
Making Sense of Genetic Information: The Promising Evolution of Clinical Stratification and Precision Oncology Using Machine Learning


Abstract
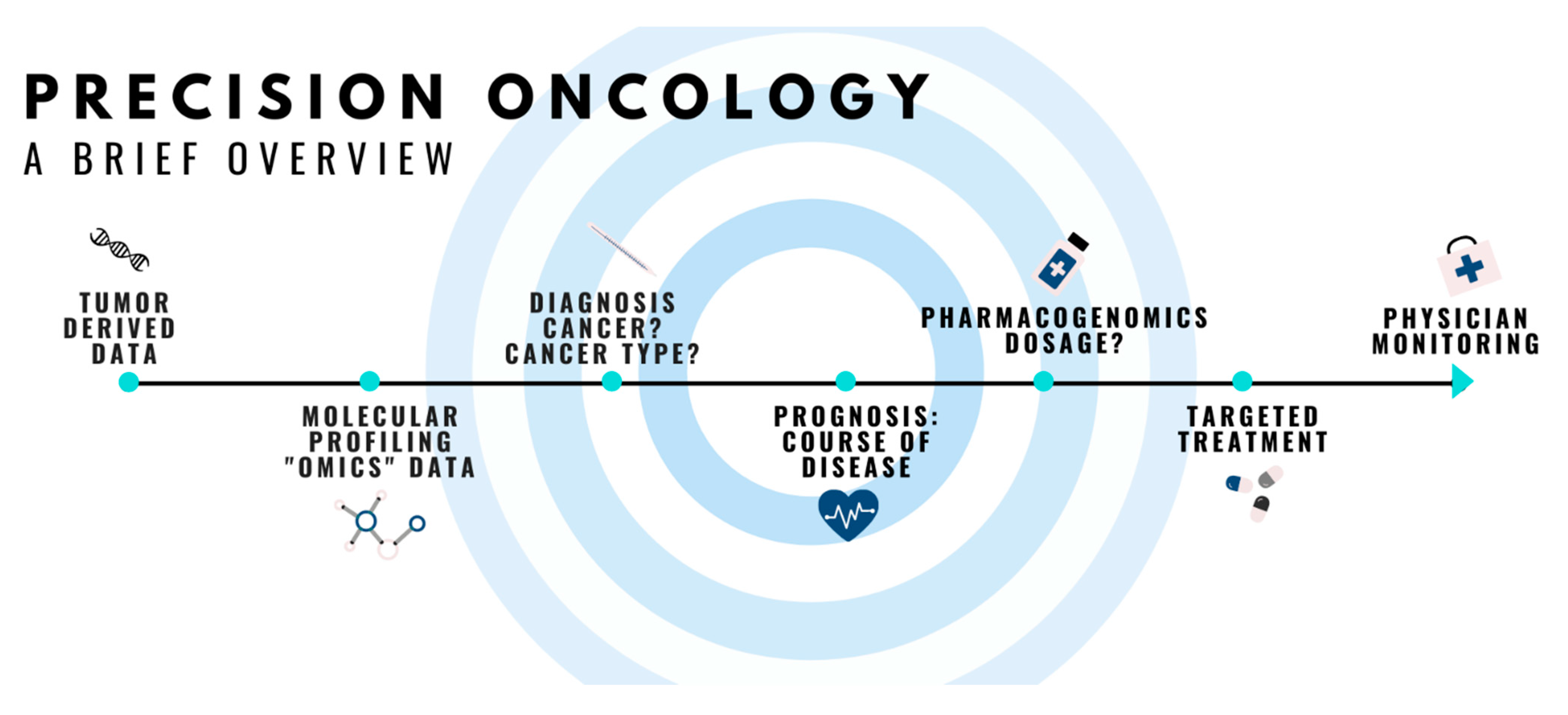
:1. Introduction
2. The Path That Precision Oncology Has Taken
2.1. Overview
2.2. Breast Cancer

{kind=link}
{kind=link}
| Type | Gene | Accession No. | Function |
|---|---|---|---|
| Breast | BRCA1 BRCA2 | NM_007294.4 NM_000059.4 | Transcriptional regulator of DNA repair genes and tumor suppressor genes. BRCA1 mutations are responsible for ~40% of inherited breast cancers and >80% of inherited breast and ovarian cancers. BRCA1 and BRCA2 variations can increase the lifetime risk of developing breast or ovarian cancer. |
| ATM | NM_000051.4 | This gene encodes a cell cycle checkpoint kinase that belongs to the PI3/PI4-kinase family. The normal function of this gene is to help repair DNA damage or kills the cell if it is unable to fix the damaged DNA. | |
| TP53 | NM_000546.6 | Halts the growth of cells with damaged DNA. TP53 mutations are associated with various human cancers. The Li-Fraumeni syndrome, a complex hereditary cancer predisposition disorder, is mainly caused by germline mutations of this gene. | |
| CHEK2 | NM_007194.4 | The CHEK2 protein is a cell cycle checkpoint regulator and a possible tumor suppressor that is known to phosphorylate BRCA1. Mutations in this gene have been correlated with the development of Li-Fraumeni syndrome. This mutation increases the likelihood of predisposition to sarcomas, breast cancer, and brain tumors. | |
| PTEN | NM_001304717.5 | Tumor suppressor gene that is mutated in a large quantity of cancers at high frequency. Helps regulate cell growth. | |
| CDH1 | NM_001317185.2 | Encodes epithelial cadherin or E-cadherin. When individuals inherit the mutated form of this gene, it causes hereditary diffuse gastric cancer, which can increase the risk of developing invasive lobular breast cancer in women. Mutations in this gene can also cause colorectal, thyroid, and ovarian cancers. Loss of function of CDH1 increases tumor proliferation, invasion, and/or metastasis. | |
| STK11 or LKB1 | NM_000455.5 | Encodes serine/threonine kinase 11 that regulates cell polarity and acts as a tumor suppressor. Mutations in STK11 are associated with Peutz-Jeghers syndrome, which is characterized by the growth of polyps in the gastrointestinal tract, pigmented macules on the skin and mouth, and other neoplasms. | |
| PALB2 | NM_024675.4 | Encodes a protein that binds to BRCA2. PALB2 may allow the stable intranuclear localization and accumulation of BRCA2. | |
| BARD1 | NM_000465.4 | Encodes protein that interacts with the N-terminal of BRCA1. Shares homology with the two most conserved regions of BRCA1, the N-terminal RING motif and the C-terminal BRCT domain. The RING motif is typically found in proteins that regulate cell growth. The protein encoded by BARD1 may be the target of oncogenic mutations that are found in breast and ovarian cancer. | |
| BRIP1 | NM_032043.3 | The protein interacts with the BRCT repeats of BRCA1. The complex is important in the normal double-strand break repair activity of type 1 (BRCA1) breast cancers. BRIP1 may be a target of germline cancer-inducing mutations. | |
| CASP8 | NM_001372051.1 | Encodes a member of the cysteine-aspartic acid protease (caspase) family. This protein allows for the apoptosis induced by Fas. Associated with the risk of developing cancer [62]. | |
| CTLA4 | NM_005214.5 | A member of the immunoglobin gene superfamily. Encodes a protein that sends an inhibitory signal to T cells. Expressed in some cancer cells [63]. | |
| FGFR2 | NM_000141.5 | The protein encoded by this gene is a member of the fibroblast growth factor receptor family, where amino acid sequence is highly conserved. Aberrations in FGFR2 have been seen to affect FGRFR2 signaling that has been recognized in breast cancer. Amplification of FGFR2 is present in 3.6% of triple-negative breast cancers (TNBCs) [64]. | |
