
IPEX Syndrome: Genetics and Treatment Options


Abstract
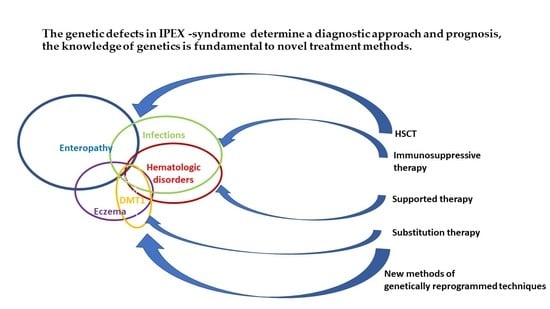
:
1. Introduction
2. Genetics of IPEX Syndrome

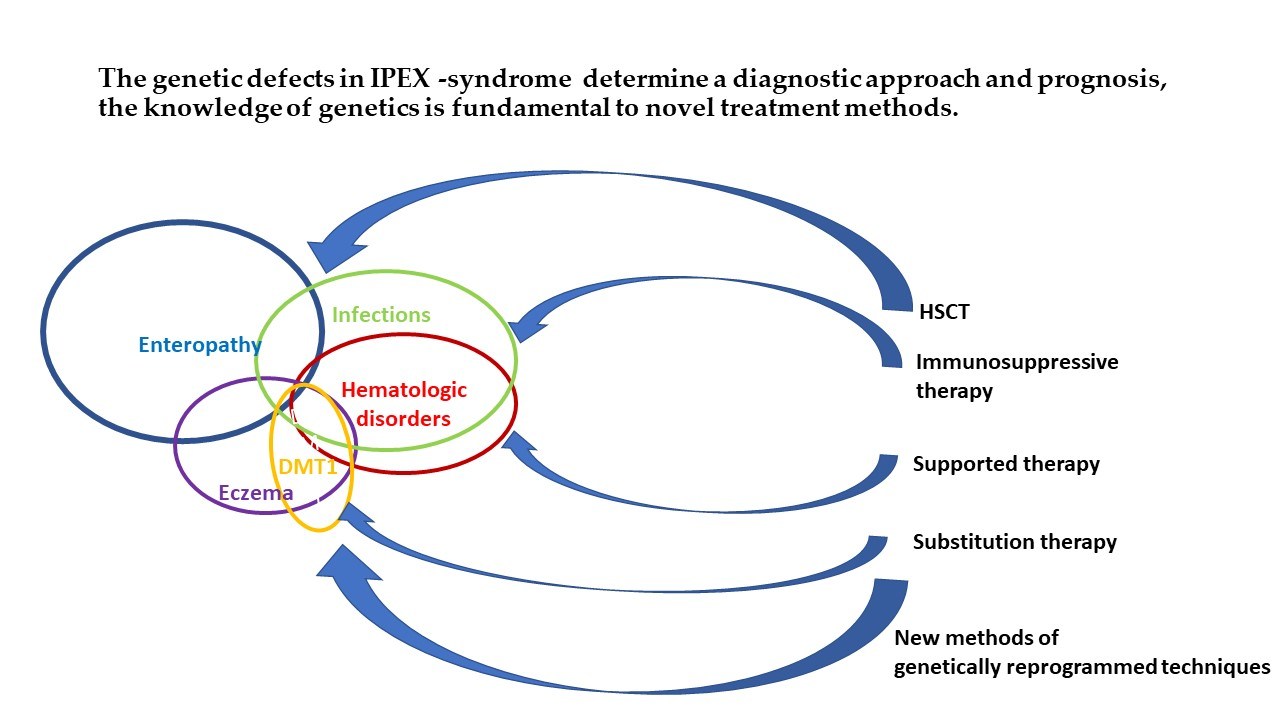{kind=link}
{kind=link}
{kind=link}
| Mutation. | Effect | Patients’ Symptoms |
|---|---|---|
| c.1150G >A [43] | Missing FKH domain | Fetal onset and severe nonviable neonatal patients survived to adolescence |
| c.1189C >T [6,44] | Located in the FKH domain | Fetal onset and severe nonviable neonates |
| c.384A >T mutation [22,45] | Lying within a helix H3 can influence DNA contact | Severe form of IPEX syndrome |
| c.319-32delTC [16] | Deleting two-base pair in the N-terminal domain | Neonatal death |
| c.227delT mutation [46] | Frameshifts and premature stop codons | |
| c.303_304delTT in exon 3 [46] | ||
| p.Glu251 del in LZ-FKH loop [47] | The LZ region is important for forkhead box P3 (FOXP3) to form dimers that impairs FOXP3′s capability to inhibit IL-2 transcription | |
| p. Glu251 del | Influences the inter-subunit salt bridge, leads to a disruption of FOXP3 homodimerization and an impaired inhibiting ability | |
| p.Lys250del [48] | ||
| F371C, F373A, and p.R347H [49,50] | The ability of FOXP3 to inhibit transcription can be impaired | |
| Mutationsoccur in the promoter and5′untranslated region of FOXP3 [6] | Milder form of IPEX syndrome | |
| g.6247–4859del [51] | Encompassing the upstream noncoding exon and the adjacent intron of FOXP3 accumulation of unsliced pre-mRNA and alternatively spliced mRNA | Enteropathy, impressive allergic phenotype |
| p.Arg114Trp, p.Arg347His, p.Lys393Met, and c.1044 +5G >A [52] | Insulin-dependent diabetes without other characteristics of IPEX syndrome | |
| c.1150G >A [53,54] | Changes in the DNA-binding site | Psoriasiform dermatitis and alopecia universalis |
| Alternativesplicing of FOXP3 | Exon 3 encoding blocking retinoic acid-related orphan receptors α and γt, exon 8 encoding a LZ motif necessary to FOXP3 function | Milder IPEX phenotype |
| exon3 as exon 2 [55] | ||
| exon 8 as exon 7 [19] |
3. Epidemiology
4. Clinical Features and Their Pathophysiology
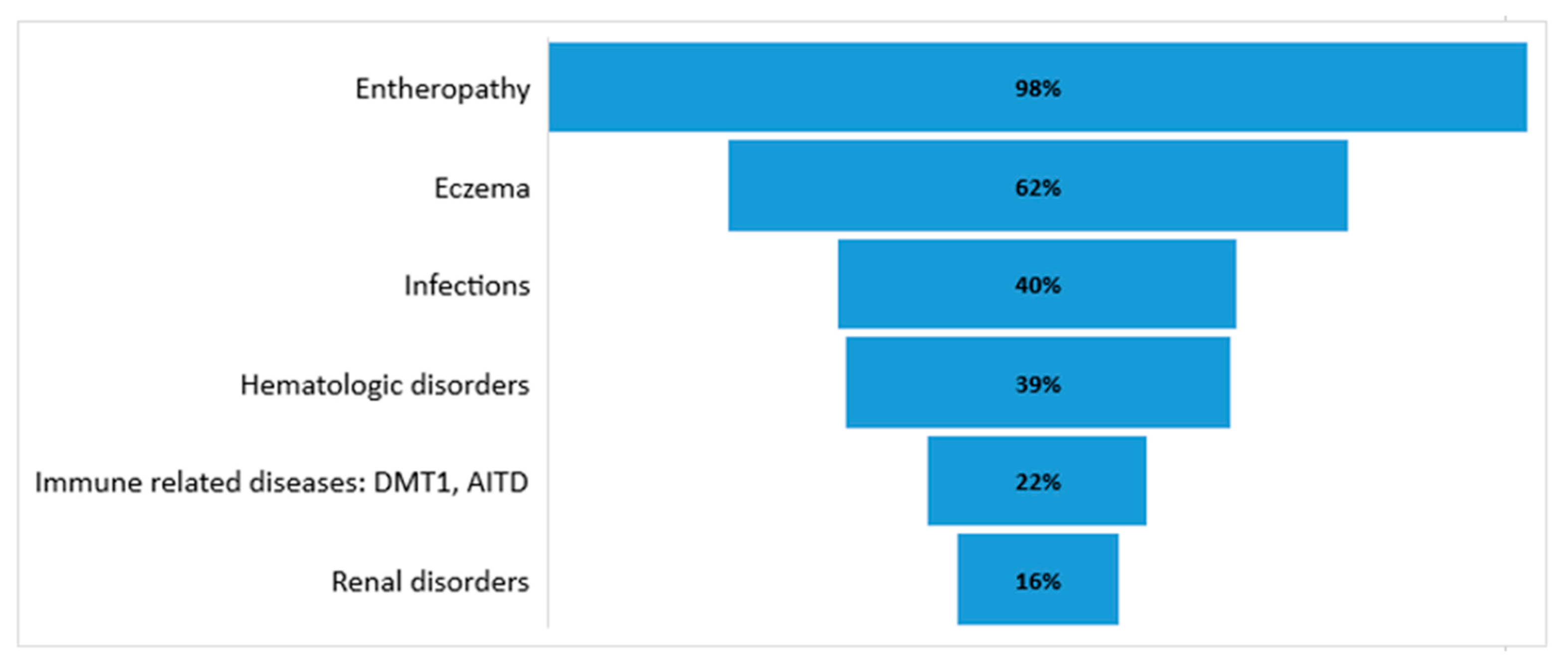
4.1. Enteropathy
4.2. Endocrinopathy
4.3. Dermatitis
4.4. Hematologic Disorders
4.5. Other Autoimmune Diseases
5. Diagnostic Methods
6. Treatment for IPEX Syndrome
New Treatment Perspectives
7. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ADP | Adenosine-5′-diphosphate |
| AITD | Autoimmune thyroid disease |
| cDNA | Complementary DNA |
| CRISPR/Cas9 RGENs | Clustered regularly interspaced short palindromic repeats (CRISPRs) and CRISPR-associated (Cas) systems. CRISPR/Cas systems can be modified and redirected to become powerful tools for genome editing in many species. |
| CTLA4 | Cytotoxic T-lymphocyte-associated protein 4 |
| DMT1 | Diabetes mellitus type 1 |
| DNA | Deoxyribonucleic acid |
| DOCK8 | Dedicator of cytokinesis 8 |
| DSB | DNA double-strand break |
| ETS | Erythroblast transformation-specific (family of transcription factors) |
| FKH domain | Forkhead domain |
| FOXP3 | Forkhead box P3 |
| GAD antibodies | Glutamic acid decarboxylase antibodies |
| GITR | Glucocorticoid-induced TNFR-related protein (belongs to the tumor necrosis factor receptor superfamily (TNFRSF)) |
| HAAs | Antiharmonin autoantibodies |
| HLA system | Human leukocyte antigen system |
| HSCT | Hematopoietic stem cell transplantation |
