
A Novel Truncating Mutation in HOMER2 Causes Nonsyndromic Progressive DFNA68 Hearing Loss in a Spanish Family

 , ,
, ,
Abstract
:1. Introduction
2. Patients and Methods
2.1. Patients Selection
2.2. Sample Collection
2.3. Targeted Next-Generation Sequencing
2.4. Sanger Sequencing
3. Results
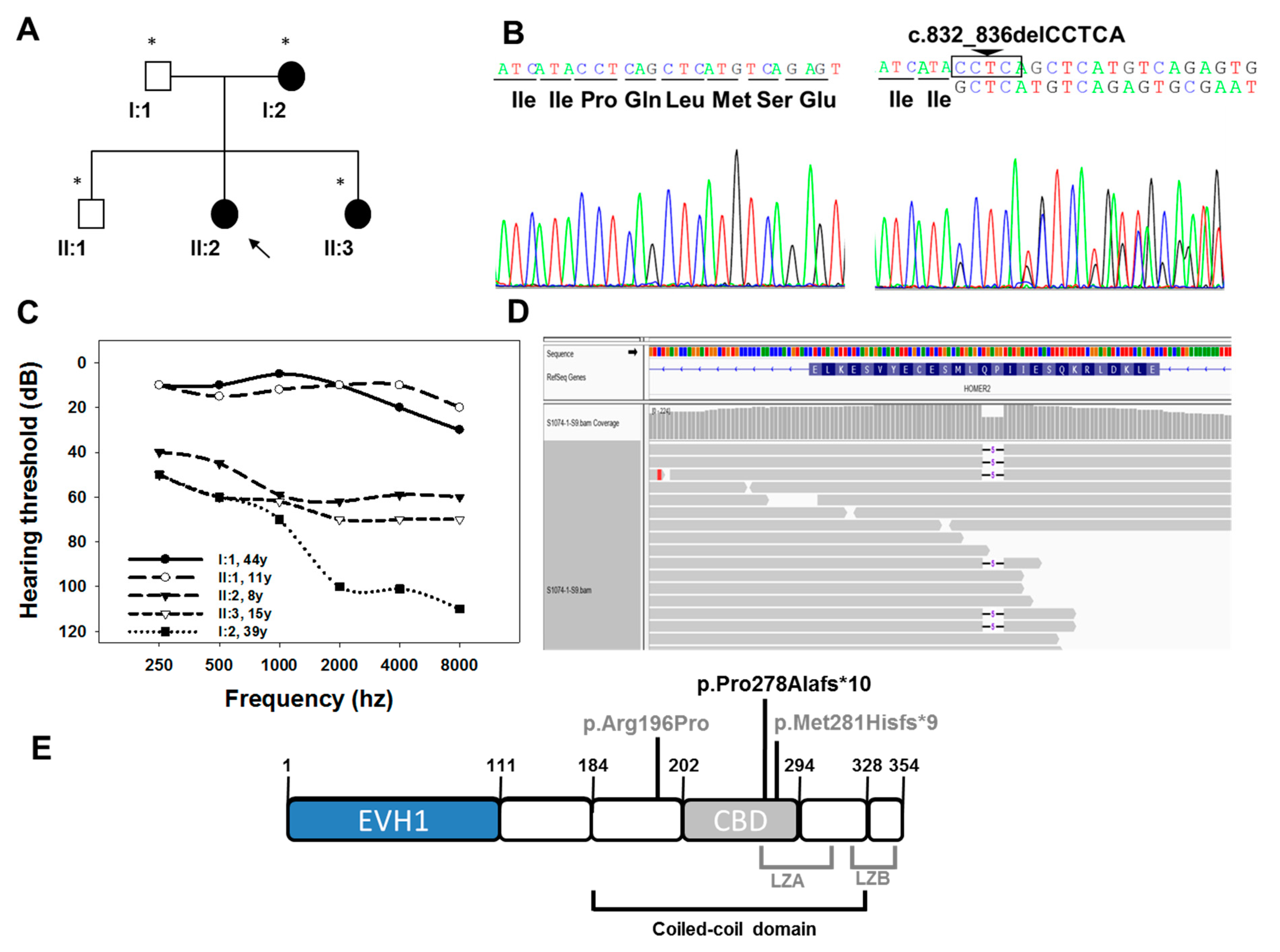
3.1. Clinical Description of the Family

3.2. Genetic Study
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Petit, C.; Levilliers, J.; Hardelin, J.P. Molecular genetics of hearing loss. Annu. Rev. Genet. 2001, 35, 589–646. [Google Scholar] [CrossRef]
- Morton, C.C.; Nance, W.E. Newborn hearing screening-a silent revolution. N. Engl. J. Med. 2006, 354, 2151–2164. [Google Scholar] [CrossRef]
- Van Camp, G.; Smith, R.J. Hereditary Hearing Loss Homepage. Available online: https://hereditaryhearingloss.org (accessed on 20 December 2020).
