
Identification and Characterization of Alternatively Spliced Transcript Isoforms of IRX4 in Prostate Cancer

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. RNA Isolation and cDNA Synthesis
2.3. Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
2.4. Relative Quantification by Real-Time Quantitative RT-PCR (qRT-PCR)
2.5. Androgen Deprivation Assay
2.6. In Silico Analysis of IRX4 Transcripts and Isoforms
2.7. RNA-seq and Genotype Data for cis-eQTL Analysis
2.8. Reprocessing PRIDE PCa Cell Line LC-MS/MS Data
2.9. DNA Sequencing
2.10. LC-MS/MS Analysis of PCa Cells
2.11. LC-MS/MS Data Analysis
2.12. Statistical Analysis
3. Results
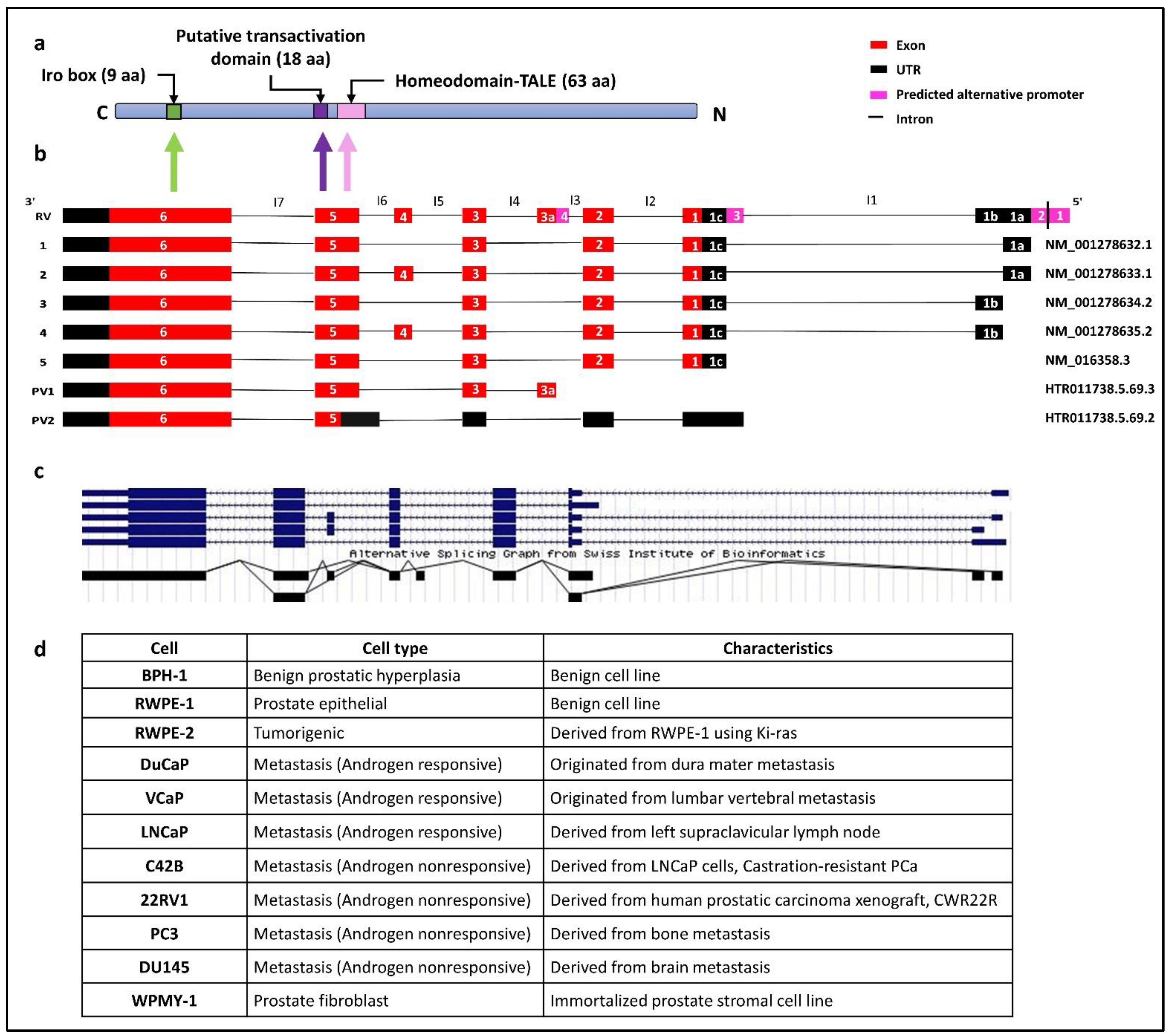
3.1. In silico identification and characterisation of human IRX4 transcripts
3.2. Identification of Human IRX4 Transcripts in PCa Cell Lines
3.3. Characterisation of Human Novel IRX4 Transcripts in a Panel of PCa Cell Lines
3.4. Androgen Regulation of IRX4 Transcripts in PCa Cell Lines
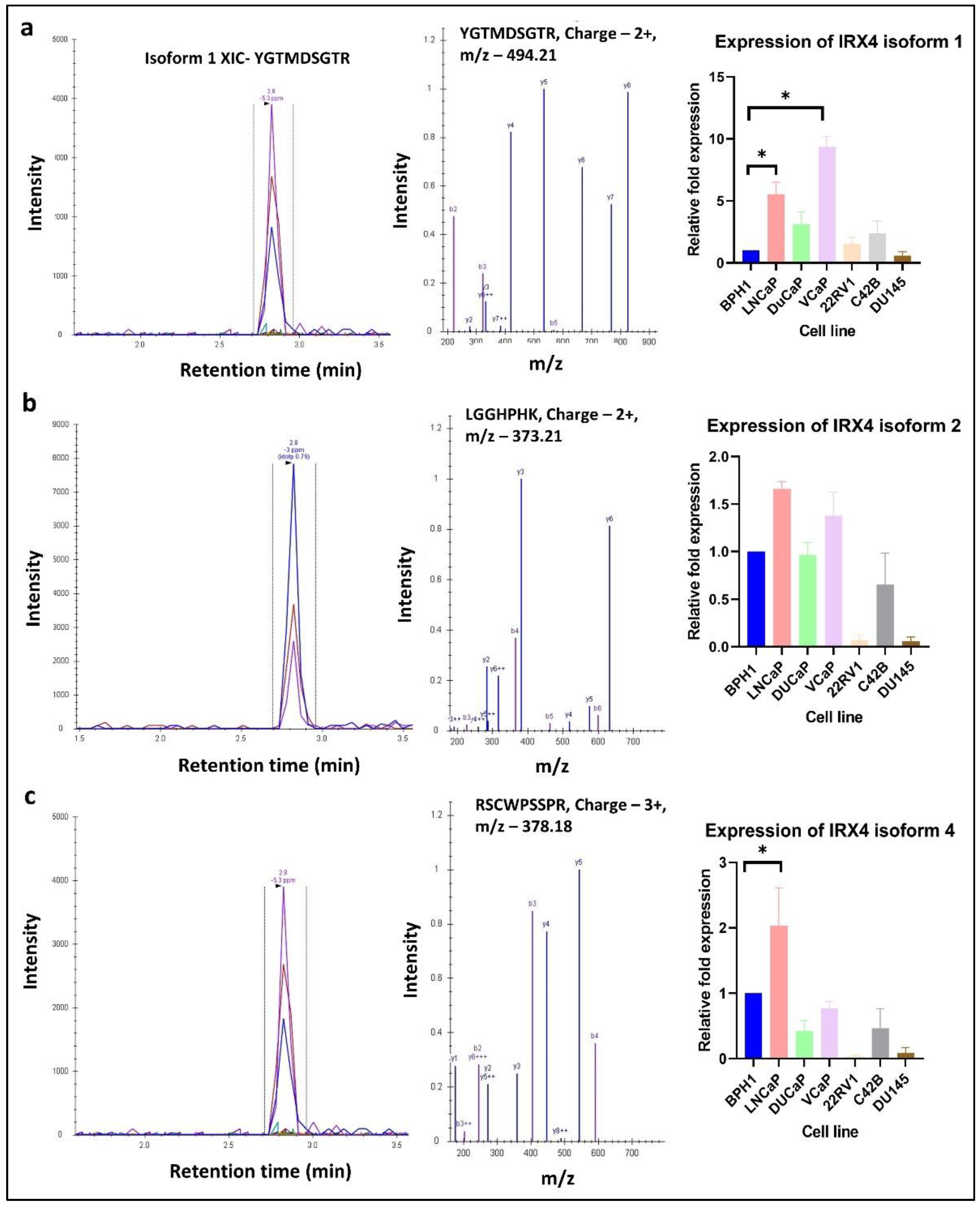
3.5. Identification and Characterisation of IRX4 Protein Isoforms by Mass Spectrometry
3.6. Validation of Expression of IRX4 Transcripts in PCa Patients
3.7. Association between PCa Risk SNP rs12653946 and Expression Levels of IRX4 Transcripts
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, Y.; Liu, J.; Huang, B.O.; Xu, Y.M.; Li, J.; Huang, L.F.; Lin, J.; Zhang, J.; Min, Q.H.; Yang, W.M.; et al. Mechanism of alternative splicing and its regulation. Biomed. Rep. 2015, 3, 152–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, B.; Eyras, E. The role of alternative splicing in cancer. Transcription 2017, 8, 91–98. [Google Scholar] [CrossRef] [Green Version]
