
Emerging Role of ODC1 in Neurodevelopmental Disorders and Brain Development


 , ,
, , {kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Coding Variant Screening
2.2. Protein Modeling and Dynamics
2.3. Ornithine Decarboxylase (ODC) Enzyme Assay
2.4. Expression Analysis
2.5. Noncoding Variant Screening
2.6. Induced Pluripotent Stem Cell (iPSC) Based Organoid RNA-Seq
3. Results
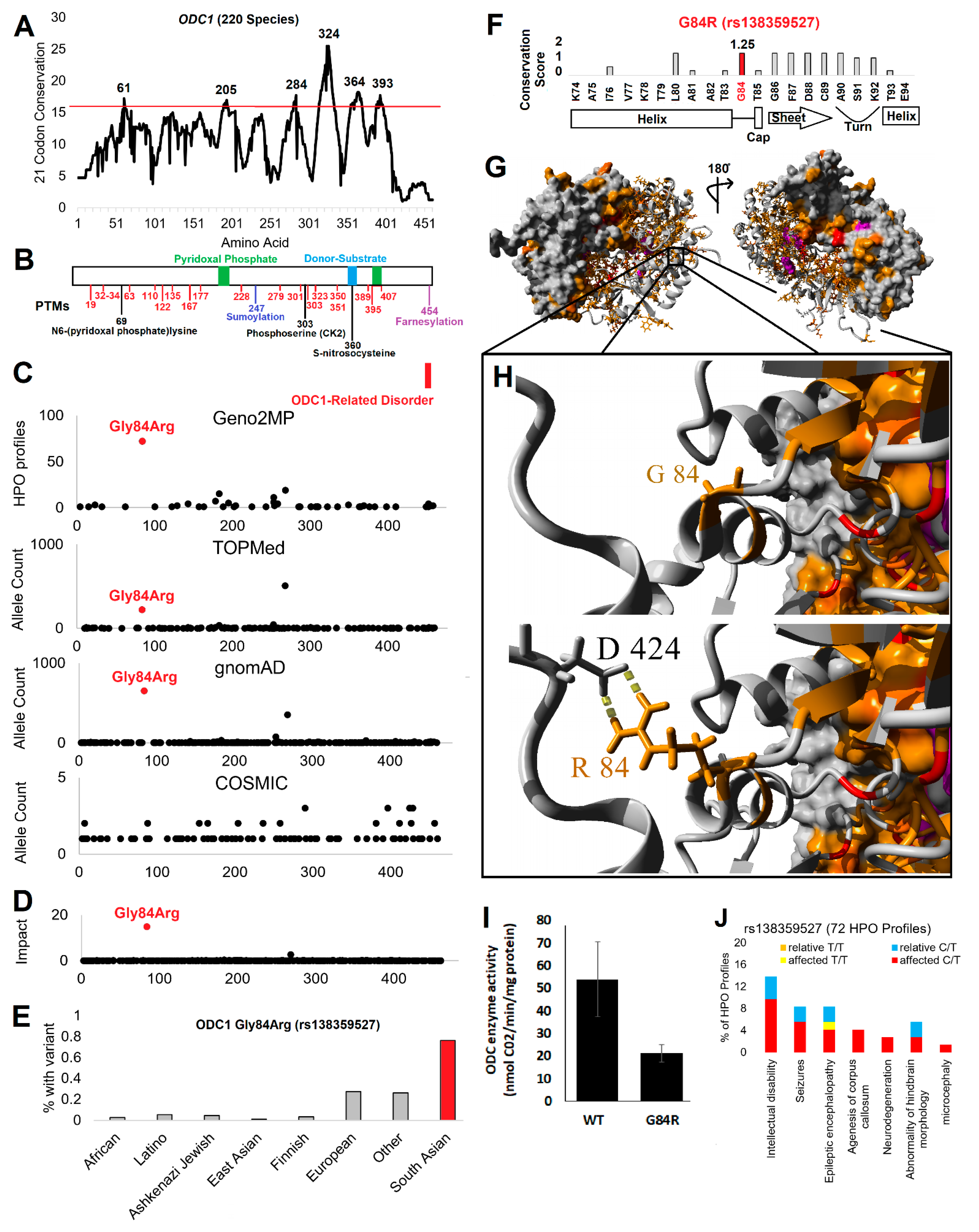
3.1. Human Variation Screen
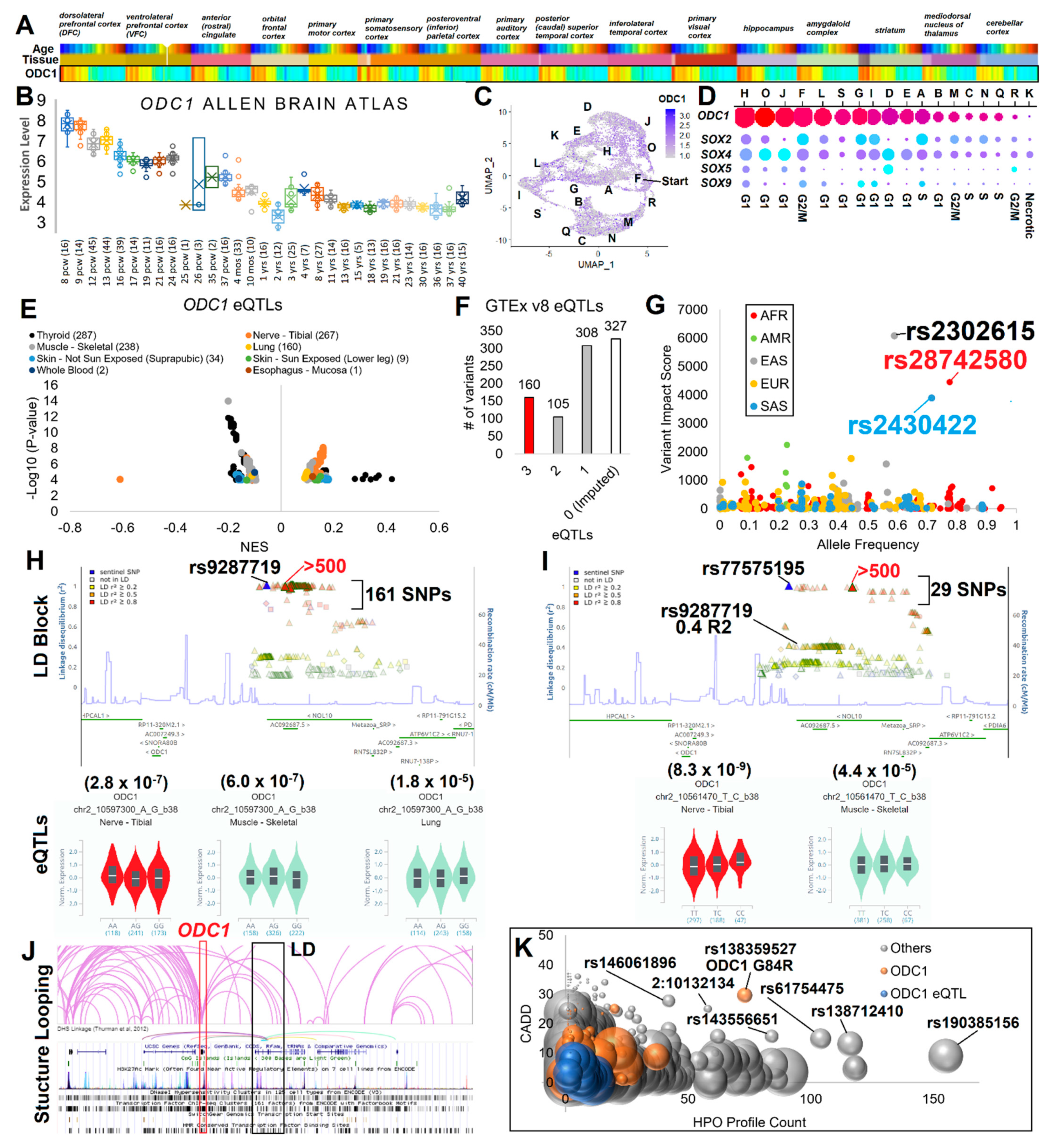
3.2. ODC1 Expression and Gene Regulation Variants
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pegg, A.E. Mammalian Polyamine Metabolism and Function. IUBMB Life 2009, 61, 880–894. [Google Scholar] [CrossRef]
- Pegg, A.E. Functions of Polyamines in Mammals. J. Biol. Chem. 2016, 291, 14904–14912. [Google Scholar] [CrossRef]
- Hogarty, M.D.; Norris, M.D.; Davis, K.; Liu, X.; Evageliou, N.F.; Hayes, C.S.; Pawel, B.; Guo, R.; Zhao, H.; Sekyere, E.; et al. ODC1 Is a Critical Determinant of MYCN Oncogenesis and a Therapeutic Target in Neuroblastoma. Cancer Res. 2008, 68, 9735–9745. [Google Scholar] [CrossRef] [PubMed]
- Bello-Fernandez, C.; Cleveland, J.L. C-Myc Transactivates the Ornithine Decarboxylase Gene. In Mechanisms in B-Cell Neoplasia 1992; Springer: Berlin/Heidelberg, Germnay, 1992; pp. 445–452. [Google Scholar] [CrossRef]
- Bachmann, A.S.; Geerts, D. Polyamine Synthesis as a Target of MYC Oncogenes. J. Biol. Chem. 2018, 293, 18757–18769. [Google Scholar] [CrossRef] [PubMed]
- Zell, J.A.; Ziogas, A.; Ignatenko, N.; Honda, J.; Qu, N.; Bobbs, A.S.; Neuhausen, S.L.; Gerner, E.W.; Anton-Culver, H. Associations of a Polymorphism in the Ornithine Decarboxylase Gene with Colorectal Cancer Survival. Clin. Cancer Res. 2009, 15, 6208–6216. [Google Scholar] [CrossRef]
- Martinez, M.E.; O’Brien, T.G.; Fultz, K.E.; Babbar, N.; Yerushalmi, H.; Qu, N.; Guo, Y.; Boorman, D.; Einspahr, J.; Alberts, D.S.; et al. Pronounced Reduction in Adenoma Recurrence Associated with Aspirin Use and a Polymorphism in the Ornithine Decarboxylase Gene. Proc. Natl. Acad. Sci. USA 2003, 100, 7859–7864. [Google Scholar] [CrossRef] [PubMed]
- Cho, L.Y.; Yang, J.J.; Ko, K.-P.; Ma, S.H.; Shin, A.; Choi, B.Y.; Kim, H.J.; Han, D.S.; Song, K.S.; Kim, Y.S.; et al. Gene Polymorphisms in the Ornithine Decarboxylase-Polyamine Pathway Modify Gastric Cancer Risk by Interaction with Isoflavone Concentrations. Gastric Cancer 2015, 18, 495–503. [Google Scholar] [CrossRef]