| H19 | NR_002196.2 | Gene only expressed from maternally inherited chromosome. Encodes a non-coding RNA that functions as a tumor suppressor. Mutations in H19 are associated with the development of Beckwith-Wiedemann Syndrome and Wilms tumorigenesis. | |
| MRE11A | NM_05591.4 | Encodes a nuclear protein involved in homologous recombination, telomere length maintenance, and DNA double-strand break repair. This protein is a member of the MRE11/RAD50 double-strand break repair complex composed of 5 proteins. | |
| NBN | NM_002485.5 | Mutations in NBN are associated with the development of Nijemegen breakage syndrome that is characterized by cancer predisposition, microcephaly, growth retardation, and immunodeficiency. The gene product of NBN has been proposed to be involved in DNA double-strand break repair and DNA damage-induced checkpoint activation. | |
| RAD51 | NM_002875.5 | Encodes a protein important for repairing damaged DNA. The protein is a member of the RAD51 family. It interacts with single-strand DNA-binding protein RPA and RAD52. This protein is also thought to be involved in homologous pairing and strand transfer of DNA. It interacts with BRCA1 and BRCA2. BRCA2 inactivation can result in the loss of RAD51 controls and be an important event resulting in genomic instability and tumorigenesis. | |
| TERT | NM_198253.3 | Encodes one subunit of the enzyme telomerase that lengthens telomeres at the end of chromosomes. The lengthening of the cancer cell telomeres allows them to continually survive. | |
| TOX3 | NM_001080430.4 | This gene encodes a protein that holds an HMG-box. This protein is possibly engaged in bending and unwinding DNA and modulating chromatin structure because of the HMG-box. This gene’s minor allele has been associated with a higher risk of developing breast cancer. | |
| Glioma | AVIL | NM_006576 | Encodes a member of gelsolin/villin family of actin regulatory proteins. May contribute to the development of ganglia. AVIL expression is increased in glioblastomas as well as glioblastoma stem/initiating cells [65]. Patients with an increased level of AVIL expression seemed to have a worse prognosis [65]. |
| MMP9 | NM_004994.3 | The matrix metalloproteinase (MMP) breaks down the extracellular matrix. MMP9 is a member of the MMP family involved in disease processes like metastasis and possibly in tumor-associated tissue remodeling. | |
| FN1 | NM_212482.4 | Encodes fibronectin, a glycoprotein present in a soluble dimeric form in plasma, and in a dimeric or multimeric form at the cell surface and in extracellular matrix. Fibronectin is known to be involved in cell adhesion and migration progresses such as metastasis. | |
| COL3A1 | NM_000090.4 | Encodes the pro-alpha1 chains of type III collagen found in skin, lungs, intestinal walls, and the walls of blood vessels. Mutations in this gene are associated with the development of Ehlers-Danlos syndrome type IV. |
2.3. Glioma
3. Machine Learning—A Keystone That Paves the Way for Precision Oncology
3.1. Overview
| Algorithm | Characteristics |
|---|---|
| K-nearest neighbor (KNN) | Often described as the simplest ML algorithm; no training phase is required. |
| Support vector machine (SVM) | Simple structure and high generalization capability; works well with insufficient training data [84]. |
| Artificial neural network (ANN) | Mimics neuronal network, in which each node changes the connection strength by experience. |
| Decision tree (DT) learning | A popular tree-based method for classification and regression, in which the learned model is represented as a decision tree. |
| Naive Bayes (NB) | Probabilistic classifier that treats each feature variable as an independent variable. |
| Bayesian network (BN) | A probabilistic graphical model in which a directed acyclic graph represents potential causal relationship between variables. |
3.2. Bayesian Networks
3.3. ML in the Treatment of Breast Cancer and Glioma
4. Future Directions
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- National Human Genome Research Institute The Human Genome Project. Available online: https://www.genome.gov/human-genome-project (accessed on 14 October 2020).