- Liyanage, C.; Fernando, A.; Batra, J. Differential roles of protease isoforms in the tumor microenvironment. Cancer Metastasis Rev. 2019, 38, 389–415. [Google Scholar] [CrossRef]
- Siegfried, Z.; Karni, R. The role of alternative splicing in cancer drug resistance. Curr. Opin. Genet. Dev. 2018, 48, 16–21. [Google Scholar] [CrossRef]
- Biamonti, G.; Infantino, L.; Gaglio, D.; Amato, A. An Intricate Connection between Alternative Splicing and Phenotypic Plasticity in Development and Cancer. Cells 2019, 9, 34. [Google Scholar] [CrossRef] [Green Version]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Blee, A.M.; Huang, H. Lineage plasticity-mediated therapy resistance in prostate cancer. Asian J. Androl. 2019, 21, 241–248. [Google Scholar] [CrossRef]
- Olender, J.; Lee, N.H. Role of Alternative Splicing in Prostate Cancer Aggressiveness and Drug Resistance in African Americans. Adv. Exp. Med. Biol. 2019, 1164, 119–139. [Google Scholar] [CrossRef] [PubMed]
- Paschalis, A.; Sharp, A.; Welti, J.C.; Neeb, A.; Raj, G.V.; Luo, J.; Plymate, S.R.; de Bono, J.S. Alternative splicing in prostate cancer. Nat. Rev. Clin. Oncol. 2018, 15, 663–675. [Google Scholar] [CrossRef]
- Bruneau, B.G.; Bao, Z.Z.; Tanaka, M.; Schott, J.J.; Izumo, S.; Cepko, C.L.; Seidman, J.G.; Seidman, C.E. Cardiac expression of the ventricle-specific homeobox gene Irx4 is modulated by Nkx2-5 and dHand. Dev. Biol. 2000, 217, 266–277. [Google Scholar] [CrossRef] [Green Version]
- Pearson, J.C.; Lemons, D.; McGinnis, W. Modulating Hox gene functions during animal body patterning. Nat. Rev. Genet. 2005, 6, 893–904. [Google Scholar] [CrossRef]
- Cavodeassi, F.; Modolell, J.; Gomez-Skarmeta, J.L. The Iroquois family of genes: From body building to neural patterning. Development 2001, 128, 2847–2855. [Google Scholar]