| IA2 antibodies | Tyrosine phosphatase-related islet antigen 2 |
| IgE | Immunoglobulin E |
| IgG | Immunoglobulin G |
| IgM | Immunoglobulin M |
| IL2RA | Interleukin 2 receptor α chain (also called CD25) |
| IL2RG | Interleukin 2 Receptor Subunit γ |
| IL4 | Interleukin 4 |
| IPEX syndrome | Immune dysregulation, polyendocinopathy, enteropathy, X-linked syndrome |
| LRBA | Lipopolysaccharide (LPS)-responsive and beige-like anchor protein |
| LZ | leucine zipper |
| mncRNAs | Micro-noncoding ribonucleic acids |
| mRNA | Messenger RNA |
| ncRNAs | Noncoding ribonucleic acids |
| NFAT | Nuclear factor of activated T cells |
| PBMC | Peripheral blood mononuclear cell |
| PRR | N-terminal proline-rich region |
| PTPN22 | Protein tyrosine phosphatase, non-receptor type 22 |
| RORγ | Retinoic acid-related orphan receptor γ (master activator of cholesterol biosynthesis) |
| RUNX | Runt-related transcription factors |
| SCID-XI | Severe combined immunodeficiency |
| STAT1 | Signal transducer and activator of transcription 1 |
| STAT3 | Signal transducer and activator of transcription 3 |
| STAT5b | Signal transducer and activator of transcription 5B |
| Tconv cells | Conventional T cells that differentiate into effector cells during immune responses |
| TG Ab | Thyroglobulin antibody |
| TGF | Transforming growth factor |
| TNFRSF18 | Tumor necrosis factor receptor superfamily member 18 (known as GITR) |
| TPO Ab | Thyroperoxodase antibody |
| Treg | Regulatory T cell |
| TTC37 | Tetratricopeptide repeat domain 37 |
| TTC7A | Tetratricopeptide repeat domain 7A |
| VAAs | Antivillin autoantibodies |
| WT-FOXP3 | Wild-type FOXP3 |
| XCI | X-chromosome inactivation |
| ZF | Central zinc finger |
| ZnT8 antibodies | Zinc transporter 8 antibodies |
References
- Fontenot, J.D.; Gavin, M.A.; Rudensky, A.Y. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat. Immunol. 2003, 4, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Fontenot, J.D.; Rasmussen, J.P.; Williams, L.M.; Dooley, J.L.; Farr, A.G.; Rudensky, A.Y. Regulatory T Cell Lineage Specification by the Forkhead Transcription Factor Foxp3. Immunity 2005, 22, 329–341. [Google Scholar] [CrossRef]
- Khattri, R.; Cox, T.; Yasayko, S.-A.; Ramsdell, F. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat. Immunol. 2003, 4, 337–342. [Google Scholar] [CrossRef]
- Hori, S.; Nomura, T.; Sakaguchi, S. Control of Regulatory T Cell Development by the Transcription Factor Foxp3. Science 2003, 299, 1057–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allenspach, E.J.; Finn, L.S.; Rendi, M.H.; Eken, A.; Singh, A.K.; Oukka, M.; Taylor, S.D.; Altman, M.C.; Fligner, C.L.; Ochs, H.D.; et al. Absence of functional fetal regulatory T cells in humans causes in utero organ-specific autoimmunity. J. Allergy Clin. Immunol. 2017, 140, 616–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gambineri, E.; Mannurita, S.C.; Hagin, D.; Vignoli, M.; Anover-Sombke, S.; DeBoer, S.; Segundo, G.R.S.; Allenspach, E.J.; Favre, C.; Ochs, H.D.; et al. Clinical, Immunological, and Molecular Heterogeneity of 173 Patients With the Phenotype of Immune Dysregulation, Polyendocrinopathy, Enteropathy, X-Linked (IPEX) Syndrome. Front. Immunol. 2018, 9, 2411. [Google Scholar] [CrossRef]
- Zhou, X.; Jeker, L.T.; Fife, B.T.; Zhu, S.; Anderson, M.S.; McManus, M.T.; Bluestone, J.A. Selective miRNA disruption in T reg cells leads to uncontrolled autoimmunity. J. Exp. Med. 2008, 205, 1983–1991. [Google Scholar] [CrossRef]
