
Shukla-Vernon Syndrome: A Second Family with a Novel Variant in the BCORL1 Gene

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
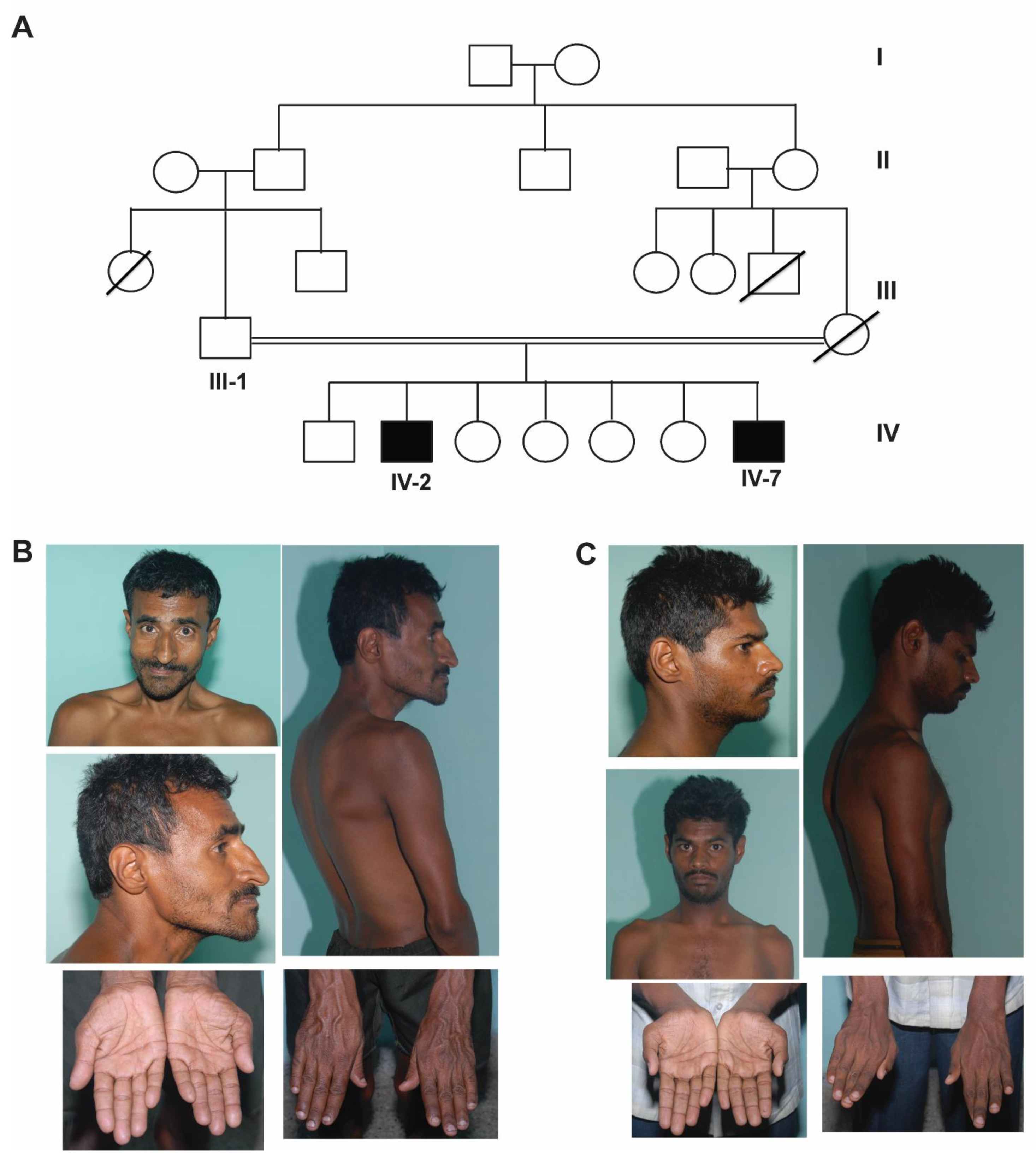
2.1. Patients
2.2. G-Banded Karyotyping and Whole Exome Sequencing
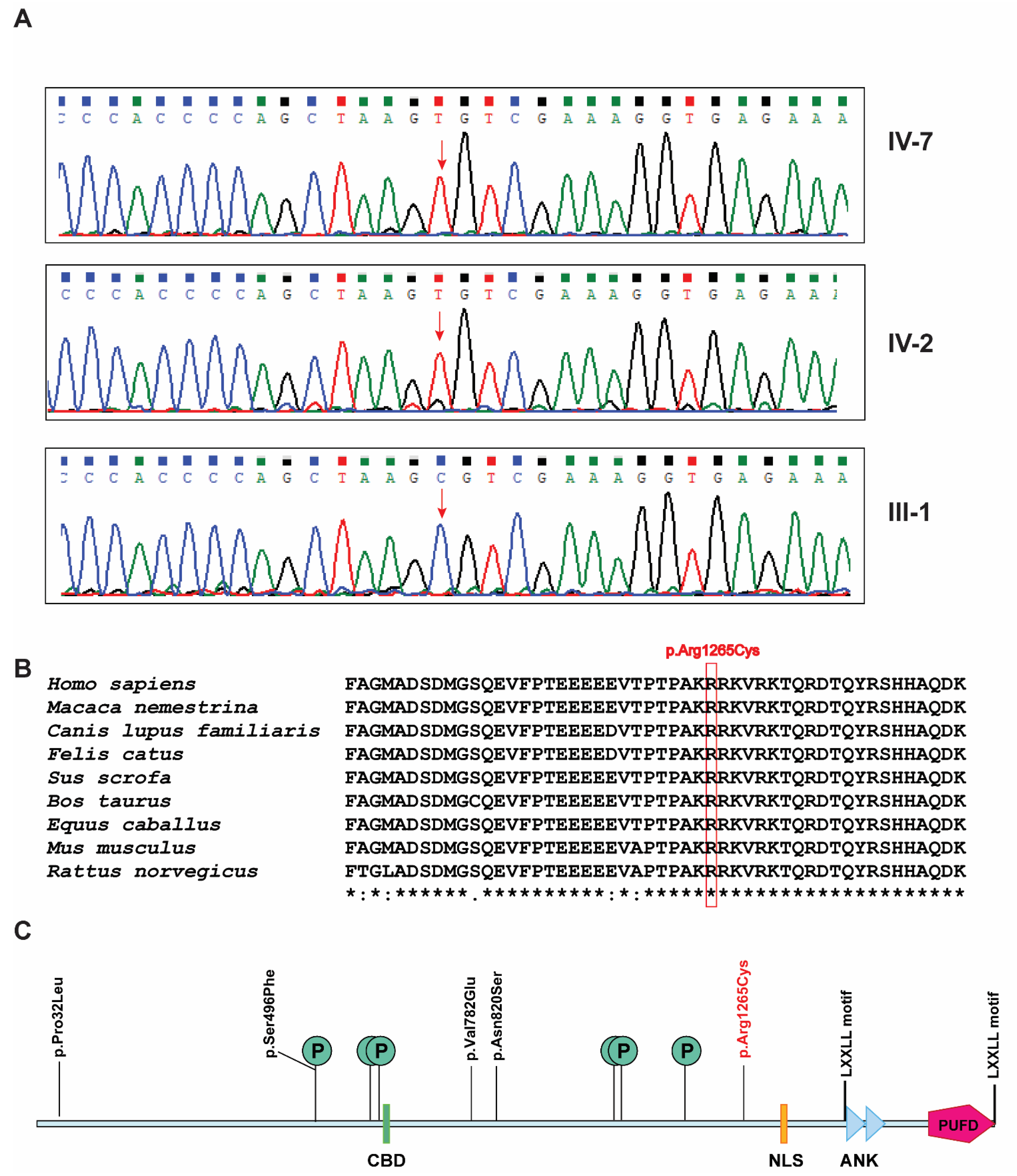
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shukla, A.; Girisha, K.M.; Somashekar, P.H.; Nampoothiri, S.; McClellan, R.; Vernon, H.J. Variants in the transcriptional corepressor BCORL1 are associated with an X-linked disorder of intellectual disability, dysmorphic features, and behavioral abnormalities. Am. J. Med. Genet. Part A 2019, 179, 870–874. [Google Scholar] [CrossRef] [PubMed]
- Schuurs-Hoeijmakers, J.H.M.; Silfhout, A.T.V.-V.; Vissers, L.E.L.M.; Vondervoort, I.I.G.M.V.D.; Van Bon, B.W.M.; De Ligt, J.; Gilissen, C.; Hehir-Kwa, J.Y.; Neveling, K.; Del Rosario, M.; et al. Identification of pathogenic gene variants in small families with intellectually disabled siblings by exome sequencing. J. Med. Genet. 2013, 50, 802–811. [Google Scholar] [CrossRef]
- Pagan, J.K.; Arnold, J.; Hanchard, K.J.; Kumar, R.; Bruno, T.; Jones, M.J.K.; Richard, D.J.; Forrest, A.; Spurdle, A.; Verdin, E.; et al. A Novel Corepressor, BCoR-L1, Represses Transcription through an Interaction with CtBP. J. Biol. Chem. 2007, 282, 15248–15257. [Google Scholar] [CrossRef] [Green Version]
- Junco, S.E.; Wang, R.; Gaipa, J.C.; Taylor, A.B.; Schirf, V.; Gearhart, M.D.; Bardwell, V.J.; Demeler, B.; Hart, P.J.; Kim, C.A. Structure of the Polycomb Group Protein PCGF1 in Complex with BCOR Reveals Basis for Binding Selectivity of PCGF Homologs. Structure 2013, 21, 665–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, S.J.; Gearhart, M.D.; Taylor, A.B.; Nanyes, D.R.; Ha, D.J.; Robinson, A.K.; Artigas, J.A.; Lee, O.J.; Demeler, B.; Hart, P.J.; et al. KDM2B Recruitment of the Polycomb Group Complex, PRC1.1, Requires Cooperation between PCGF1 and BCORL1. Structure 2016, 24, 1795–1801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ Data to High-Confidence Variant Calls: The Genome Analysis Toolkit Best Practices Pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef]
