
Full-Length Transcriptome Sequencing and Comparative Transcriptome Analysis to Evaluate Drought and Salt Stress in Iris lactea var. chinensis

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials, Growth Conditions, and Stress Treatment
2.2. Iso-Seq Library RNA Preparation, Sequencing, and Analysis
2.3. RNA-Seq Library RNA Preparation, Sequencing, and Analysis
2.4. DEGs Analysis
3. Results
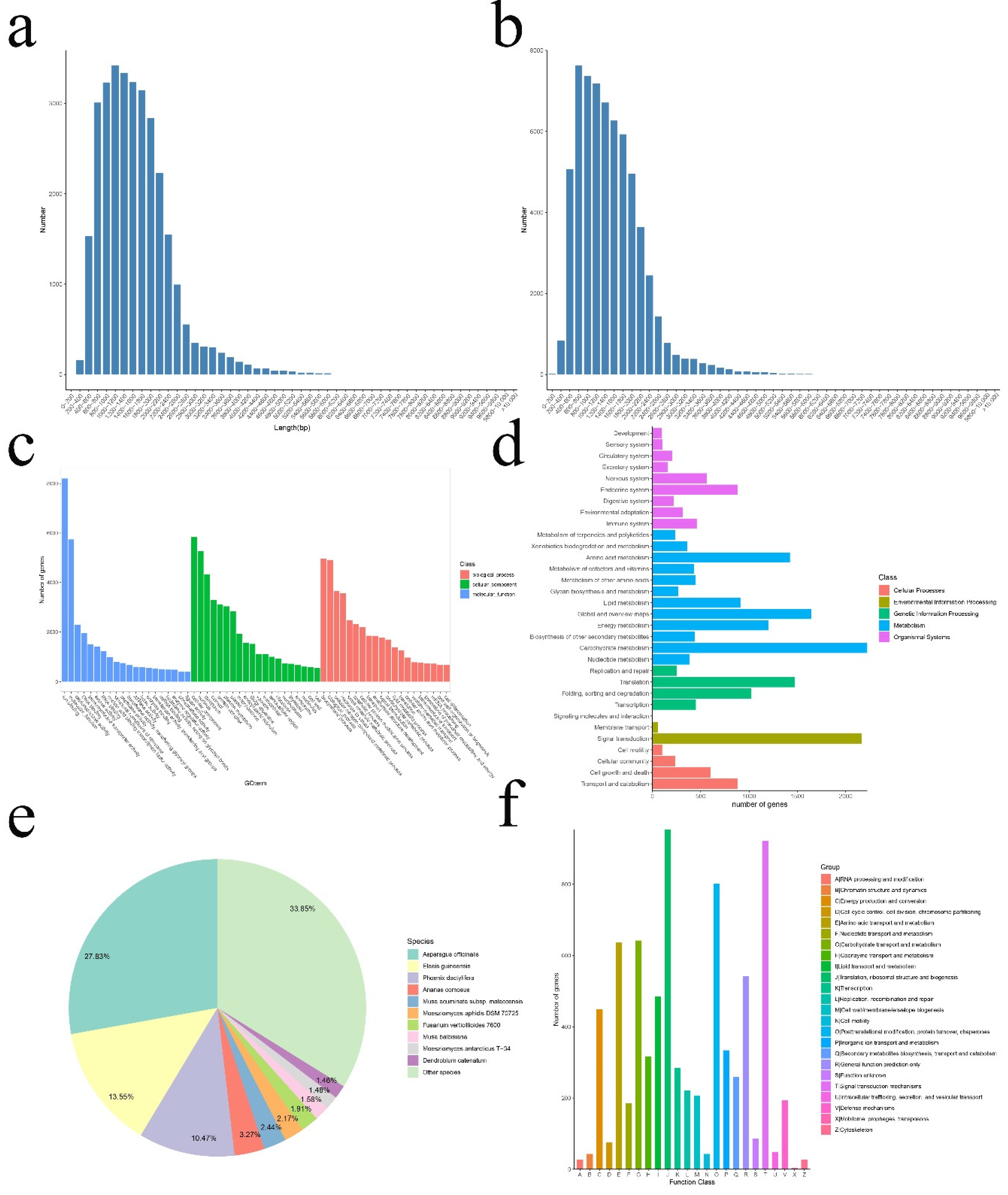
3.1. Single-Molecule Real-Time (SMRT) Sequencing, Data Processing, Annotation, and Coding Sequence (CDS) Prediction
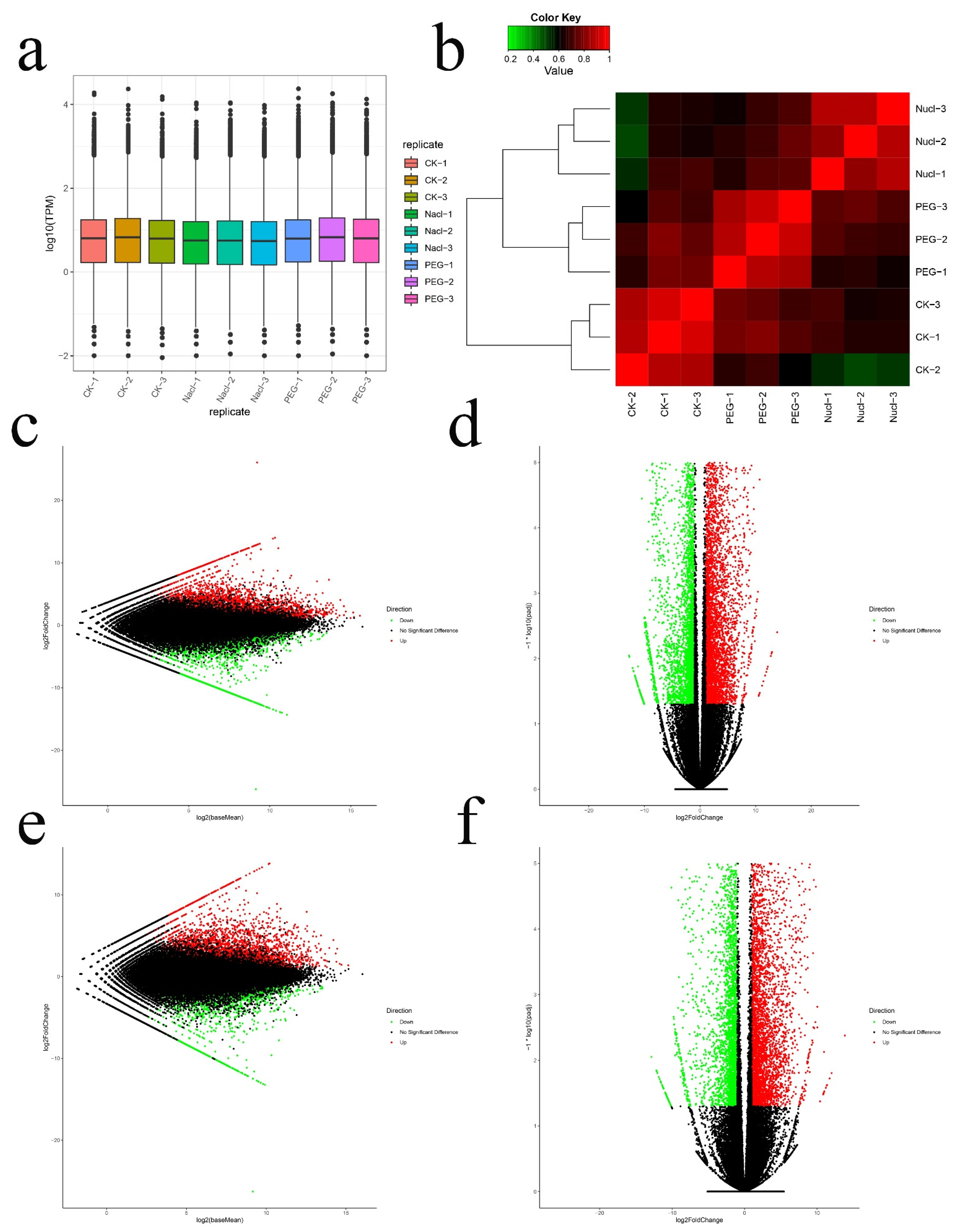
3.2. Illumina Sequencing Data Analysis and DEGs Screening
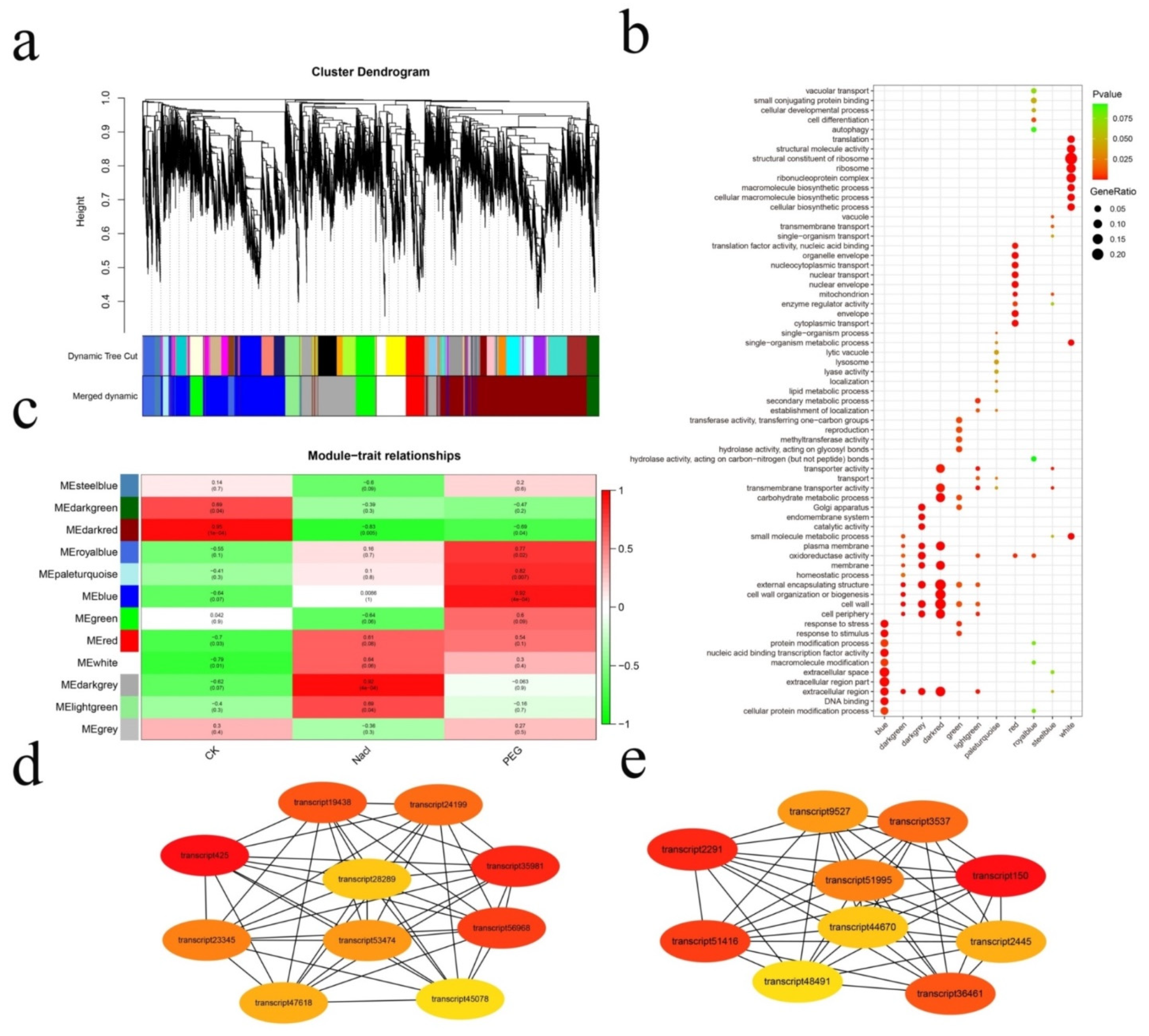
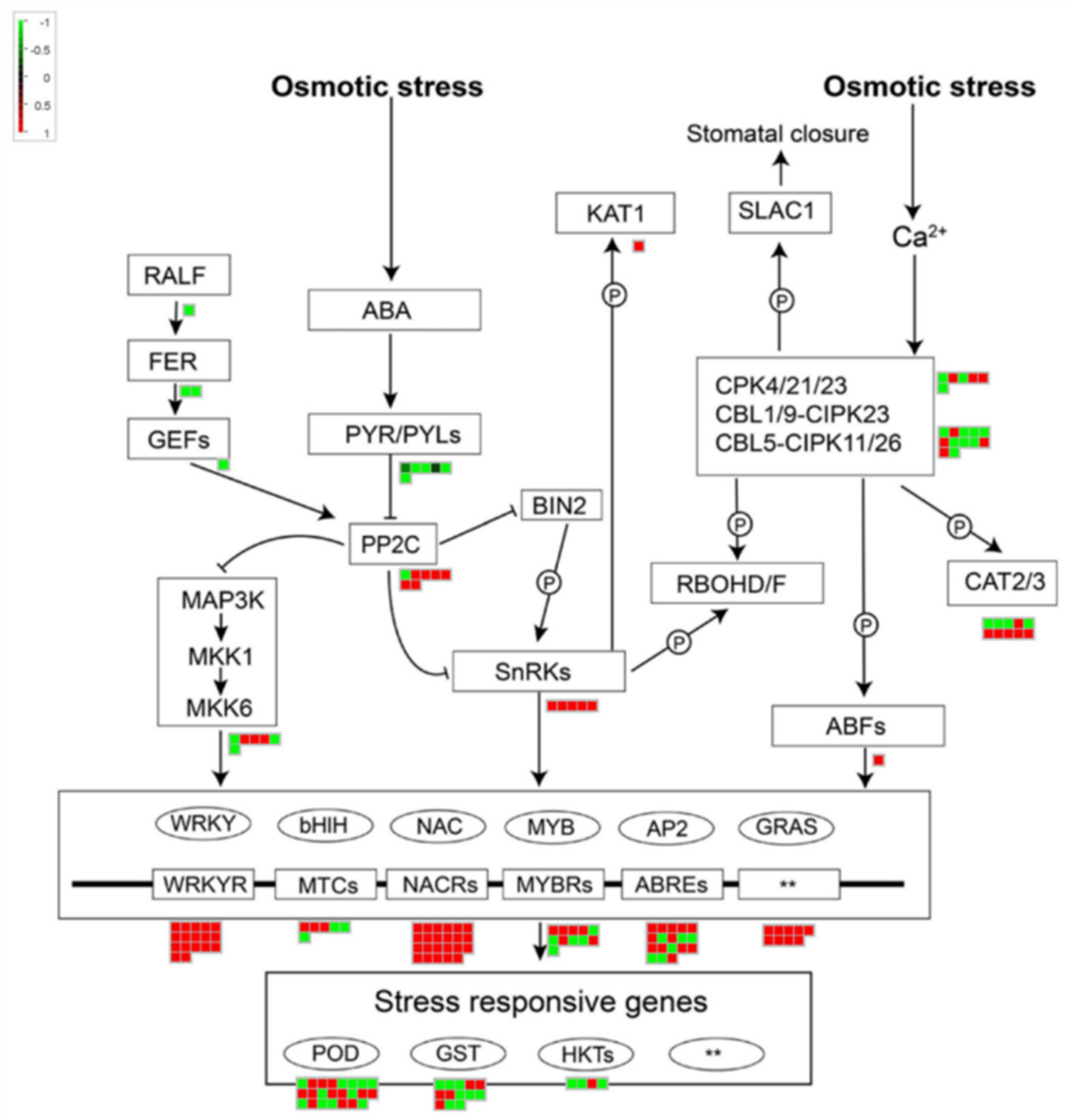
3.3. Analysis of Stress Regulation Networks
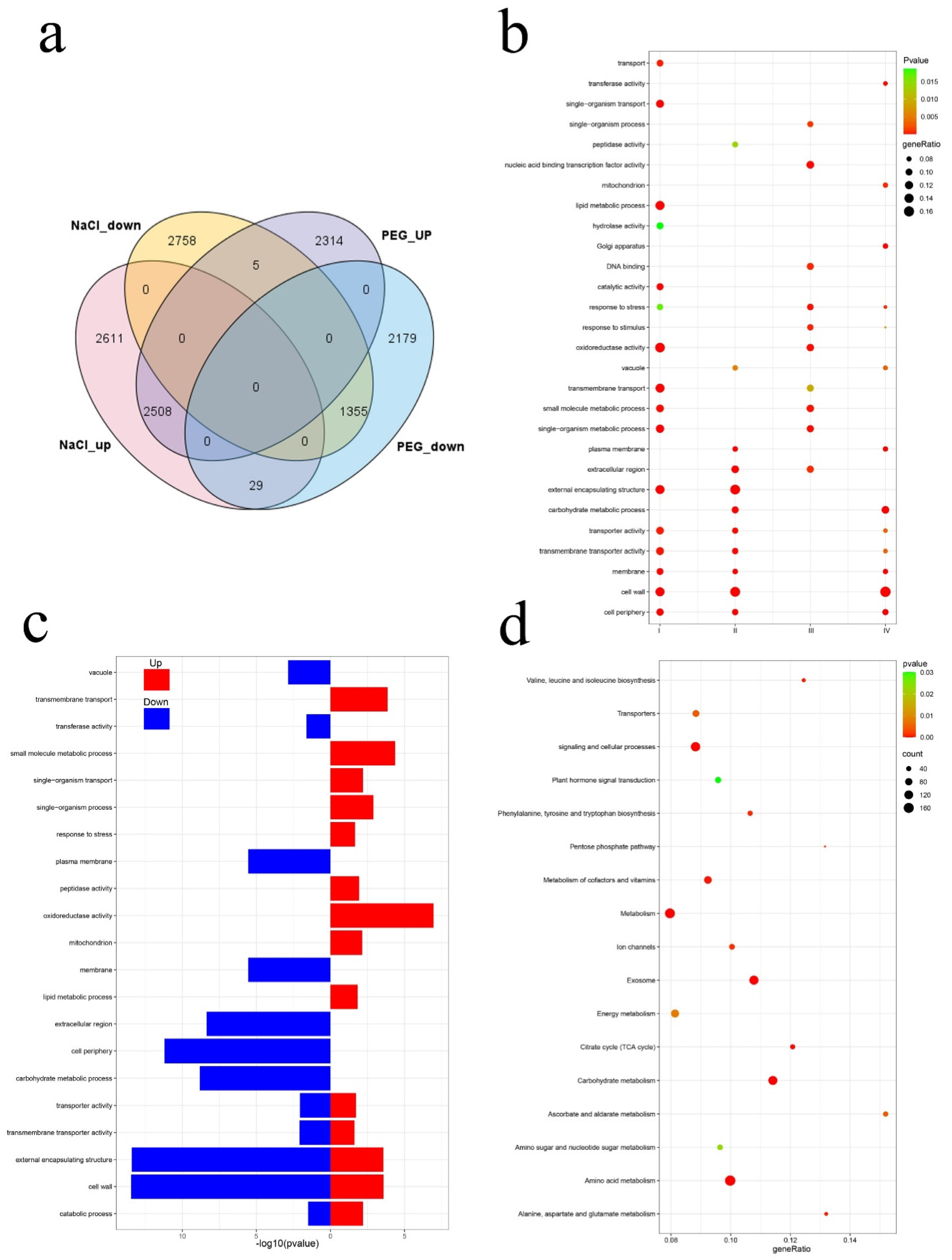
3.4. Analysis of Comparative Transcriptome
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Luo, D.; Zhou, Q.; Wu, Y.; Chai, X.; Liu, W.; Wang, Y.; Yang, Q.; Wang, Z.; Liu, Z. Full-length transcript sequencing and comparative transcriptomic analysis to evaluate the contribution of osmotic and ionic stress components towards salinity tolerance in the roots of cultivated alfalfa (Medicago sativa L.). BMC Plant Biol. 2019, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaves, M.M.; Flexas, J.; Pinheiro, C. Photosynthesis under drought and salt stress: Regulation mechanisms from whole plant to cell. Ann. Bot. 2009, 103, 551–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.-K. Abiotic Stress Signaling and Responses in Plants. Cell 2016, 167, 313–324. [Google Scholar] [CrossRef] [Green Version]
