
Genome Expression Dynamics Reveal the Parasitism Regulatory Landscape of the Root-Knot Nematode Meloidogyne incognita and a Promoter Motif Associated with Effector Genes

 , , , , and
, , , , and 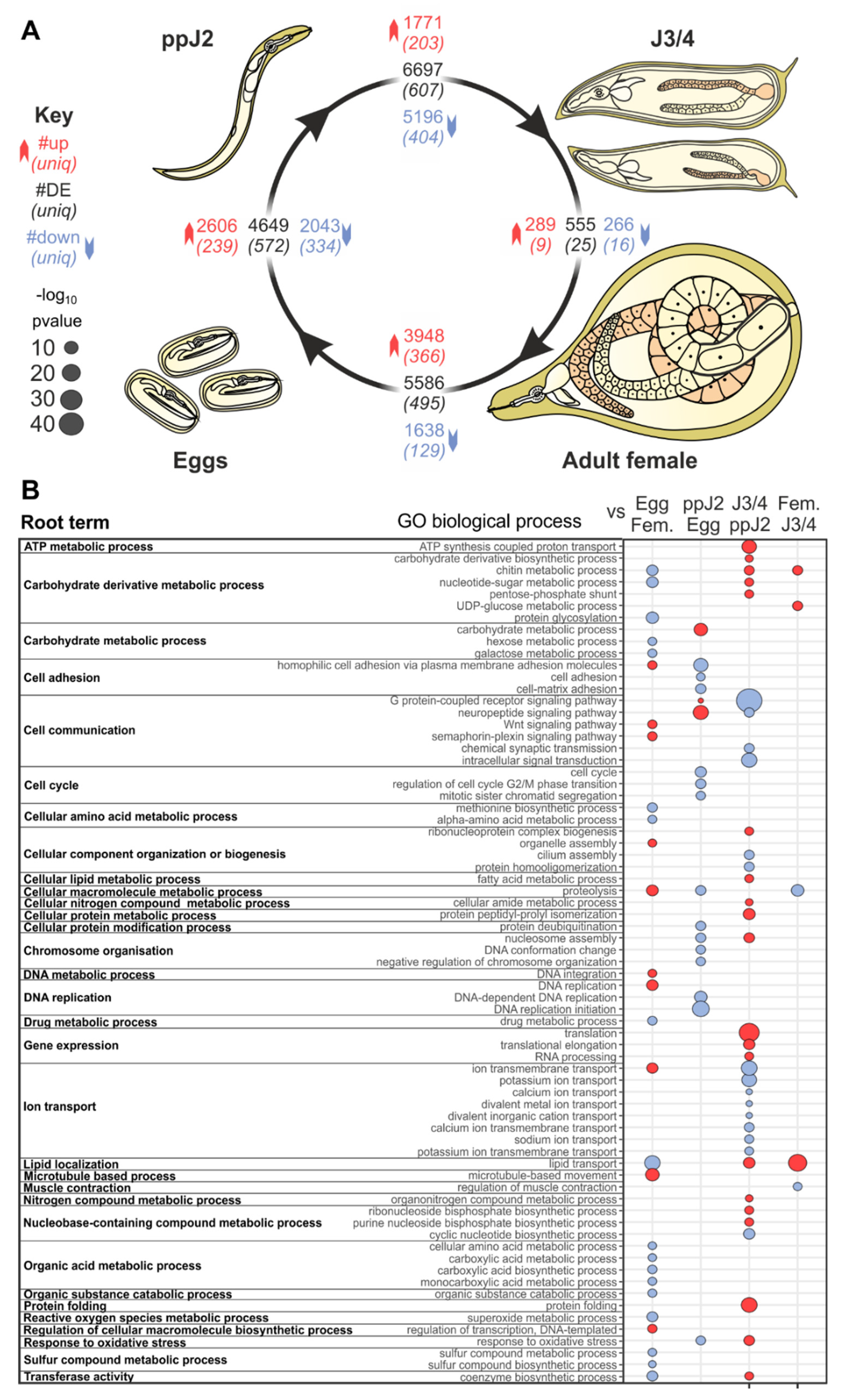{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Gene