
Transcriptional Dynamics of Genes Purportedly Involved in the Control of Meiosis, Carbohydrate, and Secondary Metabolism during Sporulation in Ganoderma lucidum

 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection at Differential Stages
2.2. Total RNA Extraction and Sequencing
2.3. Transcriptome Assembly and Gene Annotation
2.4. Transcriptional Regulatory Pathway Prediction
2.5. Quantitative Real-Time PCR (qRT-PCR) Validation
3. Results
3.1. Differences between Sporulation Stages were Evident at the Transcriptional Level
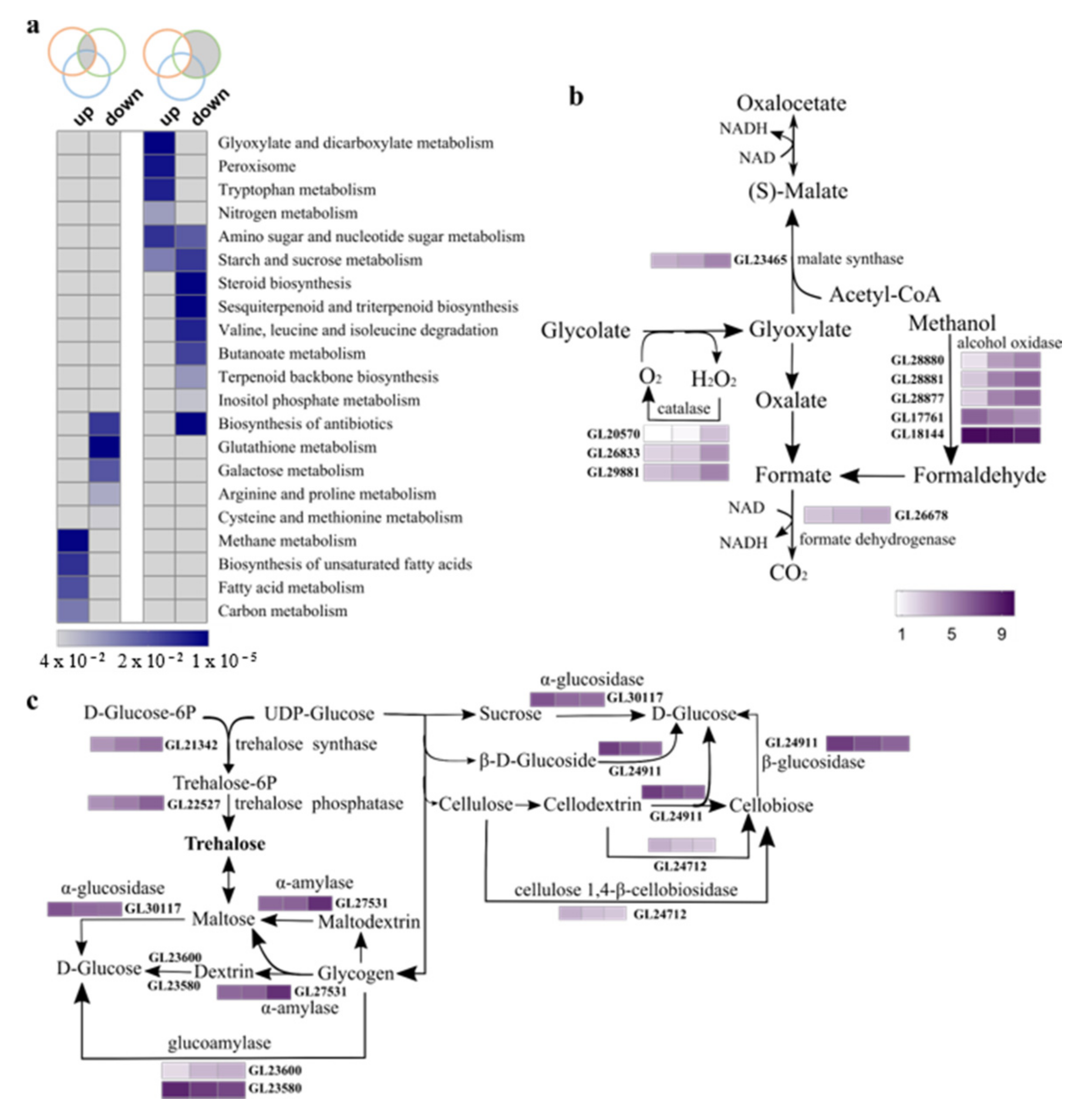
3.2. Functional Classification of Differentially Expressed Genes across Three Developmental Stages
3.3. Developmentally Regulated Transcriptional Regulators and Their Possible Regulatory Pathways
3.4. qRT-PCR Validation of the Reliability of the RNA-Seq Analysis
4. Discussions and Conclusions
4.1. Several Developmentally Regulated Transcription Regulators Contribute to Sporulation
4.2. Meiosis-Related Genes Are Necessary for Sporulation
4.3. Carbohydrate Metabolites May Play Dual Roles in Sporulation
4.4. Changes in Expression of Genes That Are Predictably Involved in Bioactive Metabolites Biosynthesis during Sporulation
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hsu, K.-D.; Cheng, K.-C. From nutraceutical to clinical trial: Frontiers in Ganoderma development. Appl. Microbiol. Biotechnol. 2018, 102, 9037–9051. [Google Scholar] [CrossRef]
- Xu, J.; Li, P. Researches and Application of Ganoderma Spores Powder. Adv. Exp. Med. Biol. 2019, 1181, 157–186. [Google Scholar] [CrossRef] [PubMed]
- Soccol, C.R.; Bissoqui, L.Y.; Rodrigues, C.; Rubel, R.; Sella, S.R.; Leifa, F.; Vandenberghe, L.P.D.S.; Soccol, V.T. Pharmacological Properties of Biocompounds from Spores of the Lingzhi or Reishi Medicinal Mushroom Ganoderma lucidum (Agaricomycetes): A Review. Int. J. Med. Mushrooms 2016, 18, 757–767. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Yan, S.; Zhang, Z.; Gong, W.; Zhu, Z.; Zhou, Y.; Yan, L.; Hu, Z.; Ai, L.; Peng, Y. Mapping the metabolic signatures of fermentation broth, mycelium, fruiting body and spores powder from Ganoderma lucidum by untargeted metabolomics. LWT 2020, 129, 109494. [Google Scholar] [CrossRef]
- Li, Z.; Zhou, J.; Lin, Z. Development and Innovation of Ganoderma Industry and Products in China. Adv. Exp. Med. Biol. 2019, 1181, 187–204. [Google Scholar] [CrossRef] [PubMed]
- Li, M. Research and development of quality standard and specifications of Ganoderma lucidum from an industry chain perspective. Edible Med. Mushroom 2015, 23, 276–279. [Google Scholar]
- Chen, S.; Xu, J.; Liu, C.; Zhu, Y.; Nelson, D.R.; Zhou, S.; Li, C.; Wang, L.; Guo, X.; Sun, Y.; et al. Genome sequence of the model medicinal mushroom Ganoderma lucidum. Nat. Commun. 2012, 3, 913. [Google Scholar] [CrossRef] [Green Version]
