1. Introduction
Iron is an essential trace element that combines with proteins or small organic molecules involved in various oxidation–reduction reactions in living organisms. Due to the scarcity of iron in nature, microorganisms secure iron by secreting siderophores and absorbing them back into the cells following the adsorption of environmental iron [
1]. Siderophores are small organic iron chelators that preferentially bind to Fe
3+ ions through the formation of a coordination complex. They are classified by the ligand structure as carboxylates, hydroxamates, catecholates, and mixed types [
2]. Siderophores, including enterobactin (catecholate), desferrioxamine (hydroxamate), and pyoverdine (mixed type), are known to be produced by bacteria [
3], whereas hydroxamate siderophores, such as ferrichromes, fusarinine C, and triacetylfusarinine C (TAFC), are the major siderophores in fungi [
1,
4].
The fungal hydroxamate siderophores are synthesized by the activity of the nonribosomal peptide synthetase complex (NRPS). In
Aspergillus fumigatus, SidC is involved in the biosynthesis of ferrichromes using N
5-acetyl-N
5-hydroxy-
l-ornithine, glycine, and serine as precursors, where SidD catalyzes the condensation of three molecules of N
5-anhydromevalonyl-N
5-hydroxy-
l-ornithine to produce fusarinine C and TAFC [
1]. Basidiomycetes also have shown to contain similar NRPSs. Sid2 and Fso1 in
Ustilago maydis [
5] and
Omphalotus olearius [
6], respectively, are NRPSs homologous to SidC in
A. fumigatus, whereas CsNPS2 in
Ceriporiopsis subvermispora, NPS1 in
Trametes versicolor, and the gene product of EAU88504 in
Coprinopsis cinerea are thought to be homologous to SidD as they show similar domain arrangement [
7].
Agaricus bisporus is reported to produce hydroxamate siderophores, such as ferrichrome, defferri-des(diserylglycyl) ferrirhodin, and fusarinine C [
8], accordingly, and putative SidC and SidD homologues are found from the genome information of
A. bisporus as described in the present study.
The expression of genes involved in the siderophore biosynthetic pathway is tightly regulated by the transcription factor
hapX, which positively regulates the expression of
sid genes and represses genes involved in iron-consuming pathways under iron-starvation conditions in
A. fumigatus [
9]. The expression of
hapX is down-regulated by another transcription factor,
sreA, under sufficient iron conditions [
10,
11]. Study on iron metabolism in basidiomycetes is rare. However,
hapX and
cir1, a homologue of
sreA, in
Cryptococcus neoformans have been extensively studied in relation to virulence in humans [
12,
13]. Similar to the ascomycetes,
hapX in
C. neoformans represses iron utilization and promotes iron uptake and
cir1 expression under iron starvation conditions [
12,
13]. Therefore, the deprivation of iron is a prerequisite for the expression of
hapX and thus for the production of siderophore in fungi.
Siderophores have multiple applications in agriculture as plant growth promotors and pathogen control agents, as well as in the removal of contaminated heavy metal ions [
14]. In medicinal applications, siderophores are employed to treat diseases associated with iron overload [
15]. Certain siderophores show antimicrobial activities against pathogenic bacteria, though siderophores mostly act as virulence factors for many pathogens [
16,
17,
18]. Drug delivery through siderophore receptors after the formation of siderophore–drug complex may be a new strategy to deliver drugs that have permeability-mediated drug resistance [
19]. Additionally, ferrichrome produced by
Lactobacillus casei has been reported to show anticancer activity against colon cancer in a mouse model [
20].
In the present study, we assess the potential of A. bisporus, a representative edible mushroom, as a host system for the production of biological molecules such as siderophores. However, A. bisporus, like other fungi, secreted siderophores only in the absence of iron, meaning that the production medium should be an iron-free minimal medium. To overcome this problem, we generated transformant strains of A. bisporus that express hapX, a master transcription factor for sid gene expression, using the constitutive glyceraldehyde 3–phosphate dehydrogenase promoter (pGPD).
