Phe | Jaijo et al., 2006 | [80] |
| USH1B | c.4697C>T | p.Thr1566Met | Nájera et al., 2002 | [79] |
| USH1B | c.4805G>A | p. Arg1602Gln | Liu et al., 1998 | [43] |
| USH1B | c.4838delA | p.Asp1613fs*1644 | Riazuddin et al., 2008 | [19] |
| USH1B | c.5146_5148delGAG | p.Glu1716del | Riazuddin et al., 2008 | [19] |
| USH1B | c.5156A>G | p.Tyr1719Cys | Janecke et al., 1999; Cuevas et al., 1999 | [75,77] |
| USH1B | c.5366+1G>A | Splice region | Riazuddin et al., 2008 | [19] |
| USH1B | c.5434G>A | p.Glu1812Lys | Roux et al., 2011 | [81] |
| USH1B | c.5464A>C | p.Thr1822Pro | Duzkale et al., 2013 | [76] |
| USH1B | c.5507T>C | p.Leu1836Pro | Jaijo et al., 2007 | [73] |
| USH1B | c.5573T>C | p.Leu1858Pro | Bharadwaj et al., 2000 | [82] |
| USH1B | c.5618G>A | p.Arg1873 Gln | Cremers et al., 2007 | [83] |
| c.5617C>T | p.Arg1873 Trp | Roux et al., 2006 | [84] | |
| USH1B | c.5648G>A | p.Arg1883Gln | Ouyang et al., 2005 | [85] |
| USH1B | c.5804T>C | p.Leu1935Pro | Duzkale et al., 2013 | [76] |
| USH1B | c.5824G>A | p.Gly1942Arg | Duman et al., 2011 | [86] |
| USH1B | c.5944G>A | p.Gly1982Arg | Riazuddin et al., 2008 | [19] |
| USH1B | c.6028G>A | p.Asp2010Asn | Chen et al., 2016 | [87] |
| USH1B | c.6062A>G | p.Lys2021Arg | Roux et al., 2011 | [81] |
| USH1B | c.6326C>T | p.Thr2109Ile | Duzkale et al., 2013. | [76] |
| USH1B | c.6487G>A | p.Gly2163Ser | Janecke et al., 1999 | [77] |
| USH1B | c.6560G>A | p.Gly2187Asp | Bharadwaj et al., 2000 | [82] |
| USH1B | c.6557T>C | p.Leu2186Pro | Bonnet et al., 2011 | [88] |
| USH1B | c.6577C> | p.Leu2193Phe | Duzkale et al., 2013 | [76] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kabahuma, R.I.; Schubert, W.-D.; Labuschagne, C.; Yan, D.; Blanton, S.H.; Pepper, M.S.; Liu, X.Z. Spectrum of MYO7A Mutations in an Indigenous South African Population Further Elucidates the Nonsyndromic Autosomal Recessive Phenotype of DFNB2 to Include Both Homozygous and Compound Heterozygous Mutations. Genes 2021, 12, 274. https://doi.org/10.3390/genes12020274
Kabahuma RI, Schubert W-D, Labuschagne C, Yan D, Blanton SH, Pepper MS, Liu XZ. Spectrum of MYO7A Mutations in an Indigenous South African Population Further Elucidates the Nonsyndromic Autosomal Recessive Phenotype of DFNB2 to Include Both Homozygous and Compound Heterozygous Mutations. Genes. 2021; 12(2):274. https://doi.org/10.3390/genes12020274
Chicago/Turabian StyleKabahuma, Rosemary Ida, Wolf-Dieter Schubert, Christiaan Labuschagne, Denise Yan, Susan Halloran Blanton, Michael Sean Pepper, and Xue Zhong Liu. 2021. "Spectrum of MYO7A Mutations in an Indigenous South African Population Further Elucidates the Nonsyndromic Autosomal Recessive Phenotype of DFNB2 to Include Both Homozygous and Compound Heterozygous Mutations" Genes 12, no. 2: 274. https://doi.org/10.3390/genes12020274
APA StyleKabahuma, R. I., Schubert, W. -D., Labuschagne, C., Yan, D., Blanton, S. H., Pepper, M. S., & Liu, X. Z. (2021). Spectrum of MYO7A Mutations in an Indigenous South African Population Further Elucidates the Nonsyndromic Autosomal Recessive Phenotype of DFNB2 to Include Both Homozygous and Compound Heterozygous Mutations. Genes, 12(2), 274. https://doi.org/10.3390/genes12020274