- Zhou, X.; Bailey-Bucktrout, S.L.; Jeker, L.T.; Penaranda, C.; Martinez-Llordella, M.; Ashby, M.; Nakayama, M.; Rosenthal, W.; Bluestone, J.A. Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nat. Immunol. 2009, 10, 1000–1007. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Yi, G.; Liu, X.; Zhu, F.; Zhao, A.; Wang, A.; Zhu, R.; Chen, Z.; Zhao, B.; Fang, S.; et al. Ring finger protein 31-mediated atypical ubiquitination stabilizes forkhead box P3 and thereby stimulates regulatory T-cell function. J. Biol. Chem. 2018, 293, 20099–20111. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Lu, Y.; Wang, S.; Han, Z.; Zhu, F.; Ni, Y.; Liang, R.; Zhang, Y.; Leng, Q.; Wei, G.; et al. USP21 prevents the generation of T-helper-1-like Treg cells. Nat. Commun. 2016, 7, 13559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Barbi, J.; Bu, S.; Yang, H.Y.; Li, Z.; Gao, Y.; Jinasena, D.; Fu, J.; Lin, F.; Chen, C.; et al. The ubiquitin ligase Stub1 negatively modulates regulatory T cell suppressive activity by promoting degradation of the transcription factor Foxp3. Immunity 2013, 39, 272–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Q.; Liu, X.; Zhang, Y.; Huang, J.; Li, D.; Li, B. Molecular feature and therapeutic perspectives of immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome. J. Genet. Genom. 2020, 47, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Seo, W.; Taniuchi, I. The Roles of RUNX Family Proteins in Development of Immune Cells. Mol. Cells 2020, 43, 107–113. [Google Scholar] [CrossRef]
- Wildin, R.S.; Smyk-Pearson, S.; Filipovich, A.H. Clinical and molecular features of the immunodysregulation, polyendocrinopathy, enteropathy, X linked (IPEX) syndrome. J. Med. Genet. 2002, 39, 537–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michels, A.W.; Gottlieb, P.A. Autoimmune polyglandular syndromes. Nat. Rev. Endocrinol. 2010, 6, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Bacchetta, R.; Barzaghi, F.; Roncarolo, M.-G. From IPEX syndrome to FOXP3 mutation: A lesson on immune dysregulation. Ann. N. Y. Acad. Sci. 2018, 1417, 5–22. [Google Scholar] [CrossRef] [PubMed]
- Verbsky, J.; Chatila, T.A. Immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) and IPEX-related disorders. Curr. Opin. Pediatr. 2013, 25, 708–714. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.L.; Yoshioka, R.; Kiyosawa, H.; Barker, D.F.; Fain, P.R.; Shigeoka, A.O.; Chance, P.F. Chance X-Linked syndrome of polyendocrinopathy, immune dysfunction, and diarrhea maps to Xp11.23-Xq13.3. Am. J. Hum. Genet. 2000, 66, 461–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mailer, R.K.; Falk, K.; Rotzschke, O. Absence of leucine zipper in the natural FOXP3Delta2Delta7 isoform does not affect dimerization but abrogates suppressive capacity. PLoS ONE 2009, 4, e6104. [Google Scholar] [CrossRef] [Green Version]
- Ziegler, S.F. FOXP3: Of mice and men. Annu. Rev. Immunol. 2006, 24, 209–226. [Google Scholar] [CrossRef]
- Lopes, J.E.; Torgerson, T.R.; Schubert, L.A.; Anover, S.D.; Ocheltree, E.L.; Ochs, H.D.; Ziegler, S.F. Analysis of FOXP3 reveals multiple domains required for its function as a transcriptional repressor. J. Immunol. 2006, 177, 3133–3142. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Borde, M.; Heissmeyer, V.; Feuerer, M.; Lapan, A.D.; Stroud, J.C.; Bates, D.L.; Guo, L.; Han, A.; Ziegler, S.F.; et al. FOXP3 Controls Regulatory T Cell Function through Cooperation with NFAT. Cell 2006, 126, 375–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konopacki, C.; Pritykin, Y.; Rubtsov, Y.; Leslie, C.S.; Rudensky, A.Y. Transcription factor Foxp1 regulates Foxp3 chromatin binding and coordinates regulatory T cell function. Nat. Immunol. 2019, 20, 232–242. [Google Scholar] [CrossRef]