2. Materials and Methods
2.1. Strains and Culture Conditions
A. bisporus NH1 was obtained from the National Institute of Horticultural and Herbal Science, Rural Development Administration (RDA; Eomsung, Korea). The mushroom strain was grown at 25 °C on compost–potato dextrose agar (C–PDA), in which PDA (Oxoid, Basingstoke, UK) was supplemented with 200 mL of compost extract per litter. For the liquid culture, compost–potato dextrose broth (C–PDB) was prepared by supplementing the compost extract (200 mL/L) with potato dextrose broth (Oxoid, Basingstoke, UK). The compost extract for the preparation of both media was prepared as follows: rice straw compost was suspended in water (100 g/L), left for a day at room temperature, autoclaved for 40 min at 121 °C, then filtered using a Miracloth (Sigma-Aldrich, St. Louis, MO, USA).
2.2. Construction of pBGgHg-hapX Vector
The
hapX gene sequence in
A. bisporus was retrieved from the MycoCosm genome database (
https://mycocosm.jgi.doe.gov/Agabi_varbisH97_2/Agabi_varbisH97_2.home.html, accessed on 16 August 2019) by BLASTP analysis using
A. fumigatus HapX protein (XP_747952.1) as a query sequence. The retrieved HapX protein contained a conserved Hap2/3/5 binding domain in front of the bZIP domain similar to
A. fumigatus HapX protein (
Supplementary Figure S1). Homologous gene sequences to
A. fumigatus sidC and
sidD were also retrieved under the gene accession numbers NW_006267370.1 and NW_006267372.1, respectively, using a similar approach.
For the isolation of
hapX gene,
A. bisporus NH1 was grown in 100 mL of C–PDB for 10 days at 25 °C. The genomic DNA was isolated from the harvested mycelia using a genomic DNA extraction kit (HiGene Genomic DNA Prep kit; BIOFACT, Daejeon, Korea). The
A. bisporus hapX gene (GenBank Accession No. XM_006454360) was amplified from the purified genomic DNA using a specific primer set (
Supplementary Table S1, hapX-F and hapX-R) and a PCR premix (nPfu-Forte; Enzynomics, Daejeon, Korea). The PCR reaction consisted of the initial denaturation at 95 °C for 5 min, followed by 25 cycles of denaturation at 95 °C for 30 s, annealing at 60 °C for 30 s, and polymerization at 72 °C for 4 min, and a final extension at 72 °C for 10 min. The resulting amplicon contained
SwaI and
SpeI restriction sites at the 5′- and 3′-ends of the
hapX gene provided by the primers, respectively. The
hapX amplicon was digested by
SwaI and
SpeI and inserted into the corresponding restriction sites in the pBGgHg vector [
21] by replacing the
eGFP gene, generating pBGgHg-hapX (
Supplementary Figure S2).
2.3. Transformation of Agrobacterium tumefaciens AGL-1 with pBGgHg-hapX
Agrobacterium tumefaciens AGL-1 was grown in 10 mL of LB (peptone 10 g/L, yeast extract 5 g/L, NaCl 5 g/L) at 28 °C until the optical density at 600 nm reached 0.6. The bacterial cells were collected by centrifugation (10,000× g, 10 min). The cell pellet was washed with 1X TE buffer and resuspended in 2 mL of 10% LB broth. Aliquots of the competent cells were stored at −70 °C until use. For the transformation, the competent cells (250 μL) were mixed with 1 μg of pBGgHg-hapX plasmid DNA by gentle pipetting and were kept on ice for 5 min. After the incubation, the tube was placed in liquid nitrogen for 5 min and then transferred to a water bath (37 °C). After 5 min of incubation, 1 mL of LB broth was added and incubated for an additional 2 h at 28 °C with vigorous agitation. The incubated cells were collected by centrifugation and were suspended in 0.2 mL LB broth. The suspension was spread on LB agar containing 50 mg/L of kanamycin. The plate was incubated for 2 days at 28 °C to obtain A. tumefaciens AGL-1 transformed with pBGgHg-hapX.