- Abecasis, G.R.; Auton, A.; Brooks, L.D.; DePristo, M.A.; Durbin, R.M.; Handsaker, R.E.; Kang, H.M.; Marth, G.T.; McVean, G.A. An integrated map of genetic variation from 1,092 human genomes. Nature 2012, 491, 56–65. [Google Scholar] [PubMed] [Green Version]
- Vaser, R.; Adusumalli, S.; Leng, S.N.; Sikic, M.; Ng, P.C. SIFT missense predictions for genomes. Nat. Protoc. 2016, 11, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting Functional Effect of Human Missense Mutations Using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, 7.20.1–7.20.41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef]
- Vigeland, M.D.; Gjøtterud, K.S.; Selmer, K.K. FILTUS: A desktop GUI for fast and efficient detection of disease-causing variants, including a novel autozygosity detector. Bioinformatics 2016, 32, 1592–1594. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Shukla et al., 2019 | Present Study | ||||||
|---|---|---|---|---|---|---|---|
| Characteristics | Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 | Patient 1 | Patient 2 |
| BCORL1 variant | c.2345T>A p.(Val782Glu) | c.1487C>T p.(Ser496Phe) | c.95C>T p.(Pro32Leu) | c.95C>T p.(Pro32Leu) | c.95C>T p.(Pro32Leu) | c.3793C>T p.Arg1265Cys | c.3793C>T p.Arg1265Cys |
| Sex | Male | Male | Male | Male | Male | Male | Male |
| Gestation | 32 weeks | 40 weeks | 40 weeks | Term | Term | Term | Term |
| Age at the time of examination | 15 years 3 months | 7 years | 15 years | 4 years | 3 years 4 months | 35 years | 24 years |
| Weight at the time of examination | 52.7 kg (−0.53 SD) | 17.8 kg (−1.9 SD) | 33 kg (−3.05 SD) | 11 kg (−3.25 SD) | 9.6 kg (−3.6 SD) | Not available | Not available |
| Height at the time of examination | 56 cm (+0.71 SD) | 53 cm (+0.74 SD) | 52 cm (−1.9 SD) | 47. 5 cm (−2 SD) | 45 cm (−3.4 SD) | 152 cm (−3.4 SD) | 161 cm (−2.1 SD) |
| Intellectual disability | Mild | No | Severe | Severe | Severe | Moderate (IQ = 39) | Moderate (IQ = 47) |
| Motor delay | Mild | Mild | Yes | No | Yes | Yes | Yes |
