
miRNAture—Computational Detection of microRNA Candidates



Abstract
:1. Introduction
2. Results
2.1. Architecture of miRNAture
2.2. Annotation of let-7 on Chordate Genomes
2.2.1. Simulation of Artificial let-7 Instances
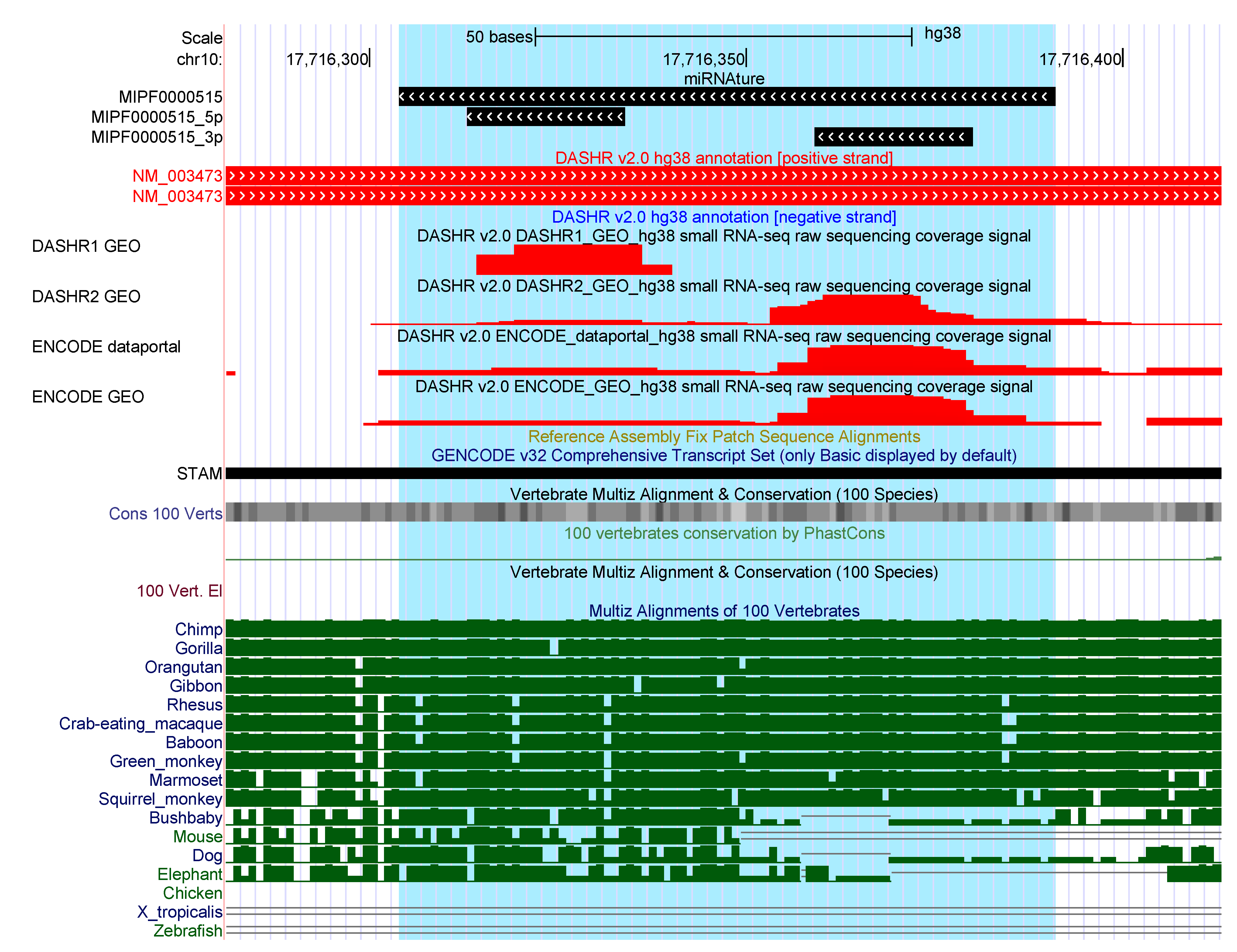
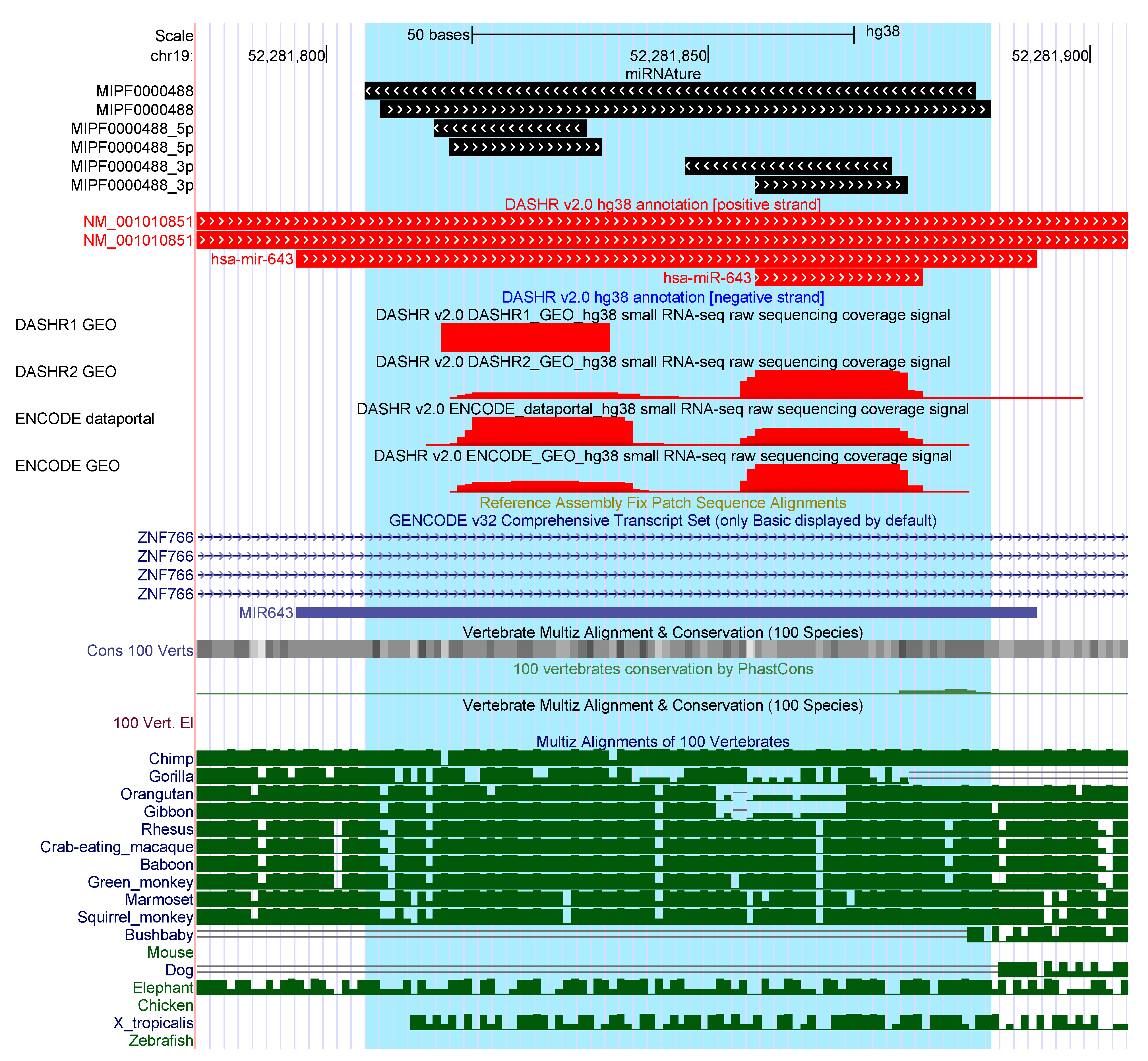
2.3. Annotation of Human Bona Fide miRNAs
2.3.1. Additional Candidates
2.3.2. Additional miRNAture Candidates without Annotation Overlaps
2.3.3. Strand-Mismatch Candidates
2.3.4. Missing Candidates
3. Discussion
4. Methods
4.1. Specific Filters on miRNAture
4.2. Genomes
4.3. Curation of the let-7 Family
4.4. Curation of Human miRNA Families
4.5. miRNA Annotation Using miRNAture
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| miRNA | microRNA |
| ncRNA | non-coding RNA |
| CM | covariance model |
| HMM | Hidden Markov Models |
References
- Ameres, S.L.; Zamore, P.D. Diversifying microRNA sequence and function. Nat. Rev. Mol. Cell Biol. 2013, 14, 475–488. [Google Scholar] [CrossRef]
- Moran, Y.; Agron, M.; Praher, D.; Technau, U. The Evolutionary Origin of Plant and Animal microRNAs. Nat. Ecol. Evol. 2017, 1, 27. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.C.; Doudna, J.A. Molecular Mechanisms of RNA Interference. Annu. Rev. Biophys. 2013, 42, 217–239. [Google Scholar] [CrossRef] [Green Version]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. miRBase: From microRNA sequences to function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef] [PubMed]
- Alles, J.; Fehlmann, T.; Fischer, U.; Backes, C.; Galata, V.; Minet, M.; Hart, M.; Abu-Halima, M.; Grässer, F.A.; Lenhof, H.P.; et al. An estimate of the total number of true human miRNAs. Nucleic Acids Res. 2019, 47, 3353–3364. [Google Scholar] [CrossRef] [Green Version]
- Fromm, B.; Billipp, T.; Peck, L.E.; Johansen, M.; Tarver, J.E.; King, B.L.; Newcomb, J.M.; Sempere, L.F.; Flatmark, K.; Hovig, E.; et al. A Uniform System for the Annotation of Vertebrate microRNA Genes and the Evolution of the Human microRNAome. Ann. Rev. Genet. 2015, 49, 213–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartel, D.P. Metazoan MicroRNAs. Cell 2018, 173, 20–51. [Google Scholar] [CrossRef] [Green Version]
- Okamura, K. Diversity of animal small RNA pathways and their biological utility. Wiley Interdiscip Rev. RNA 2012, 3, 351–368. [Google Scholar] [CrossRef] [PubMed]