- Cutler, S.R.; Rodriguez, P.L.; Finkelstein, R.R.; Abrams, S.R. Abscisic acid: Emergence of a core signaling network. Annu. Rev. Plant Biol. 2010, 61, 651–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choudhury, F.K.; Rivero, R.M.; Blumwald, E.; Mittler, R. Reactive oxygen species, abiotic stress and stress combination. Plant J. 2017, 90, 856–867. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.-K. Salt and drought stress signal transduction in plants. Annu. Rev. Plant Biol. 2002, 53, 247–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.-K. Genetic analysis of plant salt tolerance using Arabidopsis: Figure 1. Plant Physiol. 2000, 124, 941–948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, C.; Zhang, H.; Song, C.; Zhu, J.-K.; Shabala, S. Mechanisms of plant responses and adaptation to soil salinity. Innovation 2020, 1, 100017. [Google Scholar] [CrossRef]
- Jiang, Z.; Zhou, X.; Tao, M.; Yuan, F.; Liu, L.; Wu, F.; Wu, X.; Xiang, Y.; Niu, Y.; Liu, F.; et al. Plant cell-surface GIPC sphingolipids sense salt to trigger Ca2+ influx. Nature 2019, 572, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Gu, C.-S.; Liu, L.-Q.; Deng, Y.-M.; Zhang, Y.-X.; Wang, Z.-Q.; Yuan, H.-Y.; Huang, S.-Z. De novo characterization of the Iris lactea var. chinensis transcriptome and an analysis of genes under cadmium or lead exposure. Ecotoxicol. Environ. Saf. 2017, 144, 507–513. [Google Scholar] [CrossRef]
- Gu, C.; Xu, S.; Wang, Z.; Liu, L.; Zhang, Y.; Deng, Y.; Huang, S. De novo sequencing, assembly, and analysis of Iris lactea var. chinensis roots’ transcriptome in response to salt stress. Plant Physiol. Biochem. 2018, 125. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Chen, S.-Y.; Liu, G.-S. Application of next generation sequencing techniques in plant transcriptome. YiChuan 2011, 33, 1317–1326. [Google Scholar]