- Hilgert, N.; Smith, R.J.; Van Camp, G. Function and expression pattern of nonsyndromic deafness genes. Curr. Mol. Med. 2009, 9, 546–564. [Google Scholar] [CrossRef]
- Mencía, A.; Modamio-Høybjør, S.; Redshaw, N.; Morín, M.; Mayo-Merino, F.; Olavarrieta, L.; Aguirre, L.A.; del Castillo, I.; Steel, K.P.; Dalmay, T.; et al. Mutations in the seed region of human miR-96 are responsible for nonsyndromic progressive hearing loss. Nat. Genet. 2009, 41, 609–613. [Google Scholar] [CrossRef] [PubMed]
- Norton, N.; Williams, H.J.; Williams, N.M.; Spurlock, G.; Zammit, S.; Jones, G.; Jones, S.; Owen, R.; O’Donovan, M.C.; Owen, M.J. Mutation screening of the Homer gene family and association analysis in schizophrenia. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2003, 120, 18–21. [Google Scholar] [CrossRef]
- Shiraishi-Yamaguchi, Y.; Furuichi, T. The Homer family proteins. Genome Biol. 2007, 8, 206. [Google Scholar] [CrossRef] [Green Version]
- Worley, P.F.; Worley, P.F.; Zeng, W.; Huang, G.; Kim, J.Y.; Shin, D.M.; Kim, M.S.; Yuan, J.P.; Kiselyov, K.; Muallem, S. Homer proteins in Ca2+ signaling by excitable and non-excitable cells. Cell Calcium 2007, 42, 363–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.M.; Lee, J.; Jo, H.; Park, S.; Chang, I.; Muallem, S.; Shin, D.S. Homer2 protein regulates plasma membrane Ca2⁺-ATPase-mediated Ca2⁺ signaling in mouse parotid gland acinar cells. J. Biol. Chem. 2014, 289, 24971–24979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jardin, I.; López, J.J.; Berna-Erro, A.; Salido, G.M.; Rosado, J.A. Homer proteins in Ca2⁺ entry. IUBMB Life 2013, 65, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Tao-Cheng, J.H.; Thein, S.; Yang, Y.; Reese, T.S.; Gallant, P.E. Homer is concentrated at the postsynaptic density and does not redistribute after acute synaptic stimulation. Neuroscience 2014, 266, 80–90. [Google Scholar] [CrossRef] [Green Version]
- Kato, A.; Ozawa, F.; Saitoh, Y.; Fukazawa, Y.; Sugiyama, H.; Inokuchi, K. Novel members of the Vesl/Homer family of PDZ proteins that bind metabotropic glutamate receptors. J. Biol. Chem. 1998, 273, 23969–23975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barzik, M.; Carl, U.D.; Schubert, W.D.; Frank, R.; Wehland, J.; Heinz, D.W. The N-terminal domain of Homer/Vesl is a new class II EVH1 domain. J. Mol. Biol. 2001, 309, 155–169. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Tadokoro, S.; Imanaka, T.; Murakami, S.D.; Nakamura, M.; Kashiwada, K.; Ko, J.; Nishida, W.; Sobue, K. Isolation of PSD-Zip45, a novel Homer/vesl family protein containing leucine zipper motifs, from rat brain. FEBS Lett. 1998, 437, 304–308. [Google Scholar] [CrossRef] [Green Version]