- Mu, D.; Shi, L.; Ren, A.; Li, M.; Wu, F.; Jiang, A.; Zhao, M. The Development and Application of a Multiple Gene Co-Silencing System Using Endogenous URA3 as a Reporter Gene in Ganoderma lucidum. PLoS ONE 2012, 7, e43737. [Google Scholar] [CrossRef]
- Liu, K.; Sun, B.; You, H.; Tu, J.; Yu, X.; Zhao, P.; Xu, J. Dual sgRNA-directed gene deletion in basidiomycete Ganoderma lucidum using the CRISPR/Cas9 system. Microb. Biotechnol. 2019, 13, 386–396. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.-A.; Xiao, H.; Zhong, J.-J. CRISPR-Cas9 assisted functional gene editing in the mushroom Ganoderma lucidum. Appl. Microbiol. Biotechnol. 2019, 104, 1661–1671. [Google Scholar] [CrossRef]
- Hu, Y.; Xu, W.; Hu, S.; Lian, L.; Zhu, J.; Shi, L.; Ren, A.; Zhao, M. InGanoderma lucidum, Glsnf1 regulates cellulose degradation by inhibiting GlCreA during the utilization of cellulose. Environ. Microbiol. 2020, 22, 107–121. [Google Scholar] [CrossRef]
- Zhu, J.; Sun, Z.; Shi, D.; Song, S.; Lian, L.; Shi, L.; Ren, A.; Yu, H.; Zhao, M. Dual functions of AreA, a GATA transcription factor, on influencing ganoderic acid biosynthesis in Ganoderma lucidum. Environ. Microbiol. 2019, 21, 4166–4179. [Google Scholar] [CrossRef]
- Ren, A.; Shi, L.; Zhu, J.; Yu, H.; Jiang, A.; Zheng, H.; Zhao, M. Shedding light on the mechanisms underlying the environmental regulation of secondary metabolite ganoderic acid in Ganoderma lucidum using physiological and genetic methods. Fungal Genet. Biol. 2019, 128, 43–48. [Google Scholar] [CrossRef]
- Sakamoto, Y.; Nakade, K.; Sato, S.; Yoshida, K.; Miyazaki, K.; Natsume, S.; Konno, N. Lentinula edodes Genome Survey and Postharvest Transcriptome Analysis. Appl. Environ. Microbiol. 2017, 83, e02990-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, T.; Hu, C.; Xie, B.; Zhang, L.; Yan, S.; Wang, W.; Tao, Y.; Li, S. A Single Transcription Factor (PDD1) Determines Development and Yield of Winter Mushroom (Flammulina velutipes). Appl. Environ. Microbiol. 2019, 85, 85. [Google Scholar] [CrossRef] [PubMed]
- De Freitas Pereira, M.; Campos, A.N.D.R.; Anastácio, T.C.; Morin, E.; Brommonschenkel, S.H.; Martin, F.; Kohler, A.; Costa, M.D. The transcriptional landscape of basidiosporogenesis in mature Pisolithus microcarpus basidiocarp. BMC Genom. 2017, 18, 157. [Google Scholar] [CrossRef] [Green Version]