- Kim, K.H.; Rosen, A.; Bruneau, B.G.; Hui, C.C.; Backx, P.H. Iroquois homeodomain transcription factors in heart development and function. Circ. Res. 2012, 110, 1513–1524. [Google Scholar] [CrossRef] [Green Version]
- Bao, Z.Z.; Bruneau, B.G.; Seidman, J.G.; Seidman, C.E.; Cepko, C.L. Regulation of chamber-specific gene expression in the developing heart by Irx4. Science 1999, 283, 1161–1164. [Google Scholar] [CrossRef]
- Bruneau, B.G.; Bao, Z.Z.; Fatkin, D.; Xavier-Neto, J.; Georgakopoulos, D.; Maguire, C.T.; Berul, C.I.; Kass, D.A.; Kuroski-de Bold, M.L.; de Bold, A.J.; et al. Cardiomyopathy in Irx4-deficient mice is preceded by abnormal ventricular gene expression. Mol. Cell Biol. 2001, 21, 1730–1736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Z.; Wang, J.; Su, D.; Pan, H.; Huang, G.; Li, X.; Li, Z.; Shen, A.; Xie, X.; Wang, B.; et al. Two novel mutations of the IRX4 gene in patients with congenital heart disease. Hum. Genet. 2011, 130, 657–662. [Google Scholar] [CrossRef]
- Jin, Z.; Zhang, J.; Klar, A.; Chedotal, A.; Rao, Y.; Cepko, C.L.; Bao, Z.Z. Irx4-mediated regulation of Slit1 expression contributes to the definition of early axonal paths inside the retina. Development 2003, 130, 1037–1048. [Google Scholar] [CrossRef] [Green Version]
- Correa, S.; Panis, C.; Binato, R.; Herrera, A.C.; Pizzatti, L.; Abdelhay, E. Identifying Potential Markers in Breast Cancer Subtypes Using Plasma Label-Free Proteomics. J. Proteom. 2017, 151, 33–42. [Google Scholar] [CrossRef]
- Ding, L.; Su, Y.; Fassl, A.; Hinohara, K.; Qiu, X.; Harper, N.W.; Huh, S.J.; Bloushtain-Qimron, N.; Jovanovic, B.; Ekram, M.; et al. Perturbed myoepithelial cell differentiation in BRCA mutation carriers and in ductal carcinoma in situ. Nat. Commun. 2019, 10, 4182. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.L.; Qu, L.W.; Ma, L.; Zhou, Y.C.; Wang, G.Z.; Zhao, X.C.; Zhang, C.; Zhang, Y.F.; Wang, M.; Zhang, M.Y.; et al. Genome-wide identification of transcription factors that are critical to non-small cell lung cancer. Cancer Lett. 2018, 434, 132–143. [Google Scholar] [CrossRef]
- Chakma, K.; Gu, Z.D.; Motoi, F.; Unno, M.; Horii, A.; Fukushige, S. DNA hypermethylation of IRX4 is a frequent event that may confer growth advantage to pancreatic cancer cells. Cancer Res. 2019, 821–821. [Google Scholar] [CrossRef]
- Batra, J.; Lose, F.; Chambers, S.; Gardiner, R.A.; Aitken, J.; Yaxley, J.; Clements, J.A.; Spurdle, A.B.; Australian Prostate Cancer BioResource. A replication study examining novel common single nucleotide polymorphisms identified through a prostate cancer genome-wide association study in a Japanese population. Am. J. Epidemiol. 2011, 174, 1391–1395. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Liu, F.; Hsing, A.W.; Wang, X.; Shao, Q.; Qi, J.; Ye, Y.; Wang, Z.; Chen, H.; Gao, X.; et al. Replication and cumulative effects of GWAS-identified genetic variations for prostate cancer in Asians: A case-control study in the ChinaPCa consortium. Carcinogenesis 2012, 33, 356–360. [Google Scholar] [CrossRef]
- Lindstrom, S.; Schumacher, F.R.; Campa, D.; Albanes, D.; Andriole, G.; Berndt, S.I.; Bueno-de-Mesquita, H.B.; Chanock, S.J.; Diver, W.R.; Ganziano, J.M.; et al. Replication of five prostate cancer loci identified in an Asian population—Results from the NCI Breast and Prostate Cancer Cohort Consortium (BPC3). Cancer Epidemiol. Biomark. Prev. 2012, 21, 212–216. [Google Scholar] [CrossRef] [Green Version]