- Zhou, L.; Lopes, J.E.; Chong, M.M.; Ivanov, I.I.; Min, R.; Victora, G.D.; Shen, Y.; Du, J.; Rubtsov, Y.P.; Rudensky, A.Y.; et al. TGF-β-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature 2008, 453, 236–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lal, G.; Bromberg, J.S. Epigenetic mechanisms of regulation of Foxp3 expression. Blood 2009, 114, 3727–3735. [Google Scholar] [CrossRef] [Green Version]
- Kitagawa, Y.; Ohkura, N.; Sakaguchi, S. Epigenetic control of thymic Treg-cell development. Eur. J. Immunol. 2014, 45, 11–16. [Google Scholar] [CrossRef]
- Floess, S.; Freyer, J.; Siewert, C.; Baron, U.; Olek, S.; Polansky, J.; Schlawe, K.; Chang, H.-D.; Bopp, T.; Schmitt, E.; et al. Epigenetic Control of the foxp3 Locus in Regulatory T Cells. PLoS Biol. 2007, 5, e38. [Google Scholar] [CrossRef] [PubMed]
- Qin, A.; Wen, Z.; Zhou, Y.; Li, Y.; Li, Y.; Luo, J.; Ren, T.; Xu, L. MicroRNA-126 regulates the induction and function of CD4+ Foxp3+ regulatory T cells through PI3K/AKT pathway. J. Cell. Mol. Med. 2013, 17, 252–264. [Google Scholar] [CrossRef]
- Liu, X.; Robinson, S.N.; Setoyama, T.; Tung, S.S.; D’Abundo, L.; Shah, M.Y.; Yang, H.; Yvon, E.; Shah, N.; Yang, H.; et al. FOXP3 is a direct target of miR15a/16 in umbilical cord blood regulatory T cells. Bone Marrow Transpl. 2014, 49, 793–799. [Google Scholar] [CrossRef] [Green Version]
- Van Loosdregt, J.; Vercoulen, Y.; Guichelaar, T.; Gent, Y.Y.J.; Beekman, J.M.; Van Beekum, O.; Brenkman, A.B.; Hijnen, D.-J.; Mutis, T.; Kalkhoven, E.; et al. Regulation of Treg functionality by acetylation-mediated Foxp3 protein stabilization. Blood 2010, 115, 965–974. [Google Scholar] [CrossRef] [PubMed]
- Zemmour, D.; Pratama, A.; Loughhead, S.M.; Mathis, D.; Benoist, C. Flicr, a long noncoding RNA, modulates Foxp3 expression and autoimmunity. Proc. Natl. Acad. Sci. USA 2017, 114, E3472–E3480. [Google Scholar] [CrossRef] [Green Version]
- Brajic, A.; Franckaert, D.; Burton, O.; Bornschein, S.; Calvanese, A.L.; Demeyer, S.; Cools, J.; Dooley, J.; Schlenner, S.; Liston, A. The long non-coding RNA flatr anticipates Foxp3 expression in regulatory T cells. Front. Immunol. 2018, 9, 1989. [Google Scholar] [CrossRef]
- Louie, R.J.; Tan, Q.K.; Gilner, J.B.; Rogers, R.C.; Younge, N.; Wechsler, S.B.; McDonald, M.T.; Gordon, B.; Saski, C.A.; Jones, J.R.; et al. Novel pathogenic variants in FOXP3 in fetuses with echogenic bowel and skin desquamation identified by ultrasound. Am. J. Med. Genet. 2017, 173, 1219–1225. [Google Scholar] [CrossRef] [Green Version]
- Caudy, A.A.; Reddy, S.T.; Chatila, T.; Atkinson, J.P.; Verbsky, J.W. CD25 deficiency causes an immune dysregulation, polyendocrinopathy, enteropathy, X-linked–like syndrome, and defective IL-10 expression from CD4 lymphocytes. J. Allergy Clin. Immunol. 2007, 119, 482–487. [Google Scholar] [CrossRef]
- Passerini, L.; de Sio, F.R.S.; Porteus, M.H.; Bacchetta, R. Gene/Cell Therapy Approaches for Immune Dysregulation Polyendocrinopathy Enteropathy X-Linked Syndrome. Available online: https://www.researchgate.net/publication/266570661_GeneCell_Therapy_Approaches_for[_Immune_Dysregulation_Polyendocrinopathy_Enteropathy_X-Linked_Syndrome (accessed on 9 December 2020).