- Yoo, S.-I.; Lee, H.-Y.; Markkandan, K.; Moon, S.; Ahn, Y.J.; Ji, S.; Ko, J.; Kim, S.-J.; Ryu, H.; Hong, C.P. Comparative transcriptome analysis identified candidate genes involved in mycelium browning in Lentinula edodes. BMC Genom. 2019, 20, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Zhang, R.; Qiu, Y.; Wu, H.; Xiang, Q.; Yu, X.; Zhao, K.; Zhang, X.; Chen, Q.; Penttinen, P.; et al. RNA-seq Profiling Showed Divergent Carbohydrate-Active Enzymes (CAZymes) Expression Patterns in Lentinula edodes at Brown Film Formation Stage Under Blue Light Induction. Front. Microbiol. 2020, 11, 1044. [Google Scholar] [CrossRef] [PubMed]
- Almási, É.; Sahu, N.; Krizsán, K.; Bálint, B.; Kovács, G.M.; Kiss, B.; Cseklye, J.; Drula, E.; Henrissat, B.; Nagy, I.; et al. Comparative genomics reveals unique wood-decay strategies and fruiting body development in the Schizophyllaceae. New Phytol. 2019, 224, 902–915. [Google Scholar] [CrossRef]
- Krizsán, K.; Almási, É.; Merényi, Z.; Sahu, N.; Virágh, M.; Kószó, T.; Mondo, S.; Kiss, B.; Bálint, B.; Kües, U.; et al. Transcriptomic atlas of mushroom development reveals conserved genes behind complex multicellularity in fungi. Proc. Natl. Acad. Sci. USA 2019, 116, 7409–7418. [Google Scholar] [CrossRef] [Green Version]
- Tan, X.; Sun, J.; Ning, H.; Qin, Z.; Miao, Y.; Sun, T.; Zhang, X. De novo transcriptome sequencing and comprehensive analysis of the heat stress response genes in the basidiomycetes fungus Ganoderma lucidum. Gene 2018, 661, 139–151. [Google Scholar] [CrossRef]
- Ren, A.; Li, M.-J.; Shi, L.; Mu, D.-S.; Jiang, A.-L.; Han, Q.; Zhao, M.-W. Profiling and Quantifying Differential Gene Transcription Provide Insights into Ganoderic Acid Biosynthesis in Ganoderma lucidum in Response to Methyl Jasmonate. PLoS ONE 2013, 8, e65027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, G.-J.; Wang, M.; Huang, J.; Yin, Y.-L.; Chen, Y.-J.; Jiang, S.; Jin, Y.-X.; Lan, X.-Q.; Wong, B.H.C.; Liang, Y.; et al. Deep Insight into the Ganoderma lucidum by Comprehensive Analysis of Its Transcriptome. PLoS ONE 2012, 7, e44031. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-H.; Wu, H.-Y.; Wu, K.-M.; Liu, T.-T.; Liou, R.-F.; Tsai, S.-F.; Shiao, M.-S.; Ho, L.-T.; Tzean, S.-S.; Yang, U.-C. Generation and Analysis of the Expressed Sequence Tags from the Mycelium of Ganoderma lucidum. PLoS ONE 2013, 8, e61127. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Lescot, M.; Déhais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; Van De Peer, Y.; Rouzé, P.; Rombauts, S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Zhao, R.-L.; He, Y.-M. Network pharmacology analysis of the anti-cancer pharmacological mechanisms of Ganoderma lucidum extract with experimental support using Hepa1-6-bearing C57 BL/6 mice. J. Ethnopharmacol. 2018, 210, 287–295. [Google Scholar] [CrossRef]
- Wang, Q.; Xu, M.; Zhao, L.; Wang, F.; Li, Y.; Shi, G.; Ding, Z. Transcriptome dynamics and metabolite analysis revealed the candidate genes and regulatory mechanism of ganoderic acid biosynthesis during liquid superficial-static culture of Ganoderma lucidum. Microb. Biotechnol. 2021, 14, 600–613. [Google Scholar] [CrossRef]