- Jain, K.K. Personalized medicine. Curr. Opin. Mol. Ther. 2002, 4, 548. [Google Scholar]
- Novelli, G. Personalized genomic medicine. Intern. Emerg. Med. 2010, 5, 81–90. [Google Scholar] [CrossRef]
- Genomes Project Consortium; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M. A global reference for human genetic variation. Nature 2015, 526, 68. [Google Scholar] [CrossRef] [Green Version]
- Cashman, R.; Zilberberg, A.; Priel, A.; Philip, H.; Varvak, A.; Jacob, A.; Shoval, I.; Efroni, S. A single nucleotide variant of human PARP1 determines response to PARP inhibitors. NPJ Precis. Oncol. 2020, 4, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Tam, P.K.H.; International HapMap Consortium. The international HapMap project. Nature 2003, 426, 789–796. [Google Scholar]
- Consortium, T.G.P. An integrated map of genetic variation from 1092 human genomes. Nature 2012, 491, 56–65. [Google Scholar] [CrossRef] [Green Version]
- The 1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature 2010, 467, 1061. [Google Scholar] [CrossRef] [Green Version]
- Roden, D.M.; Altman, R.B.; Benowitz, N.L.; Flockhart, D.A.; Giacomini, K.M.; Johnson, J.A.; Krauss, R.M.; McLeod, H.L.; Ratain, M.J.; Relling, M.V. Pharmacogenomics: Challenges and opportunities. Ann. Intern. Med. 2006, 145, 749–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinker, K.; Chin, J.; Melsaether, A.N.; Morris, E.A.; Moy, L. Precision medicine and radiogenomics in breast cancer: New approaches toward diagnosis and treatment. Radiology 2018, 287, 732–747. [Google Scholar] [CrossRef] [PubMed]
- Agyeman, A.A.; Ofori-Asenso, R. Perspective: Does personalized medicine hold the future for medicine? J. Pharm. Bioallied. Sci. 2015, 7, 239–244. [Google Scholar] [CrossRef]
- Tao, Z.; Shi, A.; Li, R.; Wang, Y.; Wang, X.; Zhao, J. Microarray bioinformatics in cancer- a review. J. BUON 2017, 22, 838–843. [Google Scholar]
- Lu, C.-F.; Hsu, F.-T.; Hsieh, K.L.-C.; Kao, Y.-C.J.; Cheng, S.-J.; Hsu, J.B.-K.; Tsai, P.-H.; Chen, R.-J.; Huang, C.-C.; Yen, Y. Machine learning–based radiomics for molecular subtyping of gliomas. Clin. Cancer. Res. 2018, 24, 4429–4436. [Google Scholar] [CrossRef] [Green Version]
- Yau, T.O. Precision treatment in colorectal cancer: Now and the future. JGH Open 2019, 3, 361–369. [Google Scholar] [CrossRef]
- Willick, M.S. Artificial intelligence: Some legal approaches and implications. AI Mag. 1983, 4, 5. [Google Scholar]
- Luxton, D.D. An Introduction to Artificial Intelligence in Behavioral and Mental Health Care. In Artificial Intelligence in Behavioral and Mental Health Care; Luxton, D.D., Ed.; Academic Press: Cambridge, MA, USA, 2016. [Google Scholar]
- Helm, J.M.; Swiergosz, A.M.; Haeberle, H.S.; Karnuta, J.M.; Schaffer, J.L.; Krebs, V.E.; Spitzer, A.I.; Ramkumar, P.N. Machine learning and artificial intelligence: Definitions, applications, and future directions. Curr. Rev. Musculoskelet. Med. 2020, 13, 69–76. [Google Scholar] [CrossRef]
- Forrest, S.J.; Geoerger, B.; Janeway, K.A. Precision medicine in pediatric oncology. Curr. Opin. Pediatr. 2018, 30, 17. [Google Scholar] [CrossRef]
- Kang, K.-K.; Hur, H.; Byun, C.S.; Kim, Y.B.; Han, S.-U.; Cho, Y.K. Conventional cytology is not beneficial for predicting peritoneal recurrence after curative surgery for gastric cancer: Results of a prospective clinical study. J. Gastric Cancer 2014, 14, 23. [Google Scholar] [CrossRef] [Green Version]
- Cardoso, F.; van’t Veer, L.J.; Bogaerts, J.; Slaets, L.; Viale, G.; Delaloge, S.; Pierga, J.-Y.; Brain, E.; Causeret, S.; DeLorenzi, M. 70-gene signature as an aid to treatment decisions in early-stage breast cancer. N. Engl. J. Med. 2016, 375, 717–729. [Google Scholar] [CrossRef] [Green Version]
- Carrasco-Ramiro, F.; Peiro-Pastor, R.; Aguado, B. Human genomics projects and precision medicine. Gene Ther. 2017, 24, 551–561. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.H.; Bode, A.M.; Dong, Z. Addressing the challenges of applying precision oncology. NPJ Precis. Oncol. 2017, 1, 28. [Google Scholar] [CrossRef] [Green Version]