2.4. Agrobacterium tumefaciens-Mediated Transformation of Agaricus bisporus
A. bisporus was transformed by the
A. tumefaciens-mediated transformation (ATMT) method [
21] with slight modifications. For the transformation, 1 mL of freshly grown
A. tumefaciens AGL-1 harboring pBGgHg-hapX was inoculated to 100 mL of LB broth and incubated at 28 °C with agitation (170 rpm) until the optical density at 600 nm reached 0.8. The cells were collected by centrifugation (10,000×
g, 10 min) and were resuspended in 100 mL of induction medium (IM), containing 2 g/L glucose, 5 mL/L glycerol, 1 g/L NH
4Cl, 0.3 g/L MgSO
4, 0.15 g/L KCl, 0.01 g/L CaCl
2, 2.5 mg/L FeSO
4, 0.2 mM acetosyringone, 40 mM MES buffer (pH 5.3), and 50 mg/L kanamycin. The cell suspension was incubated for 6 h at 25 °C. For the infection of
A. tumefaciens to
A. bisporus, pieces of gill tissue (1 mm) were cut from the fruiting body of
A. bisporus NH1 and were co-incubated in the bacterial culture for 15 min. The infected gill tissues were transferred onto IM agar (IM with 15 g/L agar). After 3 days of incubation at 25 °C, the gill tissues were transferred to the first selection medium (C-PDA supplemented with 150 mg/L cefotaxime, 100 mg/L kanamycin, 25 mg/L chloramphenicol, gentamycin 100 mg/L, and 30 mg/L hygromycin B). After 10 days of incubation at 25 °C, the tissues showing filamentous hyphal growth were transferred to the second selection medium (C-PDA with 50 mg/L hygromycin B). The mycelia outgrowing on the second selection medium upon prolonged incubation at 25 °C were cut out from the agar medium and were subjected to further analysis.
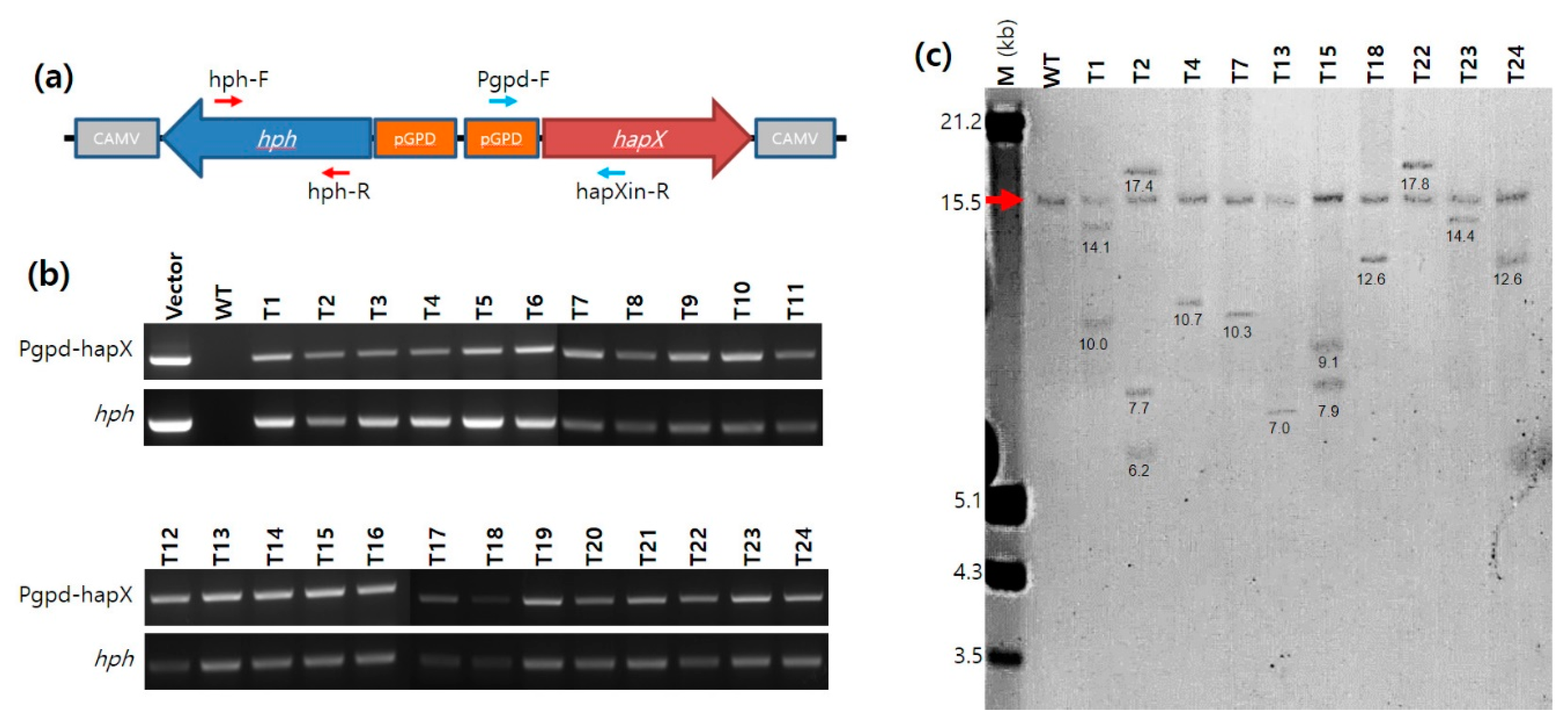
2.5. PCR and Real-Time PCR Analyses of the A. bisporus Transformants
The transformants of