- Schumacher, M.M.; Jun, D.-J.; Johnson, B.M.; DeBose-Boyd, R.A. UbiA prenyltransferase domain–containing protein-1 modulates HMG-CoA reductase degradation to coordinate synthesis of sterol and nonsterol isoprenoids. J. Biol. Chem. 2018, 293, 312–323. [Google Scholar] [CrossRef] [Green Version]
- Hollingsworth, N.M.; Goetsch, L.; Byers, B. The HOP1 gene encodes a meiosis-specific component of yeast chromosomes. Cell 1990, 61, 73–84. [Google Scholar] [CrossRef]
- Kariyazono, R.; Oda, A.; Yamada, T.; Ohta, K. Conserved HORMA domain-containing protein Hop1 stabilizes interaction between proteins of meiotic DNA break hotspots and chromosome axis. Nucleic Acids Res. 2019, 47, 10166–10180. [Google Scholar] [CrossRef] [PubMed]
- Andrianopoulos, A.; Timberlake, W.E. The Aspergillus nidulans abaA gene encodes a transcriptional activator that acts as a genetic switch to control development. Mol. Cell. Biol. 1994, 14, 2503–2515. [Google Scholar] [CrossRef] [Green Version]
- Son, H.; Kim, M.-G.; Min, K.; Seo, Y.-S.; Lim, J.Y.; Choi, G.J.; Kim, J.-C.; Chae, S.-K.; Lee, Y.-W. AbaA Regulates Conidiogenesis in the Ascomycete Fungus Fusarium graminearum. PLoS ONE 2013, 8, e72915. [Google Scholar] [CrossRef] [Green Version]
- Odenbach, D.; Breth, B.; Thines, E.; Weber, R.W.S.; Anke, H.; Foster, A.J. The transcription factor Con7p is a central regulator of infection-related morphogenesis in the rice blast fungus Magnaporthe grisea. Mol. Microbiol. 2007, 64, 293–307. [Google Scholar] [CrossRef]
- Shi, Z.; Christian, D.; Leung, H. Interactions Between Spore Morphogenetic Mutations Affect Cell Types, Sporulation, and Pathogenesis in Magnaporthe grisea. Mol. Plant. Microbe Interact. 1998, 11, 199–207. [Google Scholar] [CrossRef] [Green Version]
- Fan, C.-L.; Han, L.-T.; Jiang, S.-T.; Chang, A.-N.; Zhou, Z.-Y.; Liu, T.-B. The Cys2His2 zinc finger protein Zfp1 regulates sexual reproduction and virulence in Cryptococcus neoformans. Fungal Genet. Biol. 2019, 124, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Baudat, F.; De Massy, B. SPO11: Une activité de coupure de l’ADN indispensable à la méiose. Méd. Sci. 2004, 20, 213–218. [Google Scholar] [CrossRef] [Green Version]
- Kitamura, K.; Katayama, S.; Dhut, S.; Sato, M.; Watanabe, Y.; Yamamoto, M.; Toda, T. Phosphorylation of Mei2 and Ste11 by Pat1 Kinase Inhibits Sexual Differentiation via Ubiquitin Proteolysis and 14-3-3 Protein in Fission Yeast. Dev. Cell 2001, 1, 389–399. [Google Scholar] [CrossRef] [Green Version]
- Sanodiya, B.S.; Thakur, G.S.; Baghel, R.K.; Prasad, G.B.K.S.; Bisen, P.S. Ganoderma lucidum: A potent pharmacological macrofungus. Curr. Pharm. Biotechnol. 2009, 10, 717–742. [Google Scholar] [CrossRef]