- Sharfe, N.; Shahar, M.; Roifman, C.M. An interleukin-2 receptor γ chain mutation with normal thymus morphology. J. Clin. Investig. 1997, 100, 3036–3043. [Google Scholar] [CrossRef]
- Goudy, K.; Aydin, D.; Barzaghi, F.; Gambineri, E.; Vignoli, M.; Mannurita, S.C.; Doglioni, C.; Ponzoni, M.; Cicalese, M.P.; Assanelli, A.; et al. Human IL2RA null mutation mediates immunodeficiency with lymphoproliferation and autoimmunity. Clin. Immunol. 2013, 146, 248–261. [Google Scholar] [CrossRef] [Green Version]
- Cohen, A.C.; Nadeau, K.C.; Tu, W.; Hwa, V.; Dionis, K.; Bezrodnik, L.; Teper, A.; Gaillard, M.; Heinrich, J.; Krensky, A.M.; et al. Cutting Edge: Decreased Accumulation and Regulatory Function of CD4+CD25high T Cells in Human STAT5b Deficiency. J. Immunol. 2006, 177, 2770–2774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernasconi, A.; Marino, R.; Ribas, A.; Rossi, J.; Ciaccio, M.; Oleastro, M.; Ornani, A.; Paz, R.; Rivarola, M.A.; Zelazko, M.; et al. Characterisation of immunodeficiency in a patient with growth hormone insensitivity secondary to a novel STAT5b gene mutation. Pediatrics 2006, 118, e1584–e1592. [Google Scholar] [CrossRef] [PubMed]
- Kofoed, E.M.; Hwa, V.; Little, B.; Woods, K.A.; Buckway, C.K.; Tsubaki, J.; Pratt, K.L.; Bezrodnik, L.; Jasper, H.; Tepper, A.; et al. Growth Hormone Insensitivity Associated with aSTAT5bMutation. New Engl. J. Med. 2003, 349, 1139–1147. [Google Scholar] [CrossRef]
- Hwa, V.; Little, B.; Adiyaman, P.; Kofoed, E.M.; Pratt, K.L.; Ocal, G.; Berberoglu, M.; Rosenfeld, R.G. Severe Growth Hormone Insensitivity Resulting from Total Absence of Signal Transducer and Activator of Transcription 5b. J. Clin. Endocrinol. Metab. 2005, 90, 4260–4266. [Google Scholar] [CrossRef] [PubMed]
- Lohr, N.J.; Molleston, J.P.; Strauss, K.A.; Torres-Martinez, W.; Sherman, E.A.; Squires, R.H.; Rider, N.L.; Chikwava, K.R.; Cummings, O.W.; Morton, D.H.; et al. Human ITCH E3 ubiquitin ligas deficiency causes syndromic multisystem autoimmune disease. Am. J. Hum. Genet. 2010, 86, 447–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barzaghi, F.; Passerini, L.; Bacchetta, R. Immune dysregulation, polyendocrinopathy, enteropathy, x-linked syndrome: A paradigm of immunodeficiency with autoimmunity. Front. Immunol. 2012, 3, 211. [Google Scholar] [CrossRef] [Green Version]
- Nieves, D.S.; Phipps, R.P.; Pollock, S.J.; Ochs, H.D.; Zhu, Q.; Scott, G.A.; Ryan, C.K.; Kobayashi, I.; Rossi, T.M.; Goldsmith, L.A. Dermatologic and Immunologic Findings in the Immune Dysregulation, Polyendocrinopathy, Enteropathy, X-linked Syndrome. Arch. Derm. 2004, 140, 466–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zama, D. Late-onset of immunodysregulation, polyendocrinopathy, enteropathy, x-linked syndrome (IPEX) with intractable diarrhea. Ital. J. Pediatr. 2014, 40, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, I.; Shiari, R.; Yamada, M.; Kawamura, N.; Okano, M.; Yara, A.; Iguchi, A.; Ishikawa, N.; Ariga, T.; Sakiyama, Y.; et al. Novel mutations of FOXP3 in two Japanese patients with immune dysregulation, polyendocrinopathy, enteropathy, X linked syndrome (IPEX). J. Med. Genet. 2001, 38, 874–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Samanta, A.; Song, X.; Iacono, K.T.; Brennan, P.; Chatila, T.A.; Roncador, G.; Banham, A.H.; Riley, J.L.; Wang, Q.; et al. FOXP3 is a homo-oligomer and a component of a supramolecular regulatory complex disabled in the human XLAAD/IPEX autoimmune disease. Int. Immunol. 2007, 19, 825–835. [Google Scholar] [CrossRef]