- Zhu, Z.; Chen, H.; Xie, K.; Liu, C.; Li, L.; Liu, L.; Han, X.; Jiao, C.; Wan, Z.; Sha, A. Characterization of drought-responsive transcriptome during seed germination in Adzuki bean (L.) by PacBio SMRT and Illumina Sequencing. Front. Genet. 2020, 11, 996. [Google Scholar] [CrossRef]
- Sun, M.; Huang, D.; Zhang, A.; Khan, I.; Yan, H.; Wang, X.; Zhang, X.; Zhang, J.; Huang, L. Transcriptome analysis of heat stress and drought stress in pearl millet based on Pacbio full-length transcriptome sequencing. BMC Plant Biol. 2020, 20. [Google Scholar] [CrossRef]
- Zhang, G.; Sun, M.; Wang, J.; Lei, M.; Li, C.; Zhao, D.; Huang, J.; Li, W.; Li, S.; Li, J.; et al. PacBio full-length cDNA sequencing integrated with RNA-seq reads drastically improves the discovery of splicing transcripts in rice. Plant J. 2019, 97, 296–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Li, W.; Chen, Z.-J.; Wei, F.-G.; Liu, Y.-L.; Gao, L.-Z. SMRT- and Illumina-based RNA-seq analyses unveil the ginsinoside biosynthesis and transcriptomic complexity in Panax notoginseng. Sci. Rep. 2020, 10, 15310. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [Green Version]
- Wagner, G.P.; Kin, K.; Lynch, V.J. Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theory Biosci. 2012, 131, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Schurch, N.J.; Schofield, P.; Gierliński, M.; Cole, C.; Sherstnev, A.; Singh, V.; Wrobel, N.; Gharbi, K.; Simpson, G.G.; Owen-Hughes, T.; et al. How many biological replicates are needed in an RNA-seq experiment and which differential expression tool should you use? RNA 2016, 22, 839–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [Green