- Shiraishi, Y.; Mizutani, A.; Bito, H.; Fujisawa, K.; Narumiya, S.; Mikoshiba, K.; Furuichi, T. Cupidin, an isoform of Homer/Vesl, interacts with the actin cytoskeleton and activated rho family small GTPases and is expressed in developing mouse cerebellar granule cells. J. Neurosci. 1999, 19, 8389–8400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiraishi, Y.; Mizutani, A.; Yuasa, S.; Mikoshiba, K.; Furuichi, T. Differential expression of Homer family proteins in the developing mouse brain. J. Comp. Neurol. 2004, 473, 582–599. [Google Scholar] [CrossRef] [PubMed]
- Reibring, C.G.; Hallberg, K.; Linde, A.; Gritli-Linde, A.G. Distinct and Overlapping Expression Patterns of the Homer Family of Scaffolding Proteins and Their Encoding Genes in Developing Murine Cephalic Tissues. Int. J. Mol. Sci. 2020, 21, 1264. [Google Scholar] [CrossRef] [Green Version]
- Azaiez, H.; Decker, A.R.; Booth, K.T.; Simpson, A.C.; Shearer, A.E.; Huygen, P.L.M.; Bu, F.; Hildebrand, M.S.; Ranum, P.T.; Shibata, S.B.; et al. HOMER2, a stereociliary scaffolding protein, is essential for normal hearing in humans and mice. PLoS Genet. 2015, 11, e1005137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X.; Wang, Q.; Gu, H.; Zhang, X.; Qi, Y.; Liu, Y. Whole exome sequencing identified a second pathogenic variant in HOMER2 for autosomal dominant nonsyndromic deafness. Clin. Genet. 2018, 94, 419–428. [Google Scholar] [CrossRef]
- Morín, M.; Borreguero, L.; Booth, K.T.; Lachgar, M.; Huygen, P.; Villamar, M.; Mayo, F.; Barrio, L.C.; Santos Serrão de Castro, L.; Morales, M.; et al. Insights into the pathophysiology of DFNA10 hearing loss associated with novel EYA4 variants. Sci. Rep. 2020, 10, 6213. [Google Scholar] [CrossRef]
- Shiraishi, Y.; Sato, Y.; Sakai, R.; Mizutani, A.; Knöpfel, T.; Mori, N.; Mikoshiba, K.; Furuichi, T. Interaction of Cupidin/Homer2 with two actin cytoskeletal regulators, Cdc42 small GTPase and Drebrin, in dendritic spines. BMC Neurosci. 2009, 10, 25. [Google Scholar]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Albarca Aguilera, M.; Meyer, R.; Massouras, A. VarSome: The human genomic variant search engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Peña-Chilet, M.; Roldán, G.; Perez-Florido, J.; Ortuño, J.M.; Carmona, R.; Aquino, V.; Lopez-Lopez, D.; Loucera, C.; Fernandez-Rueda, J.L.; Gallego, A.; et al. CSVS, a crowdsourcing database of the Spanish population genetic variability. Nucleic Acid Res. 2020, 49, D1130–D1137. [Google Scholar] [CrossRef] [PubMed]
- Azaiez, H.; Booth, K.T.; Ephraim, S.S.; Crone, B.; Black-Ziegelbein, E.A.; Marini, R.J.; Shearer, A.E.; Sloan-Heggen, C.M.; Kolbe, D.; Casavant, T.; et al. Genomic Landscape and Mutational Signatures of Deafness-Associated Genes. Am. J. Hum. Genet. 2018, 103, 484–497. [Google Scholar] [CrossRef] [Green Version]
- Deafness Variation Database. Available online: http://deafnessvariationdatabase.org/ (accessed on 20 December 2020).