- Qi, J.; Tian, L.; Chen, Z.; Wang, L.; Tao, S.; Gu, X.; Na, R.; Jiao, Y.; Kang, J.; Zheng, S.; et al. Genetic variants in 2q31 and 5p15 are associated with aggressive benign prostatic hyperplasia in a Chinese population. Prostate 2013, 73, 1182–1190. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.H.; Takata, R.; Akamatsu, S.; Shigemizu, D.; Tsunoda, T.; Furihata, M.; Takahashi, A.; Kubo, M.; Kamatani, N.; Ogawa, O.; et al. IRX4 at 5p15 suppresses prostate cancer growth through the interaction with vitamin D receptor, conferring prostate cancer susceptibility. Hum. Mol. Genet. 2012, 21, 2076–2085. [Google Scholar] [CrossRef] [Green Version]
- Holmquist Mengelbier, L.; Lindell-Munther, S.; Yasui, H.; Jansson, C.; Esfandyari, J.; Karlsson, J.; Lau, K.; Hui, C.C.; Bexell, D.; Hopyan, S.; et al. The Iroquois homeobox proteins IRX3 and IRX5 have distinct roles in Wilms tumour development and human nephrogenesis. J. Pathol. 2019, 247, 86–98. [Google Scholar] [CrossRef]
- Wang, P.; Zhuang, C.; Huang, D.; Xu, K. Downregulation of miR-377 contributes to IRX3 deregulation in hepatocellular carcinoma. Oncol. Rep. 2016, 36, 247–252. [Google Scholar] [CrossRef] [Green Version]
- Morey, S.R.; Smiraglia, D.J.; James, S.R.; Yu, J.; Moser, M.T.; Foster, B.A.; Karpf, A.R. DNA methylation pathway alterations in an autochthonous murine model of prostate cancer. Cancer Res. 2006, 66, 11659–11667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [Green Version]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- Sayers, E.W.; Beck, J.; Brister, J.R.; Bolton, E.E.; Canese, K.; Comeau, D.C.; Funk, K.; Ketter, A.; Kim, S.; Kimchi, A.; et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2019, 48, D9–D16. [Google Scholar] [CrossRef] [Green Version]
- The Cancer Genome Atlas Program. Available online: https://www.cancer.gov/tcga (accessed on 24 February 2021).