- Brown, I.; Halliday, S.; Greig, H.; Heys, S.D.; Wallace, H.M.; Schofield, A.C. Genetic Polymorphism in Ornithine Decarboxylase and Risk of Breast Cancer. Fam. Cancer 2009, 8, 307–311. [Google Scholar] [CrossRef]
- Visvanathan, K.; Helzlsouer, K.J.; Boorman, D.W.; Strickland, P.T.; Hoffman, S.C.; Comstock, G.W.; O’Brien, T.G.; Guo, Y. Association among an Ornithine Decarboxylase Polymorphism, Androgen Receptor Gene (CAG) Repeat Length and Prostate Cancer Risk. J. Urol. 2004, 171, 652–655. [Google Scholar] [CrossRef] [PubMed]
- Casero, R.A.; Marton, L.J. Targeting Polyamine Metabolism and Function in Cancer and Other Hyperproliferative Diseases. Nat. Rev. Drug Discov. 2007, 6, 373–390. [Google Scholar] [CrossRef]
- Wallick, C.J.; Gamper, I.; Thorne, M.; Feith, D.J.; Takasaki, K.Y.; Wilson, S.M.; Seki, J.A.; Pegg, A.E.; Byus, C.V.; Bachmann, A.S. Key Role for P27Kip1, Retinoblastoma Protein Rb, and MYCN in Polyamine Inhibitor-Induced G1 Cell Cycle Arrest in MYCN-Amplified Human Neuroblastoma Cells. Oncogene 2005, 24, 5606–5618. [Google Scholar] [CrossRef] [PubMed]
- Meyskens, F.L.; McLaren, C.E.; Pelot, D.; Fujikawa-Brooks, S.; Carpenter, P.M.; Hawk, E.; Kelloff, G.; Lawson, M.J.; Kidao, J.; McCracken, J.; et al. Difluoromethylornithine plus Sulindac for the Prevention of Sporadic Colorectal Adenomas: A Randomized Placebo-Controlled, Double-Blind Trial. Cancer Prev. Res. 2008, 1, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Sholler, G.L.S.; Gerner, E.W.; Bergendahl, G.; MacArthur, R.B.; VanderWerff, A.; Ashikaga, T.; Bond, J.P.; Ferguson, W.; Roberts, W.; Wada, R.K.; et al. A Phase I Trial of DFMO Targeting Polyamine Addiction in Patients with Relapsed/Refractory Neuroblastoma. PLoS ONE 2015, 10, e0127246. [Google Scholar] [CrossRef]
- Lewis, E.C.; Kraveka, J.M.; Ferguson, W.; Eslin, D.; Brown, V.I.; Bergendahl, G.; Roberts, W.; Wada, R.K.; Oesterheld, J.; Mitchell, D.; et al. A Subset Analysis of a Phase II Trial Evaluating the Use of DFMO as Maintenance Therapy for High-Risk Neuroblastoma. Int. J. Cancer 2020, 147, 3152–3159. [Google Scholar] [CrossRef]
- Smith, K.J.; Skelton, H. Alpha-Difluoromethylornithine, a Polyamine Inhibitor: Its Potential Role in Controlling Hair Growth and in Cancer Treatment and Chemo-Prevention. Int. J. Dermatol. 2006, 45, 337–344. [Google Scholar] [CrossRef]