- Ueyama, T.; Sakaguchi, H.; Nakamura, T.; Goto, A.; Morioka, S.; Shimizu, A.; Nakao, K.; Hishikawa, Y.; Ninoyu, Y.; Kassai, H.; et al. Maintenance of stereocilia and apical junctional complexes by Cdc42 in cochlear hair cells. J. Cell. Sci. 2014, 127, 2040–2052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirjavainen, A.; Laos, M.; Anttonen, T.; Pirvola, U. The Rho GTPase Cdc42 regulates hair cell planar polarity and cellular patterning in the developing cochlea. Biol. Open 2015, 4, 516–526. [Google Scholar] [CrossRef] [Green Version]
- Wildeman, M.; Ophuizen, E.V.; den Dunnen, J.T.; Taschner, P.E.M. Improving sequence variant descriptions in mutation databases and literature using the Mutalyzer sequence variation nomenclature checker. Hum. Mutat. 2008, 29, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Pros, E.; Larriba, S.; López, E.; Ravella, A.; Gili, M.L.; Kruyer, H.; Valls, J.; Serra, E.; Lázaro, C. NF1 mutation rather than individual genetic variability is the main determinant of the NF1-transcriptional profile of mutations affecting splicing. Hum. Mutat. 2006, 27, 1104–1114. [Google Scholar] [CrossRef]


{kind=link}
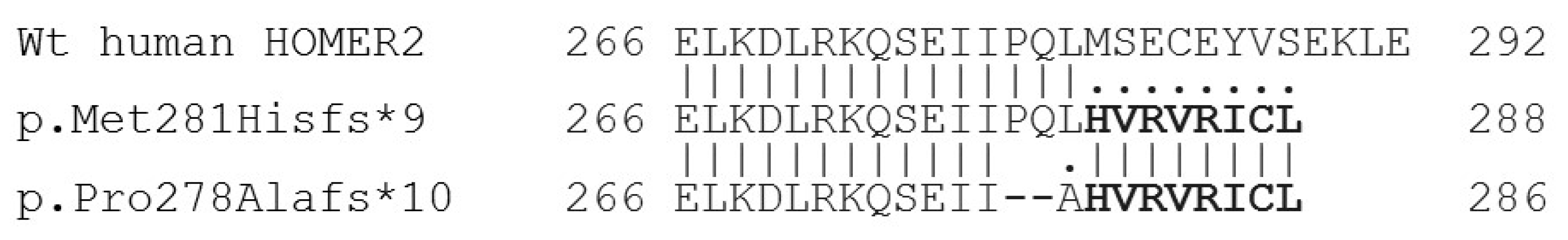{kind=link}
| Gene Transcript | Exon | c.DNA and Protein Alteration | Variant Fraction Coverage (Ref/Alt) | Coding Consequence | Pathogenicity Prediction by SOPHIA DDM | ACMG | ClinVar Rating | GnomAD | Locus |
|---|---|---|---|---|---|---|---|---|---|
| HOMER2 NM_199330 | 8 | c.832_836delCCTCA p.Pro278Alafs*10 | 34.98% (145/78) | frameshift | Highly Pathogenic | Pathogenic (PVS1, PM2, PP3) | - | 0 | DFNA68 |
| MYO7A NM_001127179 | 27 | c.3515_3536del p.Gly1172*1179del | 49.0% (582/306) | No-stop | Highly Pathogenic | Benign (PVS1, BA1, BS2, BP6) | Benign rs111033223 | 0.391 | DFNA11 DFNB2 |
| TMPRSS3 NM_001256317 | 8 | c.617-3_617-2dupTA p.? | 46.73% (421/372) | Splice acceptor | Highly Pathogenic | (PVS1, PP3, BA1, BP6) | Benign rs34966432 | 0.118 | DFNB8 DFNB10 |
| COL11A2 NM_080679 | 32 | c.2307 + 3G>A p.? | 50.43% (402/409) | splice_donor_ +3 | Potentially Pathogenic | Benign (BA1, BS2) | Benign rs970901 | 0.601 | DFNA13 DFNB53 |
| GPSM2 NM_013296 | 13 | c.1572_1574delTTC p.Ser525del | 48.55% (278/267) | inframe_3 | Potentially Pathogenic | Benign(PP3, BA1, BP3, BP6) | Benign rs35029887 | 0.291 | DFNB82 |
| BDP1 NM_018429 | 23 | c.5068G>C p.Gly1690Arg | 44.54% (335/269) | missense | Potentially Pathogenic | Likely Benign (PM2, BP1, BP4) | - rs193135814 | 7.48 × 10−5 | DFNB112 |