- Harkness, E.F.; Astley, S.M.; Evans, D.G. Risk-based breast cancer screening strategies in women. Best Pract. Res. Clin. Obstet. Gynaecol. 2020, 65, 3–17. [Google Scholar] [CrossRef]
- Busse, T.M.; Roth, J.J.; Wilmoth, D.; Wainwright, L.; Tooke, L.; Biegel, J.A. Copy number alterations determined by single nucleotide polymorphism array testing in the clinical laboratory are indicative of gene fusions in pediatric cancer patients. Genes Chromosomes Cancer 2017, 56, 730–749. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhao, Y.; Zhao, J.; Huang, T.; Wu, Y. Impact of AKAP6 polymorphisms on Glioma susceptibility and prognosis. BMC Neurol. 2019, 19, 296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasconcelos, V.C.A.; Lourenço, G.J.; Brito, A.B.C.; Vasconcelos, V.L.; Maldaun, M.V.C.; Tedeschi, H.; Marie, S.K.N.; Shinjo, S.M.O.; Lima, C.S.P. Associations of VEGFA and KDR single-nucleotide polymorphisms and increased risk and aggressiveness of high-grade gliomas. Tumor Biol. 2019, 41, 1010428319872092. [Google Scholar] [CrossRef] [Green Version]
- Mayer, D.K.; Nekhlyudov, L.; Snyder, C.F.; Merrill, J.K.; Wollins, D.S.; Shulman, L.N. American Society of Clinical Oncology clinical expert statement on cancer survivorship care planning. J. Oncol. Pract. 2014, 10, 345–351. [Google Scholar] [CrossRef]
- Rahner, N.; Steinke, V. Hereditary cancer syndromes. Dtsch. Arztebl. Int. 2008, 105, 706. [Google Scholar] [CrossRef]
- Olsson, L.; Lundin-Ström, K.B.; Castor, A.; Behrendtz, M.; Biloglav, A.; Norén-Nyström, U.; Paulsson, K.; Johansson, B. Improved cytogenetic characterization and risk stratification of pediatric acute lymphoblastic leukemia using single nucleotide polymorphism array analysis: A single center experience of 296 cases. Genes Chromosomes Cancer 2018, 57, 604–607. [Google Scholar] [CrossRef]
- Jarvis, K.B.; LeBlanc, M.; Tulstrup, M.; Nielsen, R.L.; Albertsen, B.K.; Gupta, R.; Huttunen, P.; Jónsson, Ó.G.; Rank, C.U.; Ranta, S. Candidate single nucleotide polymorphisms and thromboembolism in acute lymphoblastic leukemia–A NOPHO ALL2008 study. Thromb. Res. 2019, 184, 92–98. [Google Scholar] [CrossRef]
- Li, Y.; Li, S.; Wu, Z.; Hu, F.; Zhu, L.; Zhao, X.; Cui, B.; Dong, X.; Tian, S.; Wang, F. Polymorphisms in genes of APE1, PARP1, and XRCC1: Risk and prognosis of colorectal cancer in a northeast Chinese population. Med. Oncol. 2013, 30, 505. [Google Scholar] [CrossRef]
- Alanazi, M.; Pathan, A.A.K.; Shaik, J.P.; Amri, A.; Parine, N.R. The C Allele of a synonymous SNP (rs1805414, Ala284Ala) in PARP1 is a risk factor for susceptibility to breast cancer in Saudi patients. Asian Pac. J. Cancer Prev. 2013, 14, 3051–3056. [Google Scholar] [CrossRef] [Green Version]
- Siena, S.; Sartore-Bianchi, A.; Garcia-Carbonero, R.; Karthaus, M.; Smith, D.; Tabernero, J.; Van Cutsem, E.; Guan, X.; Boedigheimer, M.; Ang, A. Dynamic molecular analysis and clinical correlates of tumor evolution within a phase II trial of panitumumab-based therapy in metastatic colorectal cancer. Ann. Oncol. 2018, 29, 119–126. [Google Scholar] [CrossRef]
- Schwartzberg, L.; Kim, E.S.; Liu, D.; Schrag, D. Precision Oncology: Who, How, What, When, and When Not? In American Society of Clinical Oncology Educational Book; American Society of Clinical Oncology: Alexandria, VA, USA, 2017; pp. 160–169. [Google Scholar]
- Bode, A.M.; Dong, Z. Recent advances in precision oncology research. NPJ Precis. Oncol. 2018, 2, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagle, M.-C.; Kirouac, D.; Klijn, C.; Liu, B.; Mahajan, S.; Junttila, M.; Moffat, J.; Merchant, M.; Huw, L.; Wongchenko, M. A transcriptional MAPK Pathway Activity Score (MPAS) is a clinically relevant biomarker in multiple cancer types. NPJ Precis. Oncol. 2018, 2, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.T.; Kocialkowski, S.; Liu, L.; Pearson, D.M.; Bäcklund, L.M.; Ichimura, K.; Collins, V.P. Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res. 2008, 68, 8673–8677. [Google Scholar] [CrossRef] [Green Version]