- Song, X.; Li, B.; Xiao, Y.; Chen, C.; Wang, Q.; Liu, Y.; Berezov, A.; Xu, C.; Gao, Y.; Li, Z.; et al. Structural and biological features of FOXP3 dimerization relevant to regulatory T cell function. Cell Rep. 2012, 1, 665–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Chen, C.; Zhang, Z.; Liu, C.C.; Johnson, M.E.; Espinoza, C.A.; Edsal, L.E.; Ren, B.; Zhou, X.J.; Grant, S. DNA binding by FOXP3 domain-swapped dimer suggests mechanisms of long-range chromosomal interactions. Nucl. Acid. Res. 2015, 43, 1268–1282. [Google Scholar] [CrossRef] [Green Version]
- Band, H.S.; Bandukwala, H.S.; Wu, Y.; Feuerer, M.; Chen, Y.; Barboza, B.; Ghosh, S.; Stroud, J.C.; Benoist, C.; Mathis, D.; et al. Structure of a domain-swapped FOXP3 dimer on DNA and its function in regulatory T cells. Immunity 2011, 34, 479–491. [Google Scholar]
- Torgerson, T.R.; Linane, A.; Moes, N.; Anover, S.; Mateo, V.; Rieux–Laucat, F.; Hermine, O.; Vijay, S.; Gambineri, E.; Cerf–Bensussan, N.; et al. Severe Food Allergy as a Variant of IPEX Syndrome Caused by a Deletion in a Noncoding Region of the FOXP3 Gene. Gastroenterology 2007, 132, 1705–1717. [Google Scholar] [CrossRef]
- Hwang, J.L.; Park, S.-Y.; Ye, H.; Sanyoura, M.; Pastore, A.N.; Carmody, D.; Del Gaudio, D.; Wilson, J.F.; Hanis, C.L.; Liu, X.; et al. FOXP3 mutations causing early-onset insulin-requiring diabetes but without other features of immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome. Pediatr. Diabetes 2017, 19, 388–392. [Google Scholar] [CrossRef]
- Di Nunzio, S.; Cecconi, M.; Passerini, L.; McMurchy, A.N.; Baron, U.; Turbachova, I.; Vignola, S.; Valencic, E.; Tommasini, A.; Junker, A.; et al. Wild-type FOXP3 is selectively active in CD4+CD25hi regulatory T cells of healthy female carriers of different FOXP3 mutations. Blood 2009, 114, 4138–4141. [Google Scholar] [CrossRef] [PubMed]
- Rae, W.; Gao, Y.; Bunyan, D.; Holden, S.; Gilmour, K.; Patel, S.; Wellesley, D.; Williams, A. A novel FOXP3 mutation causing fetal akinesia and recurrent male miscarriages. Clin. Immunol. 2015, 161, 284–285. [Google Scholar] [CrossRef] [Green Version]
- Ge, T.; Wang, Y.; Che, Y.; Xiao, Y.; Zhang, T. Atypical late-onset immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome with intractable diarrhoea: A case report. Front. Pediatr. 2017, 5, 267. [Google Scholar] [CrossRef] [Green Version]
- Park, J.H.; Lee, K.H.; Jeon, B.; Ochs, H.D.; Lee, J.S.; Gee, H.Y.; Seo, S.; Geum, D.; Piccirillo, C.A.; Eisenhut, M.; et al. Immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome: A systematic review. Autoimmun. Rev. 2020, 19, 102526. [Google Scholar] [CrossRef]
- Lampasona, V.; Passerini, L.; Barzaghi, F.; Lombardoni, C.; Bazzigaluppi, E.; Brigatti, C.; Bacchetta, R.; Bosi, E. Autoantibodies to harmonin and villin are diagnostic markers in children with IPEX syndrome. PLoS ONE 2013, 8, e78664. [Google Scholar] [CrossRef] [PubMed]