| EPS8 NM_004447 | 20 | c.2230G>A p.Val744Ile | 44.34% (359/286) | missense | Potentially Pathogenic | Benign (BS1, BS2, BP1, BP6) | Likely Benign rs77967764 | 2.06 × 10−3 | DFNB102 |
| EPS8L2 NM_022772 | 9 | c.710G>T p.Arg237Leu | 50.22% (560/565) | missense | Potentially Pathogenic | VUS (PM2, BP4) | - | 7.1 × 10−6 | DFNB106 |
| OSBPL2 NM_014835 | 9 | c.747A>G p.Arg249Arg | 51.09% (157/164) | synonymous | Potentially Pathogenic | Likely Benign (BP4, BP7, PM2) | - rs1309059934 | 0 | DFNA67 |
| PCDH15 NM_001142771 | 21 | c.2596G>A p.Val866Met | 49.09% (446/430) | missense | Potentially Pathogenic | VUS (PM1, PM2) | Uncertain Significance rs142512524 | 6.37 × 10−4 | DFNB23 |
| DNA Change | Protein Change | Exon | Origin | Phenotype | Detection Decade | Degree | Audiogram Profile | ACMG Classification | Number of Scores Supporting Pathogenicity Pathogenicity | DVD | CSVS Allele Freq | GnomAD Allele Freq. | References |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| c.587G>C | p.Arg196Pro | 6 | European descent | SNHL | 1st | Mild-profound | Down-sloping | Uncertain significance (PM2, PP3, PP5, BP1) | 20/22 | N.A | 0 | 0 | Azaiez et al. 2015 |
| c.840_840dup | p.(Met281Hisfs*9) | 8 | Chinese | SNHL | 1st | Moderate-profound | Down-sloping | Pathogenic (PVS1, PM2, PP3, PP5) | N.A | N.A | 0 | 0 | Lu et al. 2018 |
| c.832_836delCCTCA | p.(Pro278Alafs*10) | 8 | Spanish | SNHL | 1st | Moderate-profound | Down-sloping | Pathogenic (PVS1, PM2, PP3) | N.A | N.A | 0 | 0 | This work |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lachgar, M.; Morín, M.; Villamar, M.; del Castillo, I.; Moreno-Pelayo, M.Á. A Novel Truncating Mutation in HOMER2 Causes Nonsyndromic Progressive DFNA68 Hearing Loss in a Spanish Family. Genes 2021, 12, 411. https://doi.org/10.3390/genes12030411
Lachgar M, Morín M, Villamar M, del Castillo I, Moreno-Pelayo MÁ. A Novel Truncating Mutation in HOMER2 Causes Nonsyndromic Progressive DFNA68 Hearing Loss in a Spanish Family. Genes. 2021; 12(3):411. https://doi.org/10.3390/genes12030411
Chicago/Turabian StyleLachgar, María, Matías Morín, Manuela Villamar, Ignacio del Castillo, and Miguel Ángel Moreno-Pelayo. 2021. "A Novel Truncating Mutation in HOMER2 Causes Nonsyndromic Progressive DFNA68 Hearing Loss in a Spanish Family" Genes 12, no. 3: 411. https://doi.org/10.3390/genes12030411
APA StyleLachgar, M., Morín, M., Villamar, M., del Castillo, I., & Moreno-Pelayo, M. Á. (2021). A Novel Truncating Mutation in HOMER2 Causes Nonsyndromic Progressive DFNA68 Hearing Loss in a Spanish Family. Genes, 12(3), 411. https://doi.org/10.3390/genes12030411