- Garton, R.A.; McMichael, A.J.; Sugarman, J.; Greer, K.; Setaluri, V. Association of a Polymorphism in the Ornithine Decarboxylase Gene with Male Androgenetic Alopecia. J. Am. Acad. Dermatol. 2005, 52, 535–536. [Google Scholar] [CrossRef]
- Meehan, T.F.; Conte, N.; West, D.B.; Jacobsen, J.O.; Mason, J.; Warren, J.; Chen, C.-K.; Tudose, I.; Relac, M.; Matthews, P.; et al. Disease Model Discovery from 3,328 Gene Knockouts by The International Mouse Phenotyping Consortium. Nat. Genet. 2017, 49, 1231–1238. [Google Scholar] [CrossRef]
- Carvalho-Silva, D.; Pierleoni, A.; Pignatelli, M.; Ong, C.; Fumis, L.; Karamanis, N.; Carmona, M.; Faulconbridge, A.; Hercules, A.; McAuley, E.; et al. Open Targets Platform: New Developments and Updates Two Years On. Nucleic Acids Res. 2019, 47, D1056–D1065. [Google Scholar] [CrossRef]
- Hussain, T.; Tan, B.; Ren, W.; Rahu, N.; Kalhoro, D.H.; Yin, Y. Exploring Polyamines: Functions in Embryo/Fetal Development. Anim. Nutr. 2017, 3, 7–10. [Google Scholar] [CrossRef] [PubMed]
- Coffino, P. Regulation of Cellular Polyamines by Antizyme. Nat. Rev. Mol. Cell. Biol. 2001, 2, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Heller, J.S.; Canellakis, E.S. Cellular Control of Ornithine Decarboxylase Activity by Its Antizyme. J. Cell Physiol. 1981, 107, 209–217. [Google Scholar] [CrossRef]
- Rom, E.; Kahana, C. Polyamines Regulate the Expression of Ornithine Decarboxylase Antizyme in Vitro by Inducing Ribosomal Frame-Shifting. Proc. Natl. Acad. Sci. USA 1994, 91, 3959–3963. [Google Scholar] [CrossRef]
- Suzuki, T.; He, Y.; Kashiwagi, K.; Murakami, Y.; Hayashi, S.; Igarashi, K. Antizyme Protects against Abnormal Accumulation and Toxicity of Polyamines in Ornithine Decarboxylase-Overproducing Cells. Proc. Natl. Acad. Sci. USA 1994, 91, 8930–8934. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, S.; Murakami, Y.; Matsufuji, S. Ornithine Decarboxylase Antizyme: A Novel Type of Regulatory. Protein Trends Biochem. Sci. 1996, 21, 27–30. [Google Scholar] [CrossRef]
- Fujita, K.; Murakami, Y.; Hayashi, S. A Macromolecular Inhibitor of the Antizyme to Ornithine Decarboxylase. Biochem. J. 1982, 204, 647–652. [Google Scholar] [CrossRef] [PubMed]
- Murakami, Y.; Ichiba, T.; Matsufuji, S.; Hayashi, S. Cloning of Antizyme Inhibitor, a Highly Homologous Protein to Ornithine Decarboxylase. J. Biol. Chem. 1996, 271, 3340–3342. [Google Scholar] [CrossRef] [PubMed]