- Zhang, X.; Jonassen, I. RASflow: An RNA-Seq analysis workflow with Snakemake. BMC Bioinform. 2020, 21, 110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Metzger, E.; Willmann, D.; McMillan, J.; Forne, I.; Metzger, P.; Gerhardt, S.; Petroll, K.; von Maessenhausen, A.; Urban, S.; Schott, A.K.; et al. Assembly of methylated KDM1A and CHD1 drives androgen receptor-dependent transcription and translocation. Nat. Struct. Mol. Biol. 2016, 23, 132–139. [Google Scholar] [CrossRef]
- Chambers, M.C.; Maclean, B.; Burke, R.; Amodei, D.; Ruderman, D.L.; Neumann, S.; Gatto, L.; Fischer, B.; Pratt, B.; Egertson, J.; et al. A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 2012, 30, 918–920. [Google Scholar] [CrossRef]
- Barsnes, H.; Vaudel, M. SearchGUI: A Highly Adaptable Common Interface for Proteomics Search and de Novo Engines. J. Proteome Res. 2018, 17, 2552–2555. [Google Scholar] [CrossRef]
- Vaudel, M.; Burkhart, J.M.; Zahedi, R.P.; Oveland, E.; Berven, F.S.; Sickmann, A.; Martens, L.; Barsnes, H. PeptideShaker enables reanalysis of MS-derived proteomics data sets. Nat. Biotechnol. 2015, 33, 22–24. [Google Scholar] [CrossRef]
- Kent, W.J. BLAT—The BLAST-like alignment tool. Genome Res. 2002, 12, 656–664. [Google Scholar] [CrossRef] [Green Version]
- Liyanage, C.; Adil, M.; Abeysinghe, P.; Clements, J.; Batra, J. SWATH-MS Based Proteomic Profiling of Prostate Cancer Cells Reveals Adaptive Molecular Mechanisms in Response to Anti-Androgen Therapy. Cancers 2021, 13, 715. [Google Scholar] [CrossRef]
- MacLean, B.; Tomazela, D.M.; Shulman, N.; Chambers, M.; Finney, G.L.; Frewen, B.; Kern, R.; Tabb, D.L.; Liebler, D.C.; MacCoss, M.J. Skyline: An open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010, 26, 966–968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, M.; Chang, C.Y.; Clough, T.; Broudy, D.; Killeen, T.; MacLean, B.; Vitek, O. MSstats: An R package for statistical analysis of quantitative mass spectrometry-based proteomic experiments. Bioinformatics 2014, 30, 2524–2526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Hussain, W.M.; Vijai, J.; Offit, K.; Rubin, M.A.; Demichelis, F.; Klein, R.J. Variants at IRX4 as prostate cancer expression quantitative trait loci. Eur. J. Hum. Genet. 2014, 22, 558–563. [Google Scholar] [CrossRef] [Green Version]
- Conti, D.V.; Darst, B.F.; Moss, L.C.; Saunders, E.J.; Sheng, X.; Chou, A.; Schumacher, F.R.; Olama, A.A.A.; Benlloch, S.; Dadaev, T.; et al. Publisher Correction: Trans-ancestry genome-wide association meta-analysis of prostate cancer identifies new susceptibility loci and informs genetic risk prediction. Nat. Genet. 2021, 53, 413. [Google Scholar] [CrossRef]
- Feng, H.; Jin, Z.; Liu, K.; Peng, Y.; Jiang, S.; Wang, C.; Hu, J.; Shen, X.; Qiu, W.; Cheng, X.; et al. Identification and validation of critical alternative splicing events and splicing factors in gastric cancer progression. J. Cell Mol. Med. 2020, 24, 12667–12680. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, K.; Matsushita, K.; Kobayashi, S.; Ishige, T.; Semba, T.; Kimura, A.; Kazami, T.; Ohyama, M.; Itoga, S.; Beppu, M.; et al. Alternative Splicing Detection as a Biomarker for Cancer Diagnosis: A Novel Progressive Mechanism of Acute Lymphoblastic Leukemia with Alternative Splicing as a Biomarker Candidate. Rinsho Byori 2015, 63, 1091–1102. [Google Scholar]