- Subbiah, V.; Westin, S.N.; Wang, K.; Araujo, D.; Wang, W.-L.; Miller, V.A.; Ross, J.S.; Stephens, P.J.; Palmer, G.A.; Ali, S.M. Targeted therapy by combined inhibition of the RAF and mTOR kinases in malignant spindle cell neoplasm harboring the KIAA1549-BRAF fusion protein. J. Hematol. Oncol. 2014, 7, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Jeuken, J.W.; Wesseling, P. MAPK pathway activation through BRAF gene fusion in pilocytic astrocytomas; a novel oncogenic fusion gene with diagnostic, prognostic, and therapeutic potential. J. Pathol. 2010, 222, 324–328. [Google Scholar] [CrossRef]
- Chengalvala, M.V.; Chennathukuzhi, V.M.; Johnston, D.S.; Stevis, P.E.; Kopf, G.S. Gene expression profiling and its practice in drug development. Curr. Genomics 2007, 8, 262–270. [Google Scholar] [CrossRef]
- Chang, J.C.; Wooten, E.C.; Tsimelzon, A.; Hilsenbeck, S.G.; Gutierrez, M.C.; Elledge, R.; Mohsin, S.; Osborne, C.K.; Chamness, G.C.; Allred, D.C. Gene expression profiling for the prediction of therapeutic response to docetaxel in patients with breast cancer. Lancet 2003, 362, 362–369. [Google Scholar] [CrossRef]
- Turiák, L.; Ozohanics, O.; Tóth, G.; Ács, A.; Révész, Á.; Vékey, K.; Telekes, A.; Drahos, L. High sensitivity proteomics of prostate cancer tissue microarrays to discriminate between healthy and cancerous tissue. J. Proteom. 2019, 197, 82–91. [Google Scholar] [CrossRef]
- Hu, B.; Niu, X.; Cheng, L.; Yang, L.N.; Li, Q.; Wang, Y.; Tao, S.C.; Zhou, S.M. Discovering cancer biomarkers from clinical samples by protein microarrays. Proteom. Clin. Appl. 2015, 9, 98–110. [Google Scholar] [CrossRef]
- Blau, C.A.; Liakopoulou, E. Can we deconstruct cancer, one patient at a time? Trends Genet. 2013, 29, 6–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tavassoly, I.; Hu, Y.; Zhao, S.; Mariottini, C.; Boran, A.; Chen, Y.; Li, L.; Tolentino, R.E.; Jayaraman, G.; Goldfarb, J. Genomic signatures defining responsiveness to allopurinol and combination therapy for lung cancer identified by systems therapeutics analyses. Mol. Oncol. 2019, 13, 1725–1743. [Google Scholar] [CrossRef] [Green Version]
- Sandhu, C.; Qureshi, A.; Emili, A. Panomics for precision medicine. Trends Mol. Med. 2018, 24, 85–101. [Google Scholar] [CrossRef] [PubMed]
- Yakhini, Z.; Jurisica, I. Cancer computational biology. BMC Bioinform. 2011, 12, 120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nussinov, R.; Jang, H.; Tsai, C.-J.; Cheng, F. Precision medicine and driver mutations: Computational methods, functional assays and conformational principles for interpreting cancer drivers. PLoS Comp. Biol. 2019, 15, e1006658. [Google Scholar]
- Wahba, H.A.; El-Hadaad, H.A. Current approaches in treatment of triple-negative breast cancer. Cancer Biol. Med. 2015, 12, 106. [Google Scholar]
- Chenevix-Trench, G.; Milne, R.L.; Antoniou, A.C.; Couch, F.J.; Easton, D.F.; Goldgar, D.E. An international initiative to identify genetic modifiers of cancer risk in BRCA1 and BRCA2 mutation carriers: The Consortium of Investigators of Modifiers of BRCA1 and BRCA2 (CIMBA). Breast Cancer Res. 2007, 9, 104. [Google Scholar] [CrossRef] [Green Version]
- Stratton, M.R.; Campbell, P.J.; Futreal, P.A. The cancer genome. Nature 2009, 458, 719–724. [Google Scholar] [CrossRef] [Green Version]
- Stephens, P.J.; Tarpey, P.S.; Davies, H.; Van Loo, P.; Greenman, C.; Wedge, D.C.; Nik-Zainal, S.; Martin, S.; Varela, I.; Bignell, G.R. The landscape of cancer genes and mutational processes in breast cancer. Nature 2012, 486, 400–404. [Google Scholar] [CrossRef]
- Greenman, C.; Stephens, P.; Smith, R.; Dalgliesh, G.L.; Hunter, C.; Bignell, G.; Davies, H.; Teague, J.; Butler, A.; Stevens, C. Patterns of somatic mutation in human cancer genomes. Nature 2007, 446, 153–158. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61. [Google Scholar] [CrossRef] [Green Version]
- Mathioudaki, A.; Ljungström, V.; Melin, M.; Arendt, M.L.; Nordin, J.; Karlsson, Å.; Murén, E.; Saksena, P.; Meadows, J.R.; Marinescu, V.D. Targeted sequencing reveals the somatic mutation landscape in a Swedish breast cancer cohort. Sci. Rep. 2020, 10, 1–13. [Google Scholar] [CrossRef]