Expression Levels during Four Developmental Life Stages of M. incognita
2.2. Identification of Differentially Expressed Genes between Life Stages
2.3. Clustering of Differentially Expressed Genes
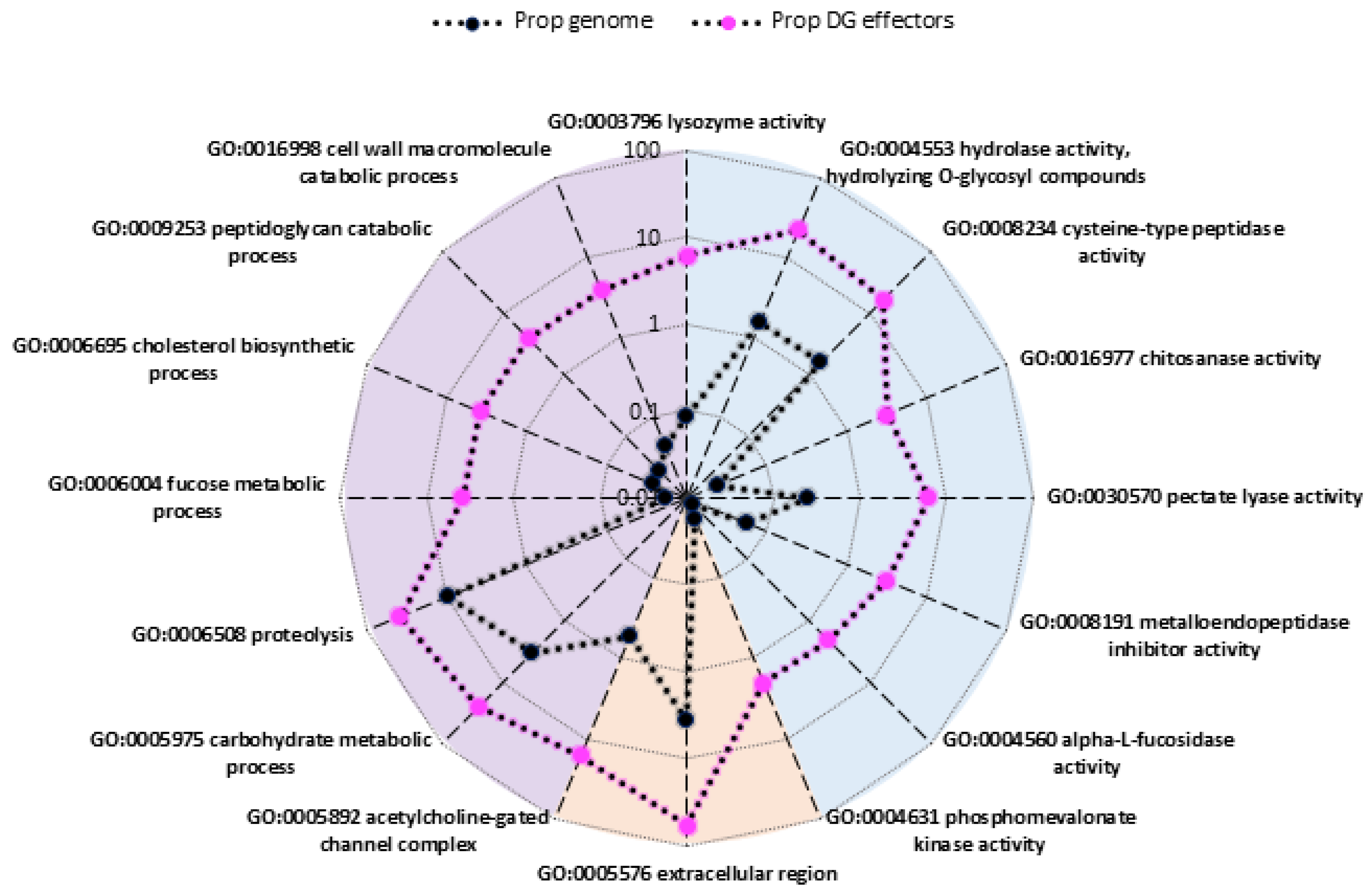
2.4. Identification of Overrepresented Gene Ontology Terms