- Fromm, B.; Domanska, D.; Høye, E.; Ovchinnikov, V.; Kang, W.; Aparicio-Puerta, E.; Johansen, M.; Flatmark, K.; Mathelier, A.; Hovig, E.; et al. MirGeneDB 2.0: The metazoan microRNA complement. Nucleic Acids Res. 2019, 48, D132–D141. [Google Scholar] [CrossRef] [Green Version]
- Velandia Huerto, C.A.; Yazbeck, A.M.; Schor, J.; Stadler, P.F. Evolution and Phylogeny of MicroRNAs—Protocols, Pitfalls, and Problems. In miRNomics: MicroRNA Biology and Computational Analysis, 2nd ed.; Allmer, J., Yousef, M., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2021; Volume 2257, in press. [Google Scholar]
- Price, N.; Cartwright, R.A.; Sabath, N.; Graur, D.; Azevedo, R.B. Neutral evolution of robustness in Drosophila microRNA precursors. Mol. Biol. Evol. 2011, 28, 2115–2123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hertel, J.; Lindemeyer, M.; Missal, K.; Fried, C.; Tanzer, A.; Flamm, C.; Hofacker, I.L.; Stadler, P.F.; The Students of Bioinformatics Computer Labs 2004 and 2005. The Expansion of the Metazoan MicroRNA Repertoire. BMC Genom. 2006, 7, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sempere, L.F.; Cole, C.N.; McPeek, M.A.; Peterson, K.J. The phylogenetic distribution of metazoan microRNAs: Insights into evolutionary complexity and constraint. J. Exp. Zool. B Mol. Dev. Evol. 2006, 306B, 575–588. [Google Scholar] [CrossRef] [PubMed]
- Heimberg, A.M.; Cowper-Sal·lari, R.; Sémon, M.; Donoghue, P.C.; Peterson, K.J. MicroRNAs reveal the interrelationships of hagfish, lampreys, and gnathostomes and the nature of the ancestral vertebrate. Proc. Natl. Acad. Sci. USA 2010, 107, 19379–19383. [Google Scholar] [CrossRef] [Green Version]
- Wheeler, B.M.; Heimberg, A.M.; Moy, V.N.; Sperling, E.A.; Holstein, T.W.; Heber, S.; Peterson, K.J. The deep evolution of metazoan microRNAs. Evol. Dev. 2009, 11, 50–68. [Google Scholar] [CrossRef]
- Tarver, J.E.; Donoghue, P.C.J.; Peterson, K.J. Do miRNAs have a deep evolutionary history? Bioessays 2012, 34, 857–866. [Google Scholar] [CrossRef]
- Tarver, J.E.; Taylor, R.S.; Puttick, M.N.; Lloyd, G.T.; Pett, W.; Fromm, B.; Schirrmeister, B.E.; Pisani, D.; Peterson, K.J.; Donoghue, P.C.J. Well-Annotated microRNAomes Do Not Evidence Pervasive miRNA Loss. Genome Biol. Evol. 2018, 10, 1457–1470. [Google Scholar] [CrossRef]