- Pereira, B.; Chin, S.-F.; Rueda, O.M.; Vollan, H.-K.M.; Provenzano, E.; Bardwell, H.A.; Pugh, M.; Jones, L.; Russell, R.; Sammut, S.-J. The somatic mutation profiles of 2,433 breast cancers refine their genomic and transcriptomic landscapes. Nat. Commun. 2016, 7, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Ahearn, T.U.; Lecarpentier, J.; Barnes, D.; Beesley, J.; Qi, G.; Jiang, X.; O’Mara, T.A.; Zhao, N.; Bolla, M.K. Genome-wide association study identifies 32 novel breast cancer susceptibility loci from overall and subtype-specific analyses. Nat. Genet. 2020, 52, 572–581. [Google Scholar] [CrossRef]
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; Van De Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81. [Google Scholar] [CrossRef] [PubMed]
- Low, S.K.; Zembutsu, H.; Nakamura, Y. Breast cancer: The translation of big genomic data to cancer precision medicine. Cancer Sci. 2018, 109, 497–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van’t Veer, L.J.; Dai, H.; Van De Vijver, M.J.; He, Y.D.; Hart, A.A.; Mao, M.; Peterse, H.L.; Van Der Kooy, K.; Marton, M.J.; Witteveen, A.T. Gene expression profiling predicts clinical outcome of breast cancer. Nature 2002, 415, 530–536. [Google Scholar] [CrossRef] [Green Version]
- Park, H.L.; Ziogas, A.; Chang, J.; Desai, B.; Bessonova, L.; Garner, C.; Lee, E.; Neuhausen, S.L.; Wang, S.S.; Ma, H. Novel polymorphisms in caspase-8 are associated with breast cancer risk in the California Teachers Study. BMC Cancer 2016, 16, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Navarrete-Bernal, M.G.; Cervantes-Badillo, M.G.; Martínez-Herrera, J.F.; Lara-Torres, C.O.; Gerson-Cwilich, R.; Zentella-Dehesa, A.; Ibarra-Sánchez, M.d.J.; Esparza-López, J.; Montesinos, J.J.; Cortés-Morales, V.A. Biological Landscape of Triple Negative Breast Cancers Expressing CTLA-4. Front. Oncol. 2020, 10, 1206. [Google Scholar] [CrossRef]
- Lei, H.; Deng, C.-X. Fibroblast growth factor receptor 2 signaling in breast cancer. Int. J. Biol. Sci. 2017, 13, 1163. [Google Scholar] [CrossRef] [Green Version]
- Xie, Z.; Janczyk, P.Ł.; Zhang, Y.; Liu, A.; Shi, X.; Singh, S.; Facemire, L.; Kubow, K.; Li, Z.; Jia, Y. A cytoskeleton regulator AVIL drives tumorigenesis in glioblastoma. Nat. Commun. 2020, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Vigneswaran, K.; Neill, S.; Hadjipanayis, C.G. Beyond the World Health Organization grading of infiltrating gliomas: Advances in the molecular genetics of glioma classification. Ann. Transl. Med. 2015, 3, 95. [Google Scholar]
- Varlet, P.; Le Teuff, G.; Le Deley, M.-C.; Giangaspero, F.; Haberler, C.; Jacques, T.S.; Figarella-Branger, D.; Pietsch, T.; Andreiuolo, F.; Deroulers, C. WHO grade has no prognostic value in the pediatric high-grade glioma included in the HERBY trial. Neuro-Oncology 2020, 22, 116–127. [Google Scholar] [CrossRef]
- Brandes, A.A.; Tosoni, A.; Franceschi, E.; Reni, M.; Gatta, G.; Vecht, C. Glioblastoma in adults. Crit. Rev. Oncol. Hematol. 2008, 67, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Ohgaki, H.; Kleihues, P. Epidemiology and etiology of gliomas. Acta Neuropathol. 2005, 109, 93–108. [Google Scholar] [CrossRef] [PubMed]
- Shamran, H.A.; Ghazi, H.F.; Ahmed, A.-S.; Al-Juboory, A.A.; Taub, D.D.; Price, R.L.; Nagarkatti, M.; Nagarkatti, P.S.; Singh, U.P. Single nucleotide polymorphisms in IL-10, IL-12p40, and IL-13 genes and susceptibility to glioma. Int. J. Med. Sci. 2015, 12, 790. [Google Scholar] [CrossRef] [Green Version]
- Monticone, M.; Daga, A.; Candiani, S.; Romeo, F.; Mirisola, V.; Viaggi, S.; Melloni, I.; Pedemonte, S.; Zona, G.; Giaretti, W. Identification of a novel set of genes reflecting different in vivo invasive patterns of human GBM cells. BMC Cancer 2012, 12, 358. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Li, H.; Chen, Z.; Fan, L.; Feng, S.; Cai, X.; Wang, H. Identification of 3 subpopulations of tumor-infiltrating immune cells for malignant transformation of low-grade glioma. Cancer Cell Int. 2019, 19, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.-C.; Hu, Z.-Q.; Long, J.-H.; Zhu, G.-M.; Wang, Y.; Jia, Y.; Zhou, J.; Ouyang, Y.; Zeng, Z. Clinical implications of tumor-infiltrating immune cells in breast cancer. J. Cancer 2019, 10, 6175. [Google Scholar] [CrossRef]