2.5. Mapping of Genes Known to Be Specifically Expressed in Secretory Gland Cells on Meloidogyne Genomes
2.6. Identification of Specific Motifs in the Upstream Regions of Genes Expressed in Secretory Gland Cells
2.7. Identification of Putative Secreted Proteins and Effectors
2.8. Evolutionary Origin of M. incognita Effectors
3. Results
3.1. Predicted Functions of Differentially-Expressed Genes Are Consistent with Transitions between Developmental Stages
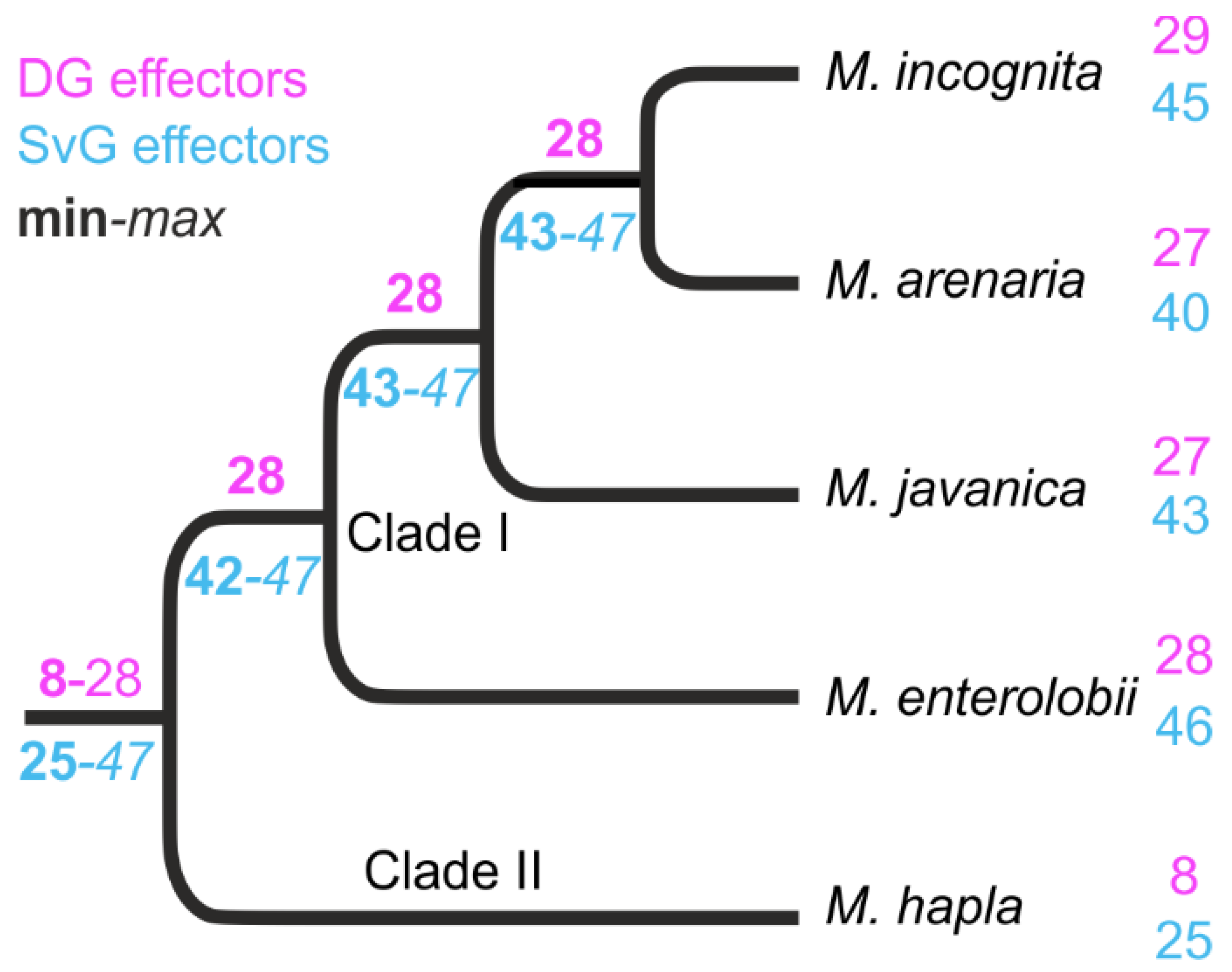
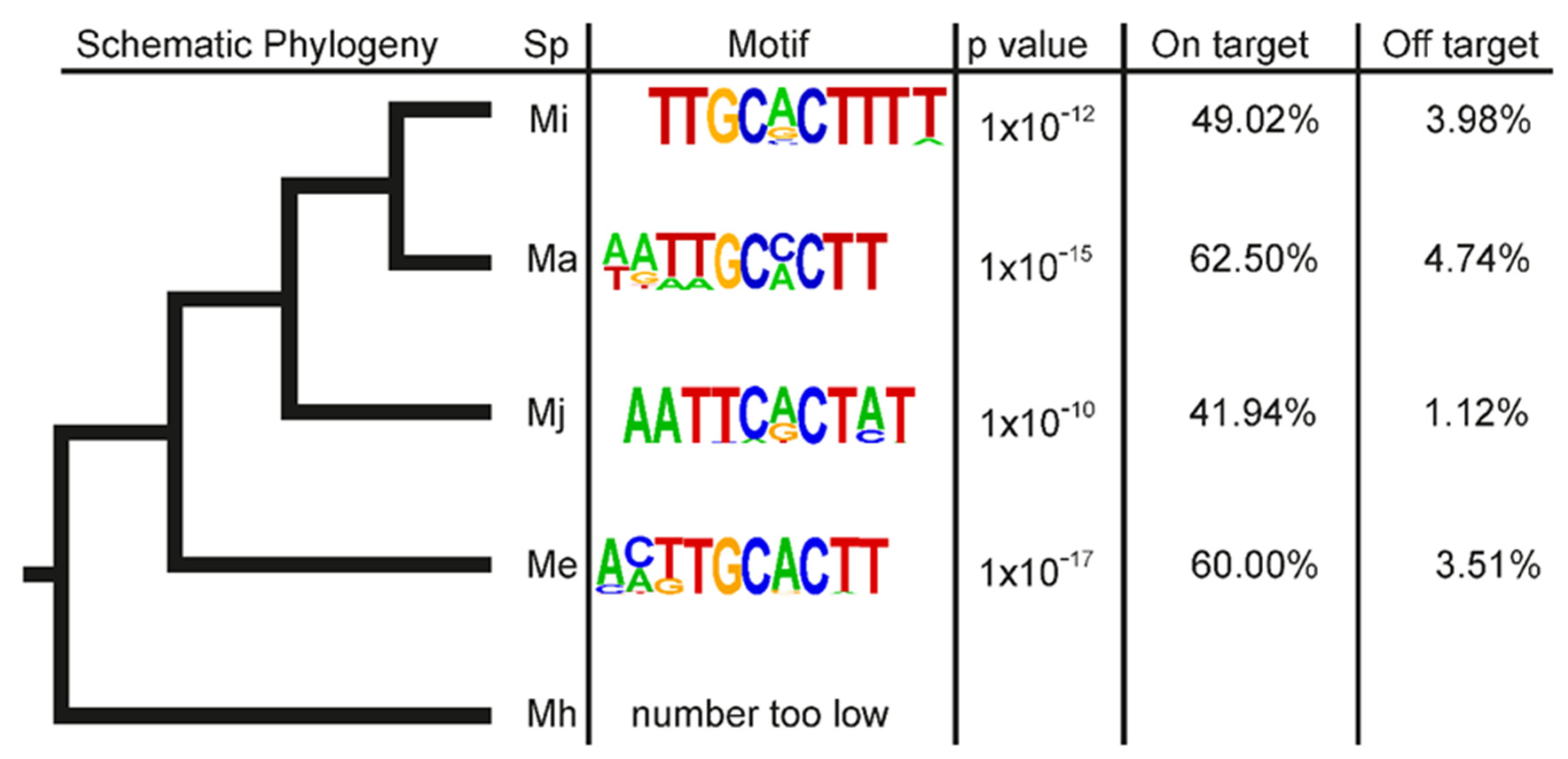