A. bisporus obtained from ATMT were analyzed by PCR using primer sets targeting an internal sequence region in the
hph gene (731 bp; hph-F and hph-R) and a sequence region ranging from pGPD to the internal
hapX sequence (725 bp; Pgpd-F and hapXin-R) (
Figure 1a and
Supplementary Table S1). The PCR conditions were the same as those described above, except for the polymerization time (30 s in this case). The expression of the
hapX gene in the transformants was investigated by quantitative real-time PCR (qPCR) analysis. For the qPCR, 1 mL of actively grown mycelia in C-PDB were inoculated in 50 mL of minimal medium (KCl 0.2 g/L, KH
2PO
4 0.14 g/L, Na
2HPO
4·12H
2O 1.9 g/L, CaCl
2·2H
2O 0.27 g/L, MgSO
4·7H
2O 0.2 g/L, ZnSO
4·7H
2O 2 mg/L, CuSO
4·6H
2O 0.1 mg/L, MnSO
4·H
2O 0.02 mg/L, (NH
4)
6Mo
7O
24·4H
2O 0.02 mg/L, H
3BO
3 0.01 mg/L, glucose 30 g/L, yeast extract 5 g/L, pH 6.8) with or without 10 μM FeCl
3. The culture was incubated for a week at 25 °C. The harvested mycelia were subjected to total RNA extraction. Total RNA was extracted using an RNeasy Plant Mini Kit (Qiagen, Hilden, Germany) from the mycelial powder (0.1 g), which was prepared by grinding with a pestle and mortar after freezing in liquid nitrogen. The extracted RNA was subjected to cDNA synthesis using a TOPscript cDNA Synthesis Kit (Enzynomics, Daejeon, Korea), followed by qPCR using FastStart Universal SYBR Green Master (Sigma-Aldrich) and Lightcycler Nano (Roche, Germany). The primers used for qPCR are shown in
Supplementary Table S1. The qPCR results were analyzed using the expression of the β-tubulin gene as a reference. Relative gene repression was calculated using the 2
−∆∆Cq value. All data were obtained in triplicate from three independent experiments. The statistical significance between data sets was analyzed using a one-way ANOVA test.