- Sequerra, E.B.; Gardino, P.; Hedin-Pereira, C.; de Mello, F.G. Putrescine as an Important Source of GABA in the Postnatal Rat Subventricular Zone. Neuroscience 2007, 146, 489–493. [Google Scholar] [CrossRef]
- Halonen, T.; Sivenius, J.; Miettinen, R.; Halmekytö, M.; Kauppinen, R.; Sinervirta, R.; Alakuijala, L.; Alhonen, L.; MacDonald, E.; Jänne, J. Elevated Seizure Threshold and Impaired Spatial Learning in Transgenic Mice with Putrescine Overproduction in the Brain. Eur. J. Neurosci. 1993, 5, 1233–1239. [Google Scholar] [CrossRef]
- Ganapathi, M.; Padgett, L.R.; Yamada, K.; Devinsky, O.; Willaert, R.; Person, R.; Au, P.-Y.B.; Tagoe, J.; McDonald, M.; Karlowicz, D.; et al. Recessive Rare Variants in Deoxyhypusine Synthase, an Enzyme Involved in the Synthesis of Hypusine, Are Associated with a Neurodevelopmental Disorder. Am. J. Hum. Genet. 2019, 104, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Bupp, C.P.; Schultz, C.R.; Uhl, K.L.; Rajasekaran, S.; Bachmann, A.S. Novel de Novo Pathogenic Variant in the ODC1 Gene in a Girl with Developmental Delay, Alopecia, and Dysmorphic Features. Am. J. Med. Genet. Part A 2018, 176, 2548–2553. [Google Scholar] [CrossRef]
- Rodan, L.H.; Anyane-Yeboa, K.; Chong, K.; Wassink-Ruiter, J.S.K.; Wilson, A.; Smith, L.; Kothare, S.V.; Rajabi, F.; Blaser, S.; Ni, M.; et al. Gain-of-Function Variants in the ODC1 Gene Cause a Syndromic Neurodevelopmental Disorder Associated with Macrocephaly, Alopecia, Dysmorphic Features, and Neuroimaging Abnormalities. Am. J. Med. Genet. Part A 2018, 176, 2554–2560. [Google Scholar] [CrossRef]
- Schultz, C.R.; Bupp, C.P.; Rajasekaran, S.; Bachmann, A.S. Biochemical Features of Primary Cells from a Pediatric Patient with a Gain-of-Function ODC1 Genetic Mutation. Biochem. J. 2019, 476, 2047–2057. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De Novo Transcript Sequence Reconstruction from RNA-Seq Using the Trinity Platform for Reference Generation and Analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X Version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]
- Prokop, J.W.; Lazar, J.; Crapitto, G.; Smith, D.C.; Worthey, E.A.; Jacob, H.J. Molecular Modeling in the Age of Clinical Genomics, the Enterprise of the next Generation. J. Mol. Model. 2017, 23, 75. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. ClinVar: Public Archive of Relationships among Sequence Variation and Human Phenotype. Nucl. Acids Res. 2014, D980–D985. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The Mutational Constraint Spectrum Quantified from Variation in 141,456 Humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Forbes, S.A.; Bindal, N.; Bamford, S.; Cole, C.; Kok, C.Y.; Beare, D.; Jia, M.; Shepherd, R.; Leung, K.; Menzies, A.; et al. COSMIC: Mining Complete Cancer Genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2010, 39, D945–D950. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Koraimann, G.; Vriend, G. Increasing the Precision of Comparative Models with YASARA NOVA--a Self-Parameterizing Force Field. Proteins Struct. Funct. Bioinform. 2002, 47, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M.C.; Xiong, G.; Zhang, W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T.; et al. A Point-Charge Force Field for Molecular Mechanics Simulations of Proteins Based on Condensed-Phase Quantum Mechanical Calculations. J. Comput. Chem. 2003, 24, 1999–2012. [Google Scholar] [CrossRef]
- Ashkenazy, H.; Erez, E.; Martz, E.; Pupko, T.; Ben-Tal, N. ConSurf 2010: Calculating Evolutionary Conservation in Sequence and Structure of Proteins and Nucleic Acids. Nucleic Acids Res. 2010, 38, W529–W533. [Google Scholar] [CrossRef]
- GTEx Consortium; Laboratory, Data Analysis &Coordinating Center (LDACC)—Analysis Working Group; Statistical Methods groups—Analysis Working Group; Enhancing GTEx (eGTEx) Groups; NIH Common Fund; NIH/NCI; NIH/NHGRI; NIH/NIMH; NIH/NIDA; Biospecimen Collection Source Site—NDRI; et al. Genetic Effects on Gene Expression across Human Tissues. Nature 2017, 550, 204–213. [Google Scholar] [CrossRef]
- Uhlen, M.; Oksvold, P.; Fagerberg, L.; Lundberg, E.; Jonasson, K.; Forsberg, M.; Zwahlen, M.; Kampf, C.; Wester, K.; Hober, S.; et al. Towards a Knowledge-Based Human Protein Atlas. Nat. Biotechnol. 2010, 28, 1248–1250. [Google Scholar] [CrossRef] [PubMed]
- Franzén, O.; Gan, L.-M.; Björkegren, J.L.M. PanglaoDB: A Web Server for Exploration of Mouse and Human Single-Cell RNA Sequencing Data. Database 2019. [Google Scholar] [CrossRef]
- Tabula Muris Consortium; Overall coordination; Logistical coordination; Organ collection and processing; Library preparation and sequencing; Computational data analysis; Cell type annotation; Writing group; Supplemental text writing group; Principal investigators. Single-Cell Transcriptomics of 20 Mouse Organs Creates a Tabula Muris. Nature 2018, 562, 367–372. [Google Scholar] [CrossRef]