3.2. Most of the Known M. incognita Effectors Are Conserved in Multiple Meloidogyne Genomes and Were Probably Inherited from a Common Ancestor
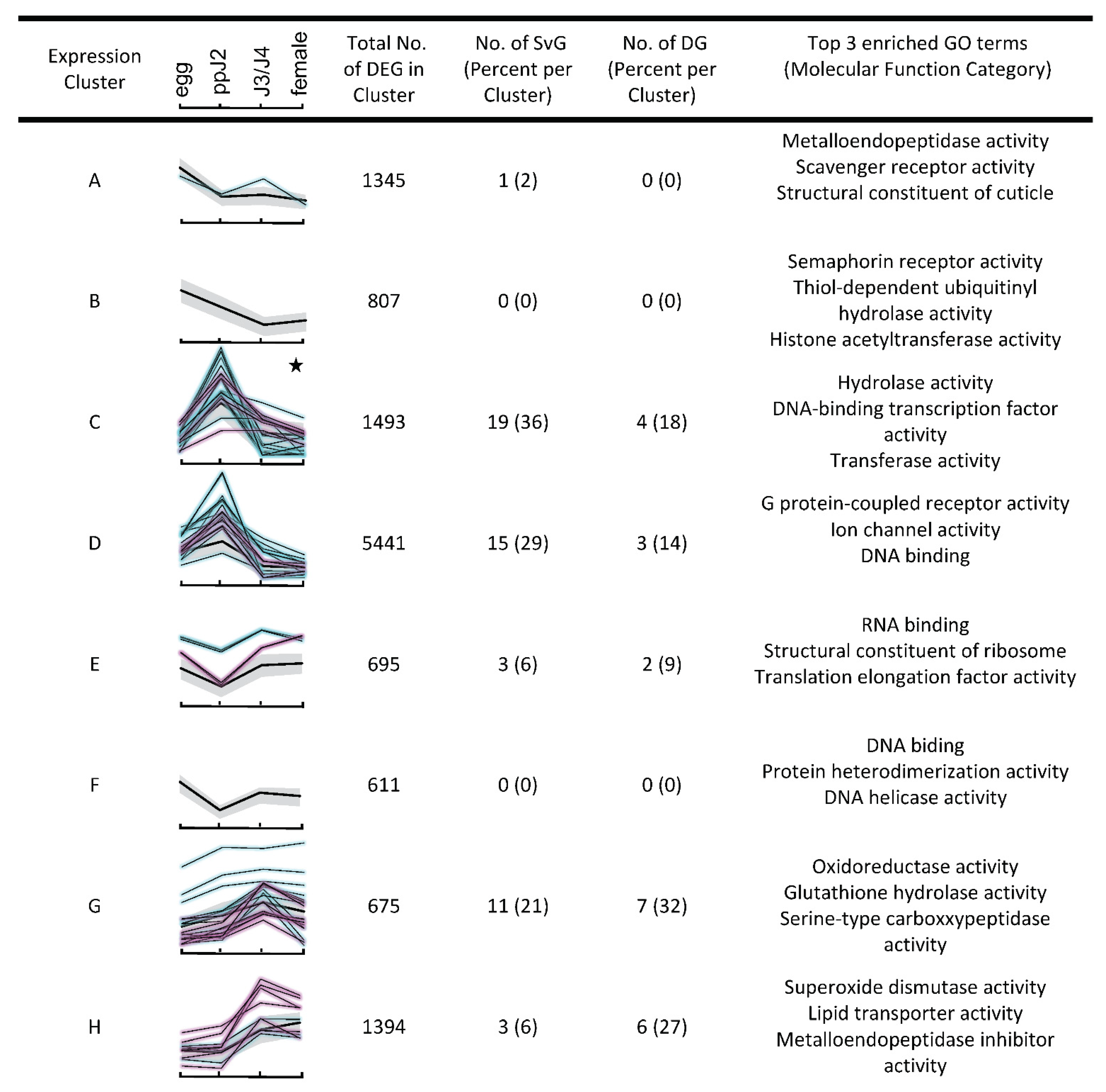
3.3. Hierarchical Clustering Analysis Highlights Changes in Expression of Genes Related to Parasitism as a Whole
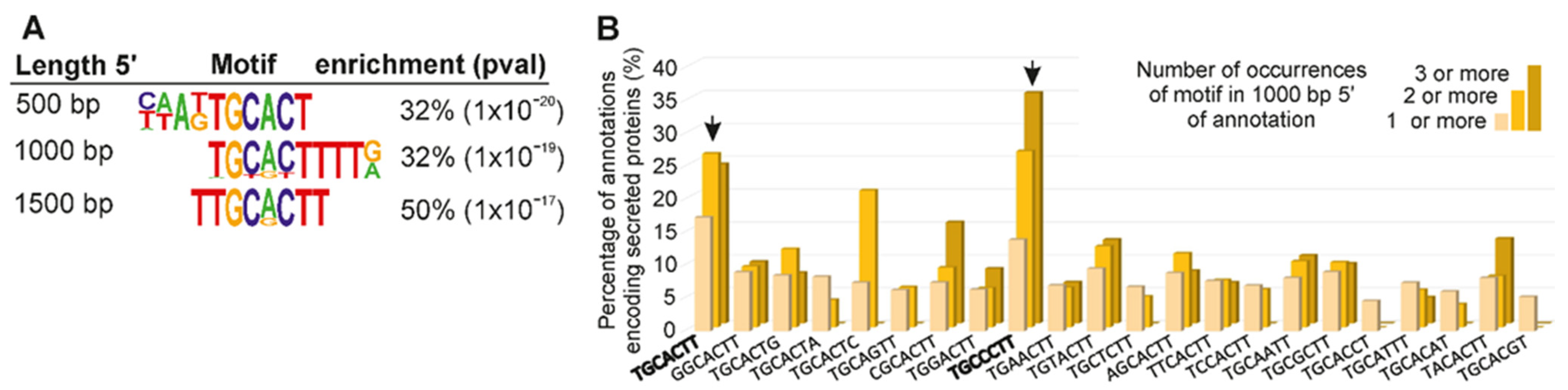