- Yazbeck, A.M.; Tout, K.R.; Stadler, P.F.; Hertel, J. Towards a Consistent, Quantitative Evaluation of MicroRNA Evolution. J. Integr. Bioinf. 2017, 14, 20160013. [Google Scholar] [CrossRef] [PubMed]
- Yazbeck, A.M.; Stadler, P.F.; Tout, K.; Fallmann, J. Automatic Curation of Large Comparative Animal MicroRNA Data Sets. Bioinformatics 2019, 35, 4553–4559. [Google Scholar] [CrossRef] [PubMed]
- Eddy, S.R.; Durbin, R. RNA sequence analysis using covariance models. Nucleic Acids Res. 1994, 22, 2079–2088. [Google Scholar] [CrossRef] [Green Version]
- Gardner, P.P. The use of covariance models to annotate RNAs in whole genomes. Briefings Funct. Genom. 2009, 8, 444–450. [Google Scholar] [CrossRef]
- Nawrocki, E.P.; Eddy, S.R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 2013, 29, 2933–2935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalvari, I.; Nawrocki, E.P.; Ontiveros-Palacios, N.; Argasinska, J.; Lamkiewicz, K.; Marz, M.; Griffiths-Jones, S.; Toffano-Nioche, C.; Gautheret, D.; Weinberg, Z.; et al. Rfam 14: Expanded coverage of metagenomic, viral and microRNA families. Nucleic Acids Res. 2020, 49, D192–D200. [Google Scholar] [CrossRef]
- Zhong, X.; Heinicke, F.; Rayner, S. miRBaseMiner, a tool for investigating miRBase content. RNA Biol. 2019, 16, 1534–1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedländer, M.R.; Mackowiak, S.D.; Li, N.; Chen, W.; Rajewsky, N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012, 40, 37–52. [Google Scholar] [CrossRef]
- Hendrix, D.; Levine, M.; Shi, W. miRTRAP, a computational method for the systematic identification of miRNAs from high throughput sequencing data. Genome Biol. 2010, 11, R39. [Google Scholar] [CrossRef] [Green Version]
- Terai, G.; Okida, H.; Asai, K.; Mituyama, T. Prediction of Conserved Precursors of miRNAs and Their Mature Forms by Integrating Position-Specific Structural Features. PLoS ONE 2012, 7, e44314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menzel, P.; Gorodkin, J.; Stadler, P.F. The Tedious Task of Finding Homologous Non-coding RNA Genes. RNA 2009, 15, 2075–2082. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Wheeler, T.J.; Eddy, S.R. nhmmer: DNA homology search with profile HMMs. Bioinformatics 2013, 29, 2487–2489. [Google Scholar] [CrossRef] [Green Version]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A Program for Improved Detection of Transfer RNA Genes in Genomic Sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [Green Version]