- Kadakia, S.; Farnaes, L.; Dimmock, D.; Chowdhury, S.; Ding, Y.; Anderson, E.J.; Kingsmore, S.; Newfield, R.S. Diagnosis and treatment of a boy with IPEX syndrome presenting with diabetes in early infancy. Clin. Case Rep. 2019, 7, 2123–2127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halabi-Tawil, M.; Ruemmele, F.; Fraitag, S.; Rieux-Laucat, F.; Neven, B.; Brousse, N.; De Prost, Y.; Fischer, A.; Goulet, O.; Bodemer, C. Cutaneous manifestations of immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome. Br. J. Derm. 2009, 160, 645–651. [Google Scholar] [CrossRef]
- Bousfiha, A.; Jeddane, L.; Picard, C.; Ailal, F.; Gaspar, H.B.; Al-Herz, W.; Chatila, T.; Crow, Y.J.; Cunningham-Rundles, C.; Etzioni, A.; et al. The 2017 IUIS phenotypic classification for primary immunodeficiencies. J. Clin. Immunol. 2018, 38, 129–143. [Google Scholar] [CrossRef] [Green Version]
- Hatzimichael, E.; Tuthill, M. Hematopoietic stem cell transplantation. Stem Cells Cloning 2010, 3, 105–117. [Google Scholar]
- Lucas, K.G.; Ungar, D.; Comito, M.; Bayerl, M.; Groh, B. Submyeloablative cord blood transplantation corrects clinical defects seen in IPEX syndrome. Bone Marrow Transpl. 2006, 39, 55–56. [Google Scholar] [CrossRef]
- Zhan, H.; Sinclair, J.; Adams, S.; Cale, C.M.; Murch, S.; Perroni, L.; Davies, G.; Amrolia, P.; Qasim, W. Immune reconstitution and recovery of FOXP3 (forkhead box P3)-expressing T cells after transplantation for IPEX (immune dysregulation, polyendocrinopathy, enteropathy, X-linked) syndrome. Pediatrics 2008, 121, e998–e1002. [Google Scholar] [CrossRef] [PubMed]
- Horino, S.; Sasahara, Y.; Sato, M.; Niizuma, H.; Kumaki, S.; Abukawa, D.; Sato, A.; Imaizumi, M.; Kanegane, H.; Kamachi, Y.; et al. Selective expansion of donor-derived regulatory T cells after allogeneic bone marrow transplantation in a patient with IPEX syndrome. Pediatr. Transpl. 2014, 18, E25–E30. [Google Scholar] [CrossRef]
- Seidel, M.G.; Fritsch, G.; Lion, T.; Jürgens, B.; Heitger, A.; Bacchetta, R.; Lawitschka, A.; Peters, C.; Gadner, H.; Matthes-Martin, S. Selective engraftment of donor CD4+25high FOXP3-positive T cells in IPEX syndrome after nonmyeloablative hematopoietic stem cell transplantation. Blood 2009, 113, 5689–5691. [Google Scholar] [CrossRef] [PubMed]
- Bindl, L.; Torgerson, T.; Perroni, L.; Youssef, N.; Ochs, H.D.; Goulet, O.; Ruemmele, F.M. Successful use of the new immune-suppressor sirolimus in IPEX (immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome). J. Pediatr. 2005, 147, 256–259. [Google Scholar] [CrossRef]
- Passerini, L.; Di Nunzio, S.; Gregori, S.; Gambineri, E.; Cecconi, M.; Seidel, M.G.; Cazzola, G.; Perroni, L.; Tommasini, A.; Vignola, S.; et al. Functional type 1 regulatory T cells develop regardless of FOXP3 mutations in patients with IPEX syndrome. Eur. J. Immunol. 2011, 41, 1120–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passerini, L.; Baccheta, R. Forkhead-Box-P3 Gene Transfer in Human CD4+ T Conventional Cells for the Generation of Stable and Efficient Regulatory T Cells, Suitable for Immune Modulatory Therapy. Front. Immunol. 2017, 8, 1282. [Google Scholar] [CrossRef] [PubMed]
- Passerini, L.; Mel, E.R.; Sartirana, C.; Fousteri, G.; Bondanza, A.; Naldini, L.; Roncarolo, M.G.; Baccheta, R. CD4+ T Cells from IPEX Patients Convert into Functional and Stable Regulatory T Cells by FOXP3 Gene Transfer. Sci. Transl. Med. 2013, 5, 215ra174. [Google Scholar] [CrossRef] [PubMed]
- Aarts-Riemens, T.; Emmelot, M.E.; Verdonck, L.F.; Mutis, T. Forced overexpression of either of the two common human Foxp3 isoforms can induce regulatory T cells from CD4+CD25– cells. Eur. J. Immunol. 2008, 38, 1381–1390. [Google Scholar] [CrossRef]