- Le, K.Q.; Prabhakar, B.S.; Hong, W.J.; Li, L.C. Alternative splicing as a biomarker and potential target for drug discovery. Acta Pharmacol. Sin. 2015, 36, 1212–1218. [Google Scholar] [CrossRef]
- Matsushita, K.; Itoga, S.; Nishimura, M.; Furuta, K.; Nomura, F. Alternative Splicing Detection as Biomarker Candidates for Cancer Diagnosis and Treatment with Establishment of Clinical Biobank in Chiba University. Rinsho Byori 2015, 63, 347–360. [Google Scholar]
- Wu, P.; Zhou, D.; Wang, Y.; Lin, W.; Sun, A.; Wei, H.; Fang, Y.; Cong, X.; Jiang, Y. Identification and validation of alternative splicing isoforms as novel biomarker candidates in hepatocellular carcinoma. Oncol. Rep. 2019, 41, 1929–1937. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Si, S.; Li, X.; Sun, W.; Cui, L. Identification and validation of an alternative splicing-based prognostic signature for head and neck squamous cell carcinoma. J. Cancer 2020, 11, 4571–4580. [Google Scholar] [CrossRef]
- Narasimhan, A.; Greiner, R.; Bathe, O.F.; Baracos, V.; Damaraju, S. Differentially expressed alternatively spliced genes in skeletal muscle from cancer patients with cachexia. J. Cachexia Sarcopenia Muscle 2018, 9, 60–70. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.D.; Lee, N.H. Aberrant RNA Splicing in Cancer and Drug Resistance. Cancers 2018, 10, 458. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.D.; Ceniccola, K.; Hwang, S.; Andrawis, R.; Horvath, A.; Freedman, J.A.; Olender, J.; Knapp, S.; Ching, T.; Garmire, L.; et al. Alternative splicing promotes tumour aggressiveness and drug resistance in African American prostate cancer. Nat. Commun. 2017, 8, 15921. [Google Scholar] [CrossRef] [PubMed]
- Anand, A.U.; Bjartell, A. Re: AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. Eur. Urol. 2015, 67, 349–350. [Google Scholar] [CrossRef]
- Harries, L.W.; Perry, J.R.; McCullagh, P.; Crundwell, M. Alterations in LMTK2, MSMB and HNF1B gene expression are associated with the development of prostate cancer. BMC Cancer 2010, 10, 315. [Google Scholar] [CrossRef] [Green Version]
- Harries, L.W. Alternate mRNA processing of the hepatocyte nuclear factor genes and its role in monogenic diabetes. Expert Rev. Endocrinol. Metab. 2006, 1, 715–726. [Google Scholar] [CrossRef] [PubMed]
- Chandra, S.; Srinivasan, S.; Batra, J. Hepatocyte nuclear factor 1 beta: A perspective in cancer. Cancer Med. 2021, 10, 1791–1804. [Google Scholar] [CrossRef] [PubMed]
- Ghiasvand, S.; Bakhshinejad, B.; Mowla, S.J.; Sadeghizadeh, M. Potential roles of 5 UTR and 3 UTR regions in post-trans-criptional regulation of mouse Oct4 gene in BMSC and P19 cells. Iran. J. Basic Med. Sci. 2014, 17, 490–496. [Google Scholar]
- Lee, I.I.; Kuznik, N.C.; Rottenberg, J.T.; Brown, M.; Cato, A.C.B. BAG1L: A promising therapeutic target for androgen receptor-dependent prostate cancer. J. Mol. Endocrinol. 2019, 62, R289–R299. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, N.; Granata, I.; Capaia, M.; Piccirillo, M.; Guarracino, M.R.; Vene, R.; Brizzolara, A.; Petretto, A.; Inglese, E.; Morini, M.; et al. Adaptive phenotype drives resistance to androgen deprivation therapy in prostate cancer. Cell Commun. Signal. 2017, 15, 51. [Google Scholar] [CrossRef] [Green Version]