2.6. Southern Blot Analysis with DIG-Labeling
Genomic DNA (20 μg) was digested with
SpeI and
AgeI for 24 h at 37 °C. The digested samples were subjected to agarose gel electrophoresis in 0.8% agarose gel (15 cm × 15 cm) at 30 V for 18 h, followed by 50 V for 3 h. A digoxigenin (DIG)-labeled DNA marker (DNA molecular weight marker III, Roche, Germany) was run together for the size analysis. The DNA fragments in the agarose gel were transferred onto a nylon membrane (Amersham hybond
TM-N
+ nylon membrane, GE Healthcare, Chicago, IL, USA) using standard blotting methods. After the transfer, the DNA fragments on the membrane were cross-linked using CL-1000 Ultraviolet Crosslinker (Spectrum chemical, New Brunswick, NJ, USA). The nylon membrane was subjected to hybridization with a 10 ng/mL DIG-labeled probe specific to the
hapX gene. The DIG-labeled
hapX probe was generated by PCR using hapX-probe-fwd and hapX-probe-rev primers (
Supplementary Table S1) followed by DIG labeling using a labeling kit (Random Primed DNA Labeling Kit, Roche, Rotkreuz, Switzerland). The DIG-labeled DNA was visualized by the NBT/BCIP reaction after binding with 150 mU/mL of Anti-DIG-AP conjugate (Roche, Rotkreuz, Switzerland).
2.7. HPLC Analysis
The mycelia of A. bisporus were grown in C–PDB for 10 days and were harvested by centrifugation (1000× g, 10 min). The collected mycelia were resuspended in minimal medium and further incubated for 3 days. The culture broth was obtained through filtration with a Miracloth (Sigma-Aldrich, St. Louis, MO, USA) followed by 3 M filter paper. Final 1 mM of FeCl3 was added to the filtrate to convert siderophores to iron-bound form. The treated filtrate (2 mL) was subjected to a Sep-Pak C18 cartridge (Waters, Milford, MA, USA). The cartridge was washed with 10 mL of deionized water, and then the bound siderophore was eluted by 5 mL of methanol. The eluate was dried by a vacuum evaporator. The dried sample was dissolved in 0.5 mL of deionized water. The obtained sample (20 μL) was subjected to HPLC analysis using an HPLC system (HP1050; Hewlett-Packard, Palo Alto, CA, USA) equipped with an HC-C18(2) column (150 × 4.6 mm; Agilent, Santa Clara, CA, USA). Isocratic elution was performed with 0.1% triflouroacetic acid as a mobile phase at a flow rate of 0.6 mL/min. The chromatogram was monitored at 214 nm.
2.8. Determination of Siderophore Activity by Chrome Azurol-S Assay
Siderophore in the sample solution was determined using a modified chrome azurol-S (CAS) assay [
22]. For the formulation of the CAS reagent, 0.75 mL of 2 mM CAS was mixed with 1 mL of 10 mM FeCl
3 (in 10 mM HCl) and 0.6 mL of 10 mM hexadecyltrimethylammonium (HDTMA). The final volume of the mixture was brought up to 10 mL with deionized water after the addition of 5 mL of 1 M MES (pH5.6) to make the CAS reagent. For the determination of siderophore, the peak fraction from the HPLC analysis (0.6 mL) was treated with 60 μL of 3% (
w/
v) 8-hydroxyquinoline (Sigma-Aldrich, St. Louis, MO, USA) in chloroform. The chloroform layer was removed after centrifugation (10,000×
g, 5 min). The aqueous layer (0.5 mL) was mixed with the CAS reagent (0.5 mL) and 10 μL of 0.2 M sulfosalicylic acid. The reaction mixture was incubated for 1 h at 25 °C. The decrease in the absorbance at 650 nm (A
650) was monitored using a UV-vis spectrophotometer. The relative production of siderophore was calculated using the following equation.
where A
max stands for the A
650 value without siderophore and A
min for the A
650 value at the saturated siderophore concentration. A
sample represents the A
650 value in the presence of the siderophore sample.