- Hertel, J.; Stadler, P.F. The Expansion of Animal MicroRNA Families Revisited. Life 2015, 5, 905–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velandia-Huerto, C.A.; Gittenberger, A.; Brown, F.D.; Stadler, P.F.; Bermúdez-Santana, C.I. Automated detection of ncRNAs in the draft genome sequence of a basal chordate: The Carpet Sea Squirt Didemnum vexillum. BMC Genom. 2016, 17, 591. [Google Scholar] [CrossRef] [Green Version]
- Freyhult, E.; Gardner, P.P.; Moulton, V. A comparison of RNA folding measures. BMC Bioinform. 2005, 6, 241. [Google Scholar] [CrossRef]
- Reinhart, B.J.; Slack, F.J.; Basson, M.; Pasquinelli, A.E.; Bettinger, J.C.; Rougvie, A.E.; Horvitz, H.R.; Ruvkun, G. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature 2000, 403, 901–906. [Google Scholar] [CrossRef]
- Pasquinelli, A.E.; Reinhart, B.J.; Slack, F.; Martindale, M.Q.; Kuroda, M.I.; Maller, B.; Hayward, D.C.; Ball, E.E.; Degnan, B.; Müller, P.; et al. Conservation of the sequence and temporal expression of let-7 heterochronic regulatory RNA. Nature 2000, 408, 86–89. [Google Scholar] [CrossRef]
- Bompfünewerer, A.F.; Flamm, C.; Fried, C.; Fritzsch, G.; Hofacker, I.L.; Lehmann, J.; Missal, K.; Mosig, A.; Müller, B.; Prohaska, S.J.; et al. Evolutionary Patterns of Non-Coding RNAs. Theory Biosci. 2005, 123, 301–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roush, S.; Slack, F.J. The let-7 family of microRNAs. Trends Cell Biol. 2008, 18, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Hertel, J.; Bartschat, S.; Wintsche, A.; Otto, C.; The Students of the Bioinformatics Computer Lab 2011; Stadler, P.F. Evolution of the let-7 microRNA Family. RNA Biol. 2012, 9, 231–241. [Google Scholar] [CrossRef] [Green Version]
- Liang, T.; Yang, C.Y.; Li, P.; Liu, C.; Guo, L. Genetic Analysis of Loop Sequences in the Let-7 Gene Family Reveal a Relationship between Loop Evolution and Multiple IsomiRs. PLoS ONE 2014, 9, e113042. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.W.; Zhou, L.F.; Liu, Y.L.; Wan, S.M.; Gao, Z.X. Evolution of Fish Let-7 MicroRNAs and Their Expression Correlated to Growth Development in Blunt Snout Bream. Int. J. Mol. Sci. 2017, 18, 646. [Google Scholar] [CrossRef]