- Chu, S.; DeRisi, J.; Eisen, M.; Mulholland, J.; Botstein, D.; Brown, P.O.; Herskowitz, I. The Transcriptional Program of Sporulation in Budding Yeast. Science 1998, 282, 699–705. [Google Scholar] [CrossRef] [Green Version]
- Kassir, Y.; Granot, D.; Simchen, G. IME1, a positive regulator gene of meiosis in S. cerevisiae. Cell 1988, 52, 853–862. [Google Scholar] [CrossRef]
- Jing, Y.; Lin, R. Transcriptional regulatory network of the light signaling pathways. New Phytol. 2020, 227, 683–697. [Google Scholar] [CrossRef] [Green Version]
- Colot, H.V.; Park, G.; Turner, G.E.; Ringelberg, C.; Crew, C.M.; Litvinkova, L.; Weiss, R.L.; Borkovich, K.A.; Dunlap, J.C. A high-throughput gene knockout procedure for Neurospora reveals functions for multiple transcription factors. Proc. Natl. Acad. Sci. USA 2006, 103, 10352–10357. [Google Scholar] [CrossRef] [Green Version]
- Carrillo, A.J.; Schacht, P.; Cabrera, I.E.; Blahut, J.; Prudhomme, L.; Dietrich, S.; Bekman, T.; Mei, J.; Carrera, C.; Chen, V.; et al. Functional Profiling of Transcription Factor Genes in Neurospora crassa. G3 Genes Genomes Genet. 2017, 7, 2945–2956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Wang, F.; Lan, N.; Liu, B.; Hu, C.; Xue, W.; Zhang, Z.; Li, S. The Zn(II)2Cys6-Type Transcription Factor ADA-6 Regulates Conidiation, Sexual Development, and Oxidative Stress Response in Neurospora crassa. Front. Microbiol. 2019, 10, 750. [Google Scholar] [CrossRef] [Green Version]
- Jung, K.-W.; Yang, D.-H.; Maeng, S.; Lee, K.-T.; So, Y.-S.; Hong, J.; Choi, J.; Byun, H.-J.; Kim, H.; Bang, S.; et al. Systematic functional profiling of transcription factor networks in Cryptococcus neoformans. Nat. Commun. 2015, 6, 6757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollingsworth, N.M.; Byers, B. HOP1: A yeast meiotic pairing gene. Genetics 1989, 121, 445–462. [Google Scholar] [CrossRef] [PubMed]
- Bowman, J.L.; Sakakibara, K.; Furumizu, C.; Dierschke, T. Evolution in the Cycles of Life. Annu. Rev. Genet. 2016, 50, 133–154. [Google Scholar] [CrossRef]
- Bennett, R.J.; Turgeon, B.G. Fungal Sex: The Ascomycota. Fungal Kingd. 2017, 4, 117–145. [Google Scholar] [CrossRef]
- Coelho, M.A.; Bakkeren, G.; Sun, S.; Hood, M.E.; Giraud, T. Fungal Sex: The Basidiomycota. Fungal Kingd. 2017, 5, 147–175. [Google Scholar] [CrossRef]
- Seitz, L.C.; Tang, K.; Cummings, W.J.; Zolan, M.E. The rad9 Gene of Coprinus cinereus Encodes a Proline-Rich Protein Required for Meiotic Chromosome Condensation and Synapsis. Genetics 1996, 142, 1105–1117. [Google Scholar] [CrossRef] [PubMed]
- Gerecke, E.E.; Zolan, M.E. An mre11 mutant of Coprinus cinereus has defects in meiotic chromosome pairing, condensation and synapsis. Genetics 2000, 154, 1125–1139. [Google Scholar]