- Zhang, X.; Quan, F.; Xu, J.; Xiao, Y.; Li, X.; Li, Y. Combination of multiple tumor-infiltrating immune cells predicts clinical outcome in colon cancer. Clin. Immunol. 2020, 215, 108412. [Google Scholar] [CrossRef] [PubMed]
- Taylor, L.P. Diagnosis, treatment, and prognosis of glioma: Five new things. Neurology 2010, 75, S28–S32. [Google Scholar] [CrossRef] [Green Version]
- Tran, W.T.; Jerzak, K.; Lu, F.-I.; Klein, J.; Tabbarah, S.; Lagree, A.; Wu, T.; Rosado-Mendez, I.; Law, E.; Saednia, K. Personalized breast cancer treatments using artificial intelligence in radiomics and pathomics. J. Med. Imaging Radiat. Sci. 2019, 50, S32–S41. [Google Scholar] [CrossRef] [Green Version]
- Way, G.P.; Sanchez-Vega, F.; La, K.; Armenia, J.; Chatila, W.K.; Luna, A.; Sander, C.; Cherniack, A.D.; Mina, M.; Ciriello, G. Machine learning detects pan-cancer ras pathway activation in the cancer genome atlas. Cell Rep. 2018, 23, 172–180.e3. [Google Scholar] [CrossRef] [Green Version]
- Ekins, S.; Puhl, A.C.; Zorn, K.M.; Lane, T.R.; Russo, D.P.; Klein, J.J.; Hickey, A.J.; Clark, A.M. Exploiting machine learning for end-to-end drug discovery and development. Nat. Mater. 2019, 18, 435. [Google Scholar] [CrossRef] [PubMed]
- Bera, K.; Schalper, K.A.; Rimm, D.L.; Velcheti, V.; Madabhushi, A. Artificial intelligence in digital pathology—new tools for diagnosis and precision oncology. Nat. Rev. Clin. Oncol. 2019, 16, 703–715. [Google Scholar] [CrossRef]
- Gullo, R.L.; Daimiel, I.; Morris, E.A.; Pinker, K. Combining molecular and imaging metrics in cancer: Radiogenomics. Insights Imaging 2020, 11, 1–17. [Google Scholar] [CrossRef]
- Nasief, H.; Zheng, C.; Schott, D.; Hall, W.; Tsai, S.; Erickson, B.; Li, X.A. A machine learning based delta-radiomics process for early prediction of treatment response of pancreatic cancer. NPJ Precis. Oncol. 2019, 3, 1–10. [Google Scholar] [CrossRef]
- Liao, H.; Long, Y.; Han, R.; Wang, W.; Xu, L.; Liao, M.; Zhang, Z.; Wu, Z.; Shang, X.; Li, X. Deep learning-based classification and mutation prediction from histopathological images of hepatocellular carcinoma. Clin. Transl. Med. 2020, 10, e102. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Verma, A.; Naveed, U.; Bakhoum, S.; Khosravi, P.; Elemento, O. Deep learning predicts chromosomal instability from histopathology images. iScience 2021, 24, 102394. [Google Scholar] [CrossRef]
- Huang, S.; Cai, N.; Pacheco, P.P.; Narrandes, S.; Wang, Y.; Xu, W. Applications of support vector machine (SVM) learning in cancer genomics. Cancer Genom. Proteom. 2018, 15, 41–51. [Google Scholar]
- Arora, P.; Boyne, D.; Slater, J.J.; Gupta, A.; Brenner, D.R.; Druzdzel, M.J. Bayesian networks for risk prediction using real-world data: A tool for precision medicine. Value Health 2019, 22, 439–445. [Google Scholar] [CrossRef] [Green Version]
- Agrahari, R.; Foroushani, A.; Docking, T.R.; Chang, L.; Duns, G.; Hudoba, M.; Karsan, A.; Zare, H. Applications of Bayesian network models in predicting types of hematological malignancies. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Braden, A.; Stankowski, R.; Engel, J.; Onitilo, A. Breast cancer biomarkers: Risk assessment, diagnosis, prognosis, prediction of treatment efficacy and toxicity, and recurrence. Curr. Pharm. Des. 2014, 20, 4879–4898. [Google Scholar] [CrossRef] [PubMed]
- Nistal-Nuño, B. Tutorial of the probabilistic methods Bayesian networks and influence diagrams applied to medicine. J. Evid. Based Med. 2018, 11, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Chudasama, D.; Bo, V.; Hall, M.; Anikin, V.; Jeyaneethi, J.; Gregory, J.; Pados, G.; Tucker, A.; Harvey, A.; Pink, R. Identification of cancer biomarkers of prognostic value using specific gene regulatory networks (GRN): A novel role of RAD51AP1 for ovarian and lung cancers. Carcinogenesis 2018, 39, 407–417. [Google Scholar] [CrossRef] [Green Version]