- Antonacci, F.; Dennis, M.Y.; Huddleston, J.; Sudmant, P.H.; Steinberg, K.M.; Rosenfeld, J.A.; Miroballo, M.; Graves, T.A.; Vives, L.; Malig, M.; et al. Palindromic GOLGA8 core duplicons promote chromosome 15q13.3 microdeletion and evolutionary instability. Nat. Genet. 2014, 46, 1293–1302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maggiolini, F.A.M.; Cantsilieris, S.; D’Addabbo, P.; Manganelli, M.; Coe, B.P.; Dumont, B.L.; Sanders, A.D.; Pang, A.W.C.; Vollger, M.R.; Palumbo, O.; et al. Genomic inversions and GOLGA core duplicons underlie disease instability at the 15q25 locus. PLoS Genet. 2019, 15, e1008075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuksa, P.P.; Amlie-Wolf, A.; Katanić, Ž.; Valladares, O.; Wang, L.S.; Leung, Y.Y. DASHR 2.0: Integrated database of human small non-coding RNA genes and mature products. Bioinformatics 2018, 35, 1033–1039. [Google Scholar] [CrossRef] [Green Version]
- Edwards, C.A.; Mungall, A.J.; Matthews, L.; Ryder, E.; Gray, D.J.; Pask, A.J.; Shaw, G.; Graves, J.A.M.; Rogers, J.; SAVOIR Consortium; et al. The Evolution of the DLK1-DIO3 Imprinted Domain in Mammals. PLoS Biol. 2008, 6, e135. [Google Scholar] [CrossRef] [Green Version]
- Piriyapongsa, J.; Jordan, I.K. A Family of Human MicroRNA Genes from Miniature Inverted-Repeat Transposable Elements. PLoS ONE 2007, 2, e203. [Google Scholar] [CrossRef] [PubMed]
- Liang, T.; Guo, L.; Liu, C. Genome-Wide Analysis of mir-548 Gene Family Reveals Evolutionary and Functional Implications. BioMed Red. Int. 2012, 2012, 679563. [Google Scholar] [CrossRef] [Green Version]
- Cifuentes, D.; Xue, H.; Taylor, D.W.; Patnode, H.; Mishima, Y.; Cheloufi, S.; Ma, E.; Mane, S.; Hannon, G.J.; Lawson, N.D.; et al. A Novel miRNA Processing Pathway Independent of Dicer Requires Argonaute2 Catalytic Activity. Science 2010, 328, 1694–1698. [Google Scholar] [CrossRef] [Green Version]
- Velandia-Huerto, C.A.; Berkemer, S.J.; Hoffmann, A.; Retzlaff, N.; Romero Marroquín, L.C.; Hernández Rosales, M.; Stadler, P.F.; Bermúdez-Santana, C.I. Orthologs, turn-over, and remolding of tRNAs in primates and fruit flies. BMC Genom. 2016, 17, 617. [Google Scholar] [CrossRef] [Green Version]