4. Discussion
Mushrooms mostly belonging to the phylum Basidiomycota are an important group of fungi due to their roles as decomposers in the ecosystem. Some of them, including
A. bisporus,
Lentinula edodes,
Pleurotus ostreatus, and
Flammulina velutipes, are mainly consumed as nutritious foods, while others, such as
Ganoderma lucidum,
Phellinus linteus,
Inonotus obliquus, and
Trametes versicolor, are used as sources of biologically active compounds [
23]. Although they have a long history of human consumption, there have been few studies on the genetic (metabolic) engineering of basidiomycetes in order to use them as biological hosts to produce small organic compounds, despite the fact that they are equipped well with various metabolic pathways and diversified by their gene duplication and horizontal gene transfer [
24].
A. bisporus is one of the best-studied commercial mushrooms in genetics and molecular biology [
21,
25,
26,
27], making it a good model system for the genetic engineering of mushrooms. In the present study, we demonstrated the successful transformation of
A. bisporus to produce siderophore independently of iron, as normally,
A. bisporus only produces siderophore in the absence of iron to acquire environmental iron. In filamentous fungi,
sid genes involved in the siderophore biosynthesis are under the control of the main transcriptional activator,
hapX, the expression of which is negatively controlled by ferrous ion [
9]. For the constitutive expression of
hapX, we isolated a
hapX homolog and GPD promoter (pGPD) from the
A. bisporus chromosomal DNA and performed genomic integration of pGPD-
hapX through
A. tumefaciens-mediated transformation. The transformation efficiency (24%) was comparable to that which was previously reported (30–40%) [
21]. Southern blot analysis revealed that single-copy integration of pGPD-
hapX was prevalent; however, the transformants with 2–3 copies of pGPD-
hapX integration were also observed, similar to a previous report on pBGgHg [
21]. The multiple copies observed in certain transformants in this study do not imply multiple integrations to the chromosomes of a certain nucleus because
A. bisporus can have multiple nuclei in the cytoplasm. Nonetheless, in contrast to this previous report, which failed to detect the expression of the
eGFP gene [
21], we observed the functional expression of
hapX at the mRNA level (
Figure 2) and also at the protein level, as deduced from the constitutive production of siderophore (
Figure 3). This is conceivable because
hapX originates from
A. bisporus itself, thereby enabling normal mRNA processing and the use of host codons, in contrast to heterologous
eGFP [
21]. Additionally, we showed that integrated pGPD-
hapX is maintained stably through successive transfers in the absence of selective pressure.
A. bisporus has been known to produce three hydroxamate siderophores, including ferrichrome (FC), defferri-des(diserylglycyl) ferrirhodin (DDF), and fusarinine C (FsC), at the ratios of 10%, 30%, and 60%, respectively [
8]. The first two siderophores are known to be synthesized by the NRPS activity of the
sidC gene product using N
5-acetyl-N
5-OH-L-Orn, glycine, and serine as precursors, whereas FsC is a condensation product comprised of three molecules of N
5-anhydromevalonyl-N
5-OH-L-Orn catalyzed by the NRPS encoded by
sidD [
1]. The true nature of the siderophore produced from the pGPD-
hapX transformants was not identified; however, our RT-PCR analysis showed that only the
sidD gene was transcribed in a
hapX-dependent manner, while the expression of
sidC was not observed (data not shown), suggesting that the siderophore produced in the present study is FsC.
Since mushrooms have mostly been consumed as fresh foods or medicines, research on their molecular genetic modification is very rare. However, considering the fact that mushrooms produce various organic molecules in their different developmental stages, they have great potential as host systems for the production of useful organic compounds. In this regard, our research provides a good example of how a common mushroom can be transformed into a producer of valuable organic compounds using genetic manipulation.



{kind=link}
{kind=link}
{kind=link}
{kind=link}