Version]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Ramsak, Ž.; Baebler, Š.; Rotter, A.; Korbar, M.; Mozetic, I.; Usadel, B.; Gruden, K. GoMapMan: Integration, consolidation and visualization of plant gene annotations within the MapMan ontology. Nucleic Acids Res. 2014, 42, D1167–D1175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.-H.; Chen, S.-H.; Wu, H.-H.; Ho, C.-W.; Ko, M.-T.; Lin, C.-Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8, S11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furuno, M.; Kasukawa, T.; Saito, R.; Adachi, J.; Suzuki, H.; Baldarelli, R.; Hayashizaki, Y.; Okazaki, Y. CDS annotation in full-length cDNA sequence. Genome Res. 2003, 13, 1478–1487. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Ding, Y.; Yang, Y.; Song, C.; Wang, B.; Yang, S.; Guo, Y.; Gong, Z. Protein kinases in plant responses to drought, salt, and cold stress. J. Integr. Plant Biol. 2021. [Google Scholar] [CrossRef]
- Zhang, B.; Li, P.-F.; Fan, F.-C. Ionic relations and proline accumulation in shoots of two Chinese Iris germplasms during NaCl stress and subsequent relief. Plant Growth Regul. 2012, 68, 49–56. [Google Scholar] [CrossRef]
- Tang, J.; Liu, Q.; Yuan, H.; Zhang, Y.; Wang, W.; Huang, S. Molecular cloning and characterization of a novel salt-specific responsive WRKY transcription factor gene IlWRKY2 from the halophyte Iris lactea var. chinensis. Genes Genom. 2018, 40, 893–903. [Google Scholar] [CrossRef] [PubMed]
- Tong, H.Y.; Gu, C.S.; Yuan, H.Y.; Ma, J.J.; Huang, S.Z. Effects of salt stress on the growth, photosynthetic pigments and structure of two halophytes, Iris halophila and I. lactea var. chinensis. Fresenius Environ. Bull. 2014, 23, 84–90. [Google Scholar]