- Fontana, W.; Stadler, P.F.; Bornberg-Bauer, E.G.; Griesmacher, T.; Hofacker, I.L.; Tacker, M.; Tarazona, P.; Weinberger, E.D.; Schuster, P. RNA Folding Landscapes and Combinatory Landscapes. Phys. Rev. E 1993, 47, 2083–2099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saçar, M.D.; Allmer, J. Machine learning methods for microRNA gene prediction. Methods Mol. Biol. 2014, 1107, 177–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofacker, I.L.; Fontana, W.; Stadler, P.F.; Bonhoeffer, L.S.; Tacker, M.; Schuster, P. Fast Folding and Comparison of RNA Secondary Structures. Monatsh. Chem. 1994, 125, 167–188. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Species | Homology | Final | MIRfix Filtered | Ann. | Match | Miss | Ratio Match | Ratio Miss | Add. |
|---|---|---|---|---|---|---|---|---|---|
| Human | 26 | 20 | 6 | 14 | 13 | 1 | 0.928 | 0.07 | 7 |
| Orang-Utan | 27 | 18 | 9 | 14 | 13 | 1 | 0.928 | 0.07 | 5 |
| Gorilla | 26 | 20 | 6 | 14 | 13 | 1 | 0.928 | 0.07 | 7 |
| Chimpanzee | 30 | 24 | 6 | 14 | 13 | 1 | 0.928 | 0.07 | 11 |
| Mouse | 19 | 14 | 5 | 12 | 11 | 1 | 0.916 | 0.08 | 3 |
| Sea squirt | 7 | 6 | 1 | 5 | 5 | 0 | 1.0 | 0.0 | 1 |
| Class | Perfect | Partial | Without | Total | Additional |
|---|---|---|---|---|---|
| Accepted | 284 | 28 | 11 | 323 | 178 |
| Filtered | 27 | 0 | 0 | 27 | 5 |
| d | r | o | Total | |
|---|---|---|---|---|
| Number | 12 (13) | 73 (685) | 31 (245) | 116 (943) |
| Fraction | 0.010 (0.014) | 0.629 (0.726) | 0.267 (0.26) |
| Sequence Homology | Alignment Score | Annotation/Structure Evaluation | Consensus Evaluation | ||
|---|---|---|---|---|---|
| Pairwise (blastn) | HMMs (nhmmer) | Alignment Score Evaluation (cmsearch) | Ann. Filter (MIRfix) | Secondary Structure (MIRfix) | SS Conservation |
| E-value | E-value | E-value | Ann. mature seq. | MFE | |
| nt HSPs | Seq. Length nt | Valid CSS | |||
| Coverage % | Coverage % | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Velandia-Huerto, C.A.; Fallmann, J.; Stadler, P.F. miRNAture—Computational Detection of microRNA Candidates. Genes 2021, 12, 348. https://doi.org/10.3390/genes12030348
Velandia-Huerto CA, Fallmann J, Stadler PF. miRNAture—Computational Detection of microRNA Candidates. Genes. 2021; 12(3):348. https://doi.org/10.3390/genes12030348
Chicago/Turabian StyleVelandia-Huerto, Cristian A., Jörg Fallmann, and Peter F. Stadler. 2021. "miRNAture—Computational Detection of microRNA Candidates" Genes 12, no. 3: 348. https://doi.org/10.3390/genes12030348
APA StyleVelandia-Huerto, C. A., Fallmann, J., & Stadler, P. F. (2021). miRNAture—Computational Detection of microRNA Candidates. Genes, 12(3), 348. https://doi.org/10.3390/genes12030348