- Sunkin, S.M.; Ng, L.; Lau, C.; Dolbeare, T.; Gilbert, T.L.; Thompson, C.L.; Hawrylycz, M.; Dang, C. Allen Brain Atlas: An Integrated Spatio-Temporal Portal for Exploring the Central Nervous System. Nucleic Acids Res. 2013, 41, D996–D1008. [Google Scholar] [CrossRef] [PubMed]
- Franceschini, A.; Szklarczyk, D.; Frankild, S.; Kuhn, M.; Simonovic, M.; Roth, A.; Lin, J.; Minguez, P.; Bork, P.; von Mering, C.; et al. STRING v9.1: Protein-Protein Interaction Networks, with Increased Coverage and Integration. Nucleic Acids Res. 2013, 41, D808–D815. [Google Scholar] [CrossRef]
- Visel, A.; Thaller, C.; Eichele, G. GenePaint.Org: An Atlas of Gene Expression Patterns in the Mouse Embryo. Nucleic Acids Res. 2004, 32, D552–D556. [Google Scholar] [CrossRef] [PubMed]
- Oscanoa, J.; Sivapalan, L.; Gadaleta, E.; Dayem Ullah, A.Z.; Lemoine, N.R.; Chelala, C. SNPnexus: A Web Server for Functional Annotation of Human Genome Sequence Variation (2020 Update). Nucleic Acids Res. 2020, 48, W185–W192. [Google Scholar] [CrossRef]
- Fishilevich, S.; Nudel, R.; Rappaport, N.; Hadar, R.; Plaschkes, I.; Stein, T.I.; Rosen, N.; Kohn, A.; Twik, M.; Safran, M.; et al. GeneHancer: Genome-Wide Integration of Enhancers and Target Genes in GeneCards. Database 2017. [Google Scholar] [CrossRef]
- Boyle, A.P.; Hong, E.L.; Hariharan, M.; Cheng, Y.; Schaub, M.A.; Kasowski, M.; Karczewski, K.J.; Park, J.; Hitz, B.C.; Weng, S.; et al. Annotation of Functional Variation in Personal Genomes Using RegulomeDB. Genome Res. 2012, 22, 1790–1797. [Google Scholar] [CrossRef]
- Kumar, S.; Ambrosini, G.; Bucher, P. SNP2TFBS–a Database of Regulatory SNPs Affecting Predicted Transcription Factor Binding Site Affinity. Nucleic Acids Res. 2017, 45, D139–D144. [Google Scholar] [CrossRef]
- Bilinovich, S.M.; Uhl, K.L.; Lewis, K.; Soehnlen, X.; Williams, M.; Vogt, D.; Prokop, J.W.; Campbell, D.B. Integrated RNA Sequencing Reveals Epigenetic Impacts of Diesel Particulate Matter Exposure in Human Cerebral Organoids. Dev. Neurosci. 2021, 1–13. [Google Scholar] [CrossRef]
- Srivastava, A.; Malik, L.; Smith, T.; Sudbery, I.; Patro, R. Alevin Efficiently Estimates Accurate Gene Abundances from DscRNA-Seq Data. Genome Biol. 2019, 20, 65. [Google Scholar] [CrossRef]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M.; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive Integration of Single-Cell Data. Cell 2019, 177, 1888–1902. [Google Scholar] [CrossRef]