- Shavrukov, Y. Salt stress or salt shock: Which genes are we studying? J. Exp. Bot. 2013, 64, 119–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, S.; Li, Y.; Chu, L.; Kuang, X.; Song, J.; Sun, C. Full-length sequencing of ginkgo transcriptomes for an in-depth understanding of flavonoid and terpenoid trilactone biosynthesis. Gene 2020, 758, 144961. [Google Scholar] [CrossRef]
- Liu, X.; Wang, J.; Sun, L. Structure of the hyperosmolality-gated calcium-permeable channel OSCA1.2. Nat. Commun. 2018, 9, 5060. [Google Scholar] [CrossRef]
- Yu, X.; Han, J.; Li, L.; Zhang, Q.; Yang, G.; He, G. Wheat PP2C-a10 regulates seed germination and drought tolerance in transgenic Arabidopsis. Plant Cell Rep. 2020, 39, 635–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaidi, I.; Ebel, C.; Belgaroui, N.; Ghorbel, M.; Amara, I.; Hanin, M. The wheat MAP kinase phosphatase 1 alleviates salt stress and increases antioxidant activities in Arabidopsis. J. Plant Physiol. 2016, 193, 12–21. [Google Scholar] [CrossRef]
- Asano, T.; Hakata, M.; Nakamura, H.; Aoki, N.; Komatsu, S.; Ichikawa, H.; Hirochika, H.; Ohsugi, R. Functional characterisation of OsCPK21, a calcium-dependent protein kinase that confers salt tolerance in rice. Plant Mol. Biol. 2011, 75, 179–191. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Lee, K.; Hwang, H.; Bhatnagar, N.; Kim, D.-Y.; Yoon, I.S.; Byun, M.-O.; Kim, S.T.; Jung, K.-H.; Kim, B.-G. Overexpression of PYL5 in rice enhances drought tolerance, inhibits growth, and modulates gene expression. J. Exp. Bot. 2014, 65, 453–464. [Google Scholar] [CrossRef] [Green Version]