- Ogawa, H.; Johzuka, K.; Nakagawa, T.; Leem, S.H.; Hagihara, A.H. Functions of the yeast meiotic recombination genes, MRE11 and MRE2. Adv. Biophys. 1995, 31, 67–76. [Google Scholar] [CrossRef]
- Okuda, Y.; Murakami, S.; Honda, Y.; Matsumoto, T. AnMSH4Homolog, stpp1, from Pleurotus pulmonarius Is a “Silver Bullet” for Resolving Problems Caused by Spores in Cultivated Mushrooms. Appl. Environ. Microbiol. 2013, 79, 4520–4527. [Google Scholar] [CrossRef] [Green Version]
- Celerin, M.; Merino, S.T.; Stone, J.E.; Menzie, A.M.; Zolan, M.E. Multiple roles of Spo11 in meiotic chromosome behavior. EMBO J. 2000, 19, 2739–2750. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, A.; Sakuno, T.; Watanabe, Y.; Yamamoto, M. Analysis ofSchizosaccharomyces pombeMeiosis. Cold Spring Harb. Protoc. 2017, 2017, 079855. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M. The selective elimination of messenger RNA underlies the mitosis–meiosis switch in fission yeast. Proc. Jpn. Acad. Ser. B 2010, 86, 788–797. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, Y.; Shinozaki-Yabana, S.; Chikashige, Y.; Hiraoka, Y.; Yamamoto, M. Phosphorylation of RNA-binding protein controls cell cycle switch from mitotic to meiotic in fission yeast. Nat. Cell Biol. 1997, 386, 187–190. [Google Scholar] [CrossRef]
- Chikashige, Y.; Kurokawa, R.; Haraguchi, T.; Hiraoka, Y. Meiosis induced by inactivation of Pat1 kinase proceeds with aberrant nuclear positioning of centromeres in the fission yeast Schizosaccharomyces pombe. Genes Cells 2004, 9, 671–684. [Google Scholar] [CrossRef]
- Nag, D.K.; Pata, J.D.; Sironi, M.; Flood, D.R.; Hart, A.M. Both conserved and non-conserved regions of Spo11 are essential for meiotic recombination initiation in yeast. Mol. Genet. Genom. 2006, 276, 313–321. [Google Scholar] [CrossRef]
- Solomon, P.S.; Lee, R.C.; Wilson, T.J.G.; Oliver, R.P. Pathogenicity of Stagonospora nodorum requires malate synthase. Mol. Microbiol. 2004, 53, 1065–1073. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Wang, Y.; Zhao, Y.; Huang, Y.; Zhang, K.-Q.; Yang, J. Malate synthase gene AoMls in the nematode-trapping fungus Arthrobotrys oligospora contributes to conidiation, trap formation, and pathogenicity. Appl. Microbiol. Biotechnol. 2013, 98, 2555–2563. [Google Scholar] [CrossRef]
- Dunn, M.F.; Ramírez-Trujillo, J.A.; Hernández-Lucas, I. Major roles of isocitrate lyase and malate synthase in bacterial and fungal pathogenesis. Microbiology 2009, 155, 3166–3175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thevelein, J.M. Regulation of trehalose mobilization in fungi. Microbiol. Rev. 1984, 48, 42–59. [Google Scholar] [CrossRef] [PubMed]