| Developmental milestones | First word, 8 months Walking, 24 months | Not available | No speech attained Walking, 3 years 6 months | First word, 3 years Walking, 1 year | First word, 3 years 6 months Walking, 1 year 6 months | Walking with support, 2 years Running, 7 years Two meaningful words, >5 years Short sentences, 20 years Drinking from glass unassisted, 5 years Feeding self, 10 years Dressing self, 10 years Schooling at 4 years | Walking without support, 7 years Running, 10 years Two meaningful words, 5 years Sentence, 10 years Drinking from glass unassisted, 5 years Fully toilet-trained, 10 years Schooling at 4 years Left school |
| Seizures | No | No | At 1 year of age | At 2 years of age | At 6 months of age | No | Yes, at the age of 7 years |
| Episodes of facial weakness | No | No | No | No | No | Yes | No |
| Behavioural abnormalities | Autism spectrum disorder, impulsive behavior, mild aggressive behavior, ADHD | Autism spectrum disorder, impulsive behavior, aggressive behavior, attention-deficit hyperactivity disorder | Autism-spectrum disorder | Autism-spectrum Disorder | Autism-spectrum disorder | None; quiet and co-operative | None; quiet and co-operative |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muthusamy, B.; Bellad, A.; Girimaji, S.C.; Pandey, A. Shukla-Vernon Syndrome: A Second Family with a Novel Variant in the BCORL1 Gene. Genes 2021, 12, 452. https://doi.org/10.3390/genes12030452
Muthusamy B, Bellad A, Girimaji SC, Pandey A. Shukla-Vernon Syndrome: A Second Family with a Novel Variant in the BCORL1 Gene. Genes. 2021; 12(3):452. https://doi.org/10.3390/genes12030452
Chicago/Turabian StyleMuthusamy, Babylakshmi, Anikha Bellad, Satish Chandra Girimaji, and Akhilesh Pandey. 2021. "Shukla-Vernon Syndrome: A Second Family with a Novel Variant in the BCORL1 Gene" Genes 12, no. 3: 452. https://doi.org/10.3390/genes12030452
APA StyleMuthusamy, B., Bellad, A., Girimaji, S. C., & Pandey, A. (2021). Shukla-Vernon Syndrome: A Second Family with a Novel Variant in the BCORL1 Gene. Genes, 12(3), 452. https://doi.org/10.3390/genes12030452