- Aslam, M.; Fakher, B.; Jakada, B.H.; Zhao, L.; Cao, S.; Cheng, Y.; Qin, Y. Genome-wide identification and expression profiling of CBL-CIPK gene family in Pineapple and the role of CBL1 in abiotic and biotic stress response. Biomolecules 2019, 9, 293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Wei, W.; Pang, X.; Wang, X.; Zhang, H.; Dong, B.; Xing, Y.; Li, X.; Wang, M. Comparative transcriptome profiling of a desert evergreen shrub, Ammopiptanthus mongolicus, in response to drought and cold stresses. BMC Genom. 2014, 15, 671. [Google Scholar] [CrossRef] [Green Version]
- Wong, C.E.; Li, Y.; Labbe, A.; Guevara, D.; Nuin, P.; Whitty, B.; Diaz, C.; Golding, G.B.; Gray, G.R.; Weretilnyk, E.A.; et al. Transcriptional profiling implicates novel interactions between abiotic stress and hormonal responses in Thellungiella, a close relative of Arabidopsis. Plant Physiol. 2006, 140, 1437–1450. [Google Scholar] [CrossRef] [Green Version]
- Rabbani, M.A.; Maruyama, K.; Abe, H.; Khan, M.A.; Katsura, K.; Ito, Y.; Yoshiwara, K.; Seki, M.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Monitoring expression profiles of rice genes under cold, drought, and high-salinity stresses and abscisic acid application using cDNA microarray and RNA gel-blot analyses. Plant Physiol. 2003, 133, 1755–1767. [Google Scholar] [CrossRef] [Green Version]
- Chan, Z. Expression profiling of ABA pathway transcripts indicates crosstalk between abiotic and biotic stress responses in Arabidopsis. Genomics 2012, 100, 110–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, G.-T.; Ma, S.-L.; Bai, L.-P.; Zhang, L.; Ma, H.; Jia, P.; Liu, J.; Zhong, M.; Guo, Z.-F. Signal transduction during cold, salt, and drought stresses in plants. Mol. Biol. Rep. 2012, 39, 969–987. [Google Scholar] [CrossRef]