- Liu, Y.; Tran, D.Q.; Lindsey, J.W.; Rhoads, J.M. The Association of Gut Microbiota and Treg Dysfunction in Autoimmune Diseases. Adv. Exp. Med. Biol. 2021, 1278, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Allan, S.E.; Alstad, A.N.; Merindol, N.; Crellin, N.K.; Amendola, M.; Baccheta, R.; Naldini, L.; Roncarolo, M.G.; Soudeyns, H.; Levings, M.K. Generation of Potent and Stable Human CD4+ T Regulatory Cells by Activation-independent Expression of FOXP3. Mol. Ther. 2008, 16, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Panchal, N.; Ghosh, S.; Booth, C. T cell gene therapy to treat immunodeficiency. Br. J. Haematol. 2021, 192, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Oswald-Richter, K.; Grill, S.M.; Shariat, N.; Leelawong, M.; Sundrud, M.S.; Haas, D.W.; Unutmaz, D. HIV infection of naturally occurring and genetically reprogrammed human regulatory T-cells. PLoS Biol. 2004, 2, E198. [Google Scholar] [CrossRef] [PubMed]
- Hombach, A.A.; Kofler, D.; Rappl, G.; Abken, H. Redirecting human CD4+CD25+ regulatory T cells from the peripheral blood with pre-defined target specificity. Gene Ther. 2009, 16, 1088–1096. [Google Scholar] [CrossRef] [PubMed]
- Brusko, T.M.; Koya, R.C.; Zhu, S.; Lee, M.R.; Putnam, A.L.; McClymont, S.A.; Nishimura, M.I.; Han, S.; Chang, L.-J.; Atkinson, M.A.; et al. Human Antigen-Specific Regulatory T Cells Generated by T Cell Receptor Gene Transfer. PLoS ONE 2010, 5, e11726. [Google Scholar] [CrossRef]
- Kohn, D.B. Gene therapy for blood diseases. Curr. Opin. Biotechnol. 2019, 60, 39–45. [Google Scholar] [CrossRef]
- Genovese, P.; Schiroli, G.; Escobar, G.; Di Tomaso, T.; Firrito, C.; Calabria, A.; Moi, D.; Mazzieri, R.; Bonini, C.; Holmes, M.; et al. Targeted genome editing in human repopulating haematopoietic stem cells. Nat. Cell Biol. 2014, 510, 235–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voit, R.A.; Hendel, A.; Pruett-Miller, S.M.; Porteus, M.H. Nuclease-mediated gene editing by homologous recombination of the human globin locus. Nucleic Acids Res. 2014, 42, 1365–1378. [Google Scholar] [CrossRef] [Green Version]
- Ousterout, D.G.; Perez-Pinera, P.; Thakore, P.I.; Kabadi, A.M.; Brown, M.T.; Qin, X.; Fedrigo, O.; Mouly, V.; Tremblay, J.P.; Gersbach, C.A. Reading frame correction by targeted genome editing restores dystrophin expression in cells from Duchenne muscular dystrophy patients. Mol. Ther. 2013, 21, 1718–1726. [Google Scholar] [CrossRef] [Green Version]
- Charbonnier, L.-M.; Cui, Y.; Stephen-Victor, E.; Harb, H.; Lopez, D.; Bleesing, J.J.; Garcia-Lloret, M.I.; Chen, K.; Ozen, A.; Carmeliet, P.; et al. Functional reprogramming of regulatory T cells in the absence of Foxp3. Nat. Immunol. 2019, 20, 1208–1219. [Google Scholar] [CrossRef]
- Hsu, P.D.; Lander, E.S.; Zhang, F. Development and Applications of CRISPR-Cas9 for Genome Engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ben-Skowronek, I. IPEX Syndrome: Genetics and Treatment Options. Genes 2021, 12, 323. https://doi.org/10.3390/genes12030323
Ben-Skowronek I. IPEX Syndrome: Genetics and Treatment Options. Genes. 2021; 12(3):323. https://doi.org/10.3390/genes12030323
Chicago/Turabian StyleBen-Skowronek, Iwona. 2021. "IPEX Syndrome: Genetics and Treatment Options" Genes 12, no. 3: 323. https://doi.org/10.3390/genes12030323
APA StyleBen-Skowronek, I. (2021). IPEX Syndrome: Genetics and Treatment Options. Genes, 12(3), 323. https://doi.org/10.3390/genes12030323