- Al-Bader, N.; Vanier, G.; Liu, H.; Gravelat, F.N.; Urb, M.; Hoareau, C.M.-Q.; Campoli, P.; Chabot, J.C.; Filler, S.G.; Sheppard, D.C. Role of Trehalose Biosynthesis in Aspergillus fumigatus Development, Stress Response, and Virulence. Infect. Immun. 2010, 78, 3007–3018. [Google Scholar] [CrossRef] [Green Version]
- Botts, M.R.; Huang, M.; Borchardt, R.K.; Hull, C.M. Developmental Cell Fate and Virulence Are Linked to Trehalose Homeostasis in Cryptococcus neoformans. Eukaryot. Cell 2014, 13, 1158–1168. [Google Scholar] [CrossRef] [Green Version]
- Wahl, V.; Ponnu, J.; Schlereth, A.; Arrivault, S.; Langenecker, T.; Franke, A.; Feil, R.; Lunn, J.E.; Stitt, M.; Schmid, M. Regulation of Flowering by Trehalose-6-Phosphate Signaling in Arabidopsis thaliana. Science 2013, 339, 704–707. [Google Scholar] [CrossRef]
- Meitzel, T.; Radchuk, R.; McAdam, E.L.; Thormählen, I.; Feil, R.; Munz, E.; Hilo, A.; Geigenberger, P.; Ross, J.J.; Lunn, J.E.; et al. Trehalose 6-phosphate promotes seed filling by activating auxin biosynthesis. New Phytol. 2021, 229, 1553–1565. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Heitman, J. Chlamydospore Formation during Hyphal Growth in Cryptococcus neoformans. Eukaryot. Cell 2005, 4, 1746–1754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, P.; Chen, Y.; Zhang, L.; Xing, M. Ganoderma lucidum polysaccharide used for treating physical frailty in China. Prog. Mol. Biol. Trans. Sci. 2019, 163, 179–219. [Google Scholar] [CrossRef]
- Zhou, S.; Tang, Q.; Tang, C.; Liu, Y.; Ma, F.; Zhang, X.; Zhang, J.-S. Triterpenes and Soluble Polysaccharide Changes in Lingzhi or Reishi Medicinal Mushroom, Ganoderma lucidum (Agaricomycetes), During Fruiting Growth. Int. J. Med. Mushroom. 2018, 20, 859–871. [Google Scholar] [CrossRef]
- Liu, D.-Z.; Zhu, Y.-Q.; Li, X.-F.; Shan, W.-G.; Gao, P.-F. New Triterpenoids from the Fruiting Bodies of Ganoderma lucidumand Their Bioactivities. Chem. Biodivers. 2014, 11, 982–986. [Google Scholar] [CrossRef]
- El-Mekkawy, S.; Meselhy, M.R.; Nakamura, N.; Tezuka, Y.; Hattori, M.; Kakiuchi, N.; Shimotohno, K.; Kawahata, T.; Otake, T. Anti-HIV-1 and anti-HIV-1-protease substances from Ganoderma lucidum. Phytochemistry 1998, 49, 1651–1657. [Google Scholar] [CrossRef]
- Niedermeyer, T.H.; Lindequist, U.; Mentel, R.; Gördes, D.; Schmidt, E.; Thurow, K.; Lalk, M. Antiviral Terpenoid Constituents of Ganoderma pfeifferi. J. Nat. Prod. 2005, 68, 1728–1731. [Google Scholar] [CrossRef]
- Xu, J.-W.; Xu, Y.-N.; Zhong, J.-J. Enhancement of Ganoderic Acid Accumulation by Overexpression of an N-Terminally Truncated 3-Hydroxy-3-Methylglutaryl Coenzyme A Reductase Gene in the Basidiomycete Ganoderma lucidum. Appl. Environ. Microbiol. 2012, 78, 7968–7976. [Google Scholar] [CrossRef] [Green Version]