- Zhang, S.; Li, X.; Fan, S.; Zhou, L.; Wang, Y. Overexpression of HcSCL13, a Halostachys caspica GRAS transcription factor, enhances plant growth and salt stress tolerance in transgenic Arabidopsis. Plant Physiol. Biochem. 2020, 151, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Han, G.; Sun, C.; Sui, N. Research advances of MYB transcription factors in plant stress resistance and breeding. Plant Signal Behav. 2019, 14, 1613131. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, S.; Zhang, N.; Zhang, W.; Li, M.; Liu, B.; Shi, Z. MYB-CC transcription factor, TaMYBsm3, cloned from wheat is involved in drought tolerance. BMC Plant Biol. 2019, 19, 143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thirumalaikumar, V.P.; Devkar, V.; Mehterov, N.; Ali, S.; Ozgur, R.; Turkan, I.; Mueller-Roeber, B.; Balazadeh, S. NAC transcription factor JUNGBRUNNEN1 enhances drought tolerance in tomato. Plant Biotechnol. J. 2018, 16, 354–366. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.-Y.; Du, Y.-T.; Ma, J.; Min, D.-H.; Jin, L.-G.; Chen, J.; Chen, M.; Zhou, Y.-B.; Ma, Y.-Z.; Xu, Z.-S.; et al. The WRKY Transcription Factor GmWRKY12 Confers Drought and Salt Tolerance in Soybean. Int. J. Mol. Sci. 2018, 19, 4087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Wang, Y.; Sui, N. Transcriptional regulation of bHLH during plant response to stress. Biochem. Biophys. Res. Commun. 2018, 503, 397–401. [Google Scholar] [CrossRef]
- Xie, Z.; Nolan, T.M.; Jiang, H.; Yin, Y. AP2/ERF transcription factor regulatory networks in hormone and abiotic stress Responses. Front. Plant Sci. 2019, 10, 228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golldack, D.; Li, C.; Mohan, H.; Probst, N. Tolerance to drought and salt stress in plants: Unraveling the signaling networks. Front. Plant Sci. 2014, 5, 151. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pacbio | |