- Witteveen, A.; Nane, G.F.; Vliegen, I.M.; Siesling, S.; IJzerman, M.J. Comparison of logistic regression and Bayesian networks for risk prediction of breast cancer recurrence. Med. Decis. Making 2018, 38, 822–833. [Google Scholar] [CrossRef]
- Asri, H.; Mousannif, H.; Al Moatassime, H.; Noel, T. Using machine learning algorithms for breast cancer risk prediction and diagnosis. Procedia Comput. Sci. 2016, 83, 1064–1069. [Google Scholar] [CrossRef] [Green Version]
- Nahid, A.-A.; Kong, Y. Involvement of machine learning for breast cancer image classification: A survey. Comput. Math. Methods Med. 2017, 2017. [Google Scholar] [CrossRef]
- Nindrea, R.D.; Aryandono, T.; Lazuardi, L.; Dwiprahasto, I. Diagnostic accuracy of different machine learning algorithms for breast cancer risk calculation: A meta-analysis. Asian Pac. J. Cancer Prev. 2018, 19, 1747. [Google Scholar]
- Visvanathan, K.; Hurley, P.; Bantug, E.; Brown, P.; Col, N.F.; Cuzick, J.; Davidson, N.E.; DeCensi, A.; Fabian, C.; Ford, L. Use of pharmacologic interventions for breast cancer risk reduction: American Society of Clinical Oncology clinical practice guideline. J. Clin. Oncol. 2013, 31, 2942–2962. [Google Scholar] [CrossRef]
- Moyer, V.A. Medications to decrease the risk for breast cancer in women: Recommendations from the US Preventive Services Task Force recommendation statement. Ann. Intern. Med. 2013, 159, 698–708. [Google Scholar] [PubMed]
- Gevaert, O.; Smet, F.D.; Timmerman, D.; Moreau, Y.; Moor, B.D. Predicting the prognosis of breast cancer by integrating clinical and microarray data with Bayesian networks. Bioinformatics 2006, 22, e184–e190. [Google Scholar] [CrossRef] [Green Version]
- Niu, B.; Liang, C.; Lu, Y.; Zhao, M.; Chen, Q.; Zhang, Y.; Zheng, L.; Chou, K.-C. Glioma stages prediction based on machine learning algorithm combined with protein-protein interaction networks. Genomics 2020, 112, 837–847. [Google Scholar] [CrossRef]
- Long, H.; Liang, C.; Zhang, X.A.; Fang, L.; Wang, G.; Qi, S.; Huo, H.; Song, Y. Prediction and analysis of key genes in glioblastoma based on bioinformatics. Biomed. Red. Int. 2017, 2017, 7653101. [Google Scholar] [CrossRef]
- Leclerc, P.; Ray, C.; Mahieu-Williame, L.; Alston, L.; Frindel, C.; Brevet, P.-F.; Meyronet, D.; Guyotat, J.; Montcel, B.; Rousseau, D. Machine learning-based prediction of glioma margin from 5-ALA induced PpIX fluorescence spectroscopy. Sci. Rep. 2020, 10, 1–9. [Google Scholar] [CrossRef]
- Pirmohamed, M. Pharmacogenetics and pharmacogenomics. Br. J. Clin. Pharmacol. 2001, 52, 345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, S.H.; Bode, A.M.; Dong, Z. Precision medicine: The foundation of future cancer therapeutics. NPJ Precis. Oncol. 2017, 1, 12. [Google Scholar] [CrossRef] [PubMed]
- Booth, T.C.; Williams, M.; Luis, A.; Cardoso, J.; Ashkan, K.; Shuaib, H. Machine learning and glioma imaging biomarkers. Clin. Radiol. 2020, 75, 20–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baptiste, M.; Moinuddeen, S.S.; Soliz, C.L.; Ehsan, H.; Kaneko, G. Making Sense of Genetic Information: The Promising Evolution of Clinical Stratification and Precision Oncology Using Machine Learning. Genes 2021, 12, 722. https://doi.org/10.3390/genes12050722
Baptiste M, Moinuddeen SS, Soliz CL, Ehsan H, Kaneko G. Making Sense of Genetic Information: The Promising Evolution of Clinical Stratification and Precision Oncology Using Machine Learning. Genes. 2021; 12(5):722. https://doi.org/10.3390/genes12050722
Chicago/Turabian StyleBaptiste, Mahaly, Sarah Shireen Moinuddeen, Courtney Lace Soliz, Hashimul Ehsan, and Gen Kaneko. 2021. "Making Sense of Genetic Information: The Promising Evolution of Clinical Stratification and Precision Oncology Using Machine Learning" Genes 12, no. 5: 722. https://doi.org/10.3390/genes12050722
APA StyleBaptiste, M., Moinuddeen, S. S., Soliz, C. L., Ehsan, H., & Kaneko, G. (2021). Making Sense of Genetic Information: The Promising Evolution of Clinical Stratification and Precision Oncology Using Machine Learning. Genes, 12(5), 722. https://doi.org/10.3390/genes12050722