- Heiskala, M.; Zhang, J.; Hayashi, S.; Hölttä, E.; Andersson, L.C. Translocation of Ornithine Decarboxylase to the Surface Membrane during Cell Activation and Transformation. EMBO J. 1999, 18, 1214–1222. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; MacDonald, A.I.; Hoyt, M.A.; Coffino, P. Proteasomes Begin Ornithine Decarboxylase Digestion at the C Terminus. J. Biol. Chem. 2004, 279, 20959–20965. [Google Scholar] [CrossRef] [PubMed]
- Ellis, P.; Fagan, B.M.; Magness, S.T.; Hutton, S.; Taranova, O.; Hayashi, S.; McMahon, A.; Rao, M.; Pevny, L. SOX2, a Persistent Marker for Multipotential Neural Stem Cells Derived from Embryonic Stem Cells, the Embryo or the Adult. Dev. Neurosci. 2004, 26, 148–165. [Google Scholar] [CrossRef]
- Auvinen, M.; Paasinen, A.; Andersson, L.C.; Hölttä, E. Ornithine Decarboxylase Activity Is Critical for Cell Transformation. Nature 1992, 360, 355–358. [Google Scholar] [CrossRef] [PubMed]
- Cervelli, M.; Angelucci, E.; Germani, F.; Amendola, R.; Mariottini, P. Inflammation, Carcinogenesis and Neurodegeneration Studies in Transgenic Animal Models for Polyamine Research. Amino Acids 2014, 46, 521–530. [Google Scholar] [CrossRef]
- Krab, L.C.; Goorden, S.M.I.; Elgersma, Y. Oncogenes on My Mind: ERK and MTOR Signaling in Cognitive Diseases. Trends Genet. 2008, 24, 498–510. [Google Scholar] [CrossRef]
- Müller, R.; Slamon, D.J.; Tremblay, J.M.; Cline, M.J.; Verma, I.M. Differential Expression of Cellular Oncogenes during Pre- and Postnatal Development of the Mouse. Nature 1982, 299, 640–644. [Google Scholar] [CrossRef]
- Morrison, L.D.; Cao, X.C.; Kish, S.J. Ornithine Decarboxylase in Human Brain: Influence of Aging, Regional Distribution, and Alzheimer’s Disease. J. Neurochem. 1998, 71, 288–294. [Google Scholar] [CrossRef]
- Fiori, L.M.; Turecki, G. Implication of the Polyamine System in Mental Disorders. J. Psychiatry. Neurosci. 2008, 33, 102–110. [Google Scholar]
- Raghavendra Rao, V.L.; Dogan, A.; Bowen, K.K.; Dempsey, R.J. Ornithine Decarboxylase Knockdown Exacerbates Transient Focal Cerebral Ischemia-Induced Neuronal Damage in Rat Brain. J. Cereb. Blood Flow Metab. 2001, 21, 945–954. [Google Scholar] [CrossRef]
- Knickmeyer, R.; Baron-Cohen, S.; Raggatt, P.; Taylor, K. Foetal Testosterone, Social Relationships, and Restricted Interests in Children. J. Child Psychol. Psychiatry 2005, 46, 198–210. [Google Scholar] [CrossRef] [PubMed]