|---|---|
| Subreads base (G) | 30.89 |
| Subreads number | 21,487,251 |
| Average subreads length | 1438 |
| CCS | 258,615 |
| 5′-primer | 249,012 |
| 3′-primer | 249,046 |
| Poly-A | 232,150 |
| Flnc | 168,924 |
| Average flnc read length | 1304 |
| Consensus reads | 99,483 |
| Number of isoform | 62,717 |
| Number of unigene | 31,195 |
| Mean_length | 1564 |
| Min_length | 161 |
| Max_length | 6386 |
| ExN50 (consensus) | 1115 |
| ExN90 (consensus) | 1693 |
| Number of Genes | 31,195 |
| Number of CDS | 30,295 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ni, L.; Wang, Z.; Guo, J.; Pei, X.; Liu, L.; Li, H.; Yuan, H.; Gu, C. Full-Length Transcriptome Sequencing and Comparative Transcriptome Analysis to Evaluate Drought and Salt Stress in Iris lactea var. chinensis. Genes 2021, 12, 434. https://doi.org/10.3390/genes12030434
Ni L, Wang Z, Guo J, Pei X, Liu L, Li H, Yuan H, Gu C. Full-Length Transcriptome Sequencing and Comparative Transcriptome Analysis to Evaluate Drought and Salt Stress in Iris lactea var. chinensis. Genes. 2021; 12(3):434. https://doi.org/10.3390/genes12030434
Chicago/Turabian StyleNi, Longjie, Zhiquan Wang, Jinbo Guo, Xiaoxiao Pei, Liangqin Liu, Huogen Li, Haiyan Yuan, and Chunsun Gu. 2021. "Full-Length Transcriptome Sequencing and Comparative Transcriptome Analysis to Evaluate Drought and Salt Stress in Iris lactea var. chinensis" Genes 12, no. 3: 434. https://doi.org/10.3390/genes12030434
APA StyleNi, L., Wang, Z., Guo, J., Pei, X., Liu, L., Li, H., Yuan, H., & Gu, C. (2021). Full-Length Transcriptome Sequencing and Comparative Transcriptome Analysis to Evaluate Drought and Salt Stress in Iris lactea var. chinensis. Genes, 12(3), 434. https://doi.org/10.3390/genes12030434