- Fei, Y.; Li, N.; Zhang, D.-H.; Xu, J.-W. Increased production of ganoderic acids by overexpression of homologous farnesyl diphosphate synthase and kinetic modeling of ganoderic acid production in Ganoderma lucidum. Microb. Cell Fact. 2019, 18, 1–9. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Annotated_Database | Annotated_Number | 300 ≤ Length < 1000 b | Length ≥ 1000 | |||
|---|---|---|---|---|---|---|
| All | New-Isoform a | All | New-Isoform | All | New-Isoform | |
| COG_Annotation | 3831 | 17 | 928 | 9 | 2886 | 8 |
| GO_Annotation | 5502 | 72 | 1639 | 33 | 3779 | 39 |
| KEGG_Annotation | 3709 | 44 | 1136 | 21 | 2512 | 23 |
| KOG_Annotation | 4985 | 29 | 1268 | 12 | 3685 | 17 |
| Pfam_Annotation | 7207 | 49 | 1992 | 17 | 5164 | 32 |
| Swiss-Prot_Annotation | 5721 | 37 | 1472 | 16 | 4202 | 21 |
| eggNOG_Annotation | 9183 | 123 | 2820 | 39 | 6254 | 84 |
| NR_Annotation | 12,093 | 373 | 4318 | 146 | 7513 | 225 |
| All_Annotated | 12,109 | 375 | 4327 | 147 | 7518 | 226 |
| Annotated_Database | YW2/YW1 | YW3/YW2 | YW3/YW1 |
|---|---|---|---|
| COG | 183 | 115 | 429 |
| GO | 187 | 127 | 465 |
| KEGG | 87 | 57 | 205 |
| KOG | 139 | 105 | 369 |
| NR | 510 | 388 | 1300 |
| Pfam | 263 | 183 | 656 |
| Swiss-Prot | 212 | 142 | 507 |
| eggNOG | 352 | 256 | 885 |
| Total | 511 | 388 | 1302 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cai, M.; Liang, X.; Liu, Y.; Hu, H.; Xie, Y.; Chen, S.; Gao, X.; Li, X.; Xiao, C.; Chen, D.; et al. Transcriptional Dynamics of Genes Purportedly Involved in the Control of Meiosis, Carbohydrate, and Secondary Metabolism during Sporulation in Ganoderma lucidum. Genes 2021, 12, 504. https://doi.org/10.3390/genes12040504
Cai M, Liang X, Liu Y, Hu H, Xie Y, Chen S, Gao X, Li X, Xiao C, Chen D, et al. Transcriptional Dynamics of Genes Purportedly Involved in the Control of Meiosis, Carbohydrate, and Secondary Metabolism during Sporulation in Ganoderma lucidum. Genes. 2021; 12(4):504. https://doi.org/10.3390/genes12040504
Chicago/Turabian StyleCai, Manjun, Xiaowei Liang, Yuanchao Liu, Huiping Hu, Yizhen Xie, Shaodan Chen, Xiong Gao, Xiangmin Li, Chun Xiao, Diling Chen, and et al. 2021. "Transcriptional Dynamics of Genes Purportedly Involved in the Control of Meiosis, Carbohydrate, and Secondary Metabolism during Sporulation in Ganoderma lucidum" Genes 12, no. 4: 504. https://doi.org/10.3390/genes12040504
APA StyleCai, M., Liang, X., Liu, Y., Hu, H., Xie, Y., Chen, S., Gao, X., Li, X., Xiao, C., Chen, D., & Wu, Q. (2021). Transcriptional Dynamics of Genes Purportedly Involved in the Control of Meiosis, Carbohydrate, and Secondary Metabolism during Sporulation in Ganoderma lucidum. Genes, 12(4), 504. https://doi.org/10.3390/genes12040504