- Iglesias-Gato, D.; Wikstrom, P.; Tyanova, S.; Lavallee, C.; Thysell, E.; Carlsson, J.; Hagglof, C.; Cox, J.; Andren, O.; Stattin, P.; et al. The Proteome of Primary Prostate Cancer. Eur. Urol. 2016, 69, 942–952. [Google Scholar] [CrossRef]
- Liu, Y.; Beyer, A.; Aebersold, R. On the Dependency of Cellular Protein Levels on mRNA Abundance. Cell 2016, 165, 535–550. [Google Scholar] [CrossRef] [Green Version]
- Nelson, D.O.; Jin, D.X.; Downs, K.M.; Kamp, T.J.; Lyons, G.E. Irx4 identifies a chamber-specific cell population that contributes to ventricular myocardium development. Dev. Dyn. 2014, 243, 381–392. [Google Scholar] [CrossRef] [Green Version]
- Belluti, S.; Rigillo, G.; Imbriano, C. Transcription Factors in Cancer: When Alternative Splicing Determines Opposite Cell Fates. Cells 2020, 9, 760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gershenwald, J.E.; Sumner, W.; Calderone, T.; Wang, Z.; Huang, S.; Bar-Eli, M. Dominant-negative transcription factor AP-2 augments SB-2 melanoma tumor growth in vivo. Oncogene 2001, 20, 3363–3375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buettner, R.; Kannan, P.; Imhof, A.; Bauer, R.; Yim, S.O.; Glockshuber, R.; Van Dyke, M.W.; Tainsky, M.A. An alternatively spliced mRNA from the AP-2 gene encodes a negative regulator of transcriptional activation by AP-2. Mol. Cell Biol. 1993, 13, 4174–4185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, S.; Liu, W.; Li, Y.; Liu, P.; Li, S.; Dou, D.; Wang, Y.; Yang, R.; Xiang, R.; Liu, F. Alternative Splice Variants Modulates Dominant-Negative Function of Helios in T-Cell Leukemia. PLoS ONE 2016, 11, e0163328. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Transcript | Coding Frames | No. of Amino Acids | Predicted Protein Size | The Presence of Predicted Domains | ||
|---|---|---|---|---|---|---|
| Homeodomain | Transactivation Domain | Irobox | ||||
| 1,3,5 | Exon 1–Exon 6 | 519 | 54.4 kDa (Isoform 1) | ✓ | ✓ | ✓ |
| 2,4 | Exon 1–Exon 6 | 545 | 57 kDa (Isoform 2) | ✓ | ✓ | ✓ |
| 6,7 | Exon 3a–Exon 5 | 114 | 8.7 kDa (Isoform 4) | - | - | - |
| Exon 5–Exon 6 | 380 | 40 kDa (Isoform 3) | ✓ | ✓ | ✓ | |
| 8,9 | Exon 1–Exon 3 | 105 | 11 kDa | - | - | - |
| Exon 5–Exon 6 | 380 | 40 kDa (Isoform 3) | ✓ | ✓ | ✓ | |
| 10 | Exon 1–Exon 6 | 204 | 21 kDa | ✓ | ✓ | - |
| 11 | Exon 1–Exon 6 | 230 | 23.6 kDa | ✓ | ✓ | - |
| 12 | Exon 5–Exon 6 | 380 | 40 kDa (Isoform 3) | ✓ | ✓ | ✓ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernando, A.; Liyanage, C.; Moradi, A.; Janaththani, P.; Batra, J. Identification and Characterization of Alternatively Spliced Transcript Isoforms of IRX4 in Prostate Cancer. Genes 2021, 12, 615. https://doi.org/10.3390/genes12050615
Fernando A, Liyanage C, Moradi A, Janaththani P, Batra J. Identification and Characterization of Alternatively Spliced Transcript Isoforms of IRX4 in Prostate Cancer. Genes. 2021; 12(5):615. https://doi.org/10.3390/genes12050615
Chicago/Turabian StyleFernando, Achala, Chamikara Liyanage, Afshin Moradi, Panchadsaram Janaththani, and Jyotsna Batra. 2021. "Identification and Characterization of Alternatively Spliced Transcript Isoforms of IRX4 in Prostate Cancer" Genes 12, no. 5: 615. https://doi.org/10.3390/genes12050615
APA StyleFernando, A., Liyanage, C., Moradi, A., Janaththani, P., & Batra, J. (2021). Identification and Characterization of Alternatively Spliced Transcript Isoforms of IRX4 in Prostate Cancer. Genes, 12(5), 615. https://doi.org/10.3390/genes12050615