- Baron-Cohen, S.; Knickmeyer, R.C.; Belmonte, M.K. Sex Differences in the Brain: Implications for Explaining Autism. Science 2005, 310, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Manteuffel-Cymborowska, M.; Peska, M.; Chmurzyńska, W.; Grzelakowska-Sztabert, B. Catecholamines Are Required for Androgen-Induced ODC Expression but Not for Hypertrophy of Mouse Kidney. Biochim. Biophys. Acta Mol. Cell Res. 1997, 1356, 292–298. [Google Scholar] [CrossRef]
- Grzelakowska-Sztabert, B.; Dudkowska, M.; Manteuffel-Cymborowska, M. Nuclear and Membrane Receptor-Mediated Signalling Pathways Modulate Polyamine Biosynthesis and Interconversion. Biochem. Soc. Trans. 2007, 35, 386–390. [Google Scholar] [CrossRef]
- Kwekel, J.C.; Burgoon, L.D.; Burt, J.W.; Harkema, J.R.; Zacharewski, T.R. A Cross-Species Analysis of the Rodent Uterotrophic Program: Elucidation of Conserved Responses and Targets of Estrogen Signaling. Physiol. Genom. 2005, 23, 327–342. [Google Scholar] [CrossRef]
- Prokop, J.W.; Yeo, N.C.; Ottmann, C.; Chhetri, S.B.; Florus, K.L.; Ross, E.J.; Sosonkina, N.; Link, B.A.; Freedman, B.I.; Coppola, C.J.; et al. Characterization of Coding/Noncoding Variants ForSHROOM3in Patients with CKD. J. Am. Soc. Nephrol. 2018. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prokop, J.W.; Bupp, C.P.; Frisch, A.; Bilinovich, S.M.; Campbell, D.B.; Vogt, D.; Schultz, C.R.; Uhl, K.L.; VanSickle, E.; Rajasekaran, S.; et al. Emerging Role of ODC1 in Neurodevelopmental Disorders and Brain Development. Genes 2021, 12, 470. https://doi.org/10.3390/genes12040470
Prokop JW, Bupp CP, Frisch A, Bilinovich SM, Campbell DB, Vogt D, Schultz CR, Uhl KL, VanSickle E, Rajasekaran S, et al. Emerging Role of ODC1 in Neurodevelopmental Disorders and Brain Development. Genes. 2021; 12(4):470. https://doi.org/10.3390/genes12040470
Chicago/Turabian StyleProkop, Jeremy W., Caleb P. Bupp, Austin Frisch, Stephanie M. Bilinovich, Daniel B. Campbell, Daniel Vogt, Chad R. Schultz, Katie L. Uhl, Elizabeth VanSickle, Surender Rajasekaran, and et al. 2021. "Emerging Role of ODC1 in Neurodevelopmental Disorders and Brain Development" Genes 12, no. 4: 470. https://doi.org/10.3390/genes12040470
APA StyleProkop, J. W., Bupp, C. P., Frisch, A., Bilinovich, S. M., Campbell, D. B., Vogt, D., Schultz, C. R., Uhl, K. L., VanSickle, E., Rajasekaran, S., & Bachmann, A. S. (2021). Emerging Role of ODC1 in Neurodevelopmental Disorders and Brain Development. Genes, 12(4), 470. https://doi.org/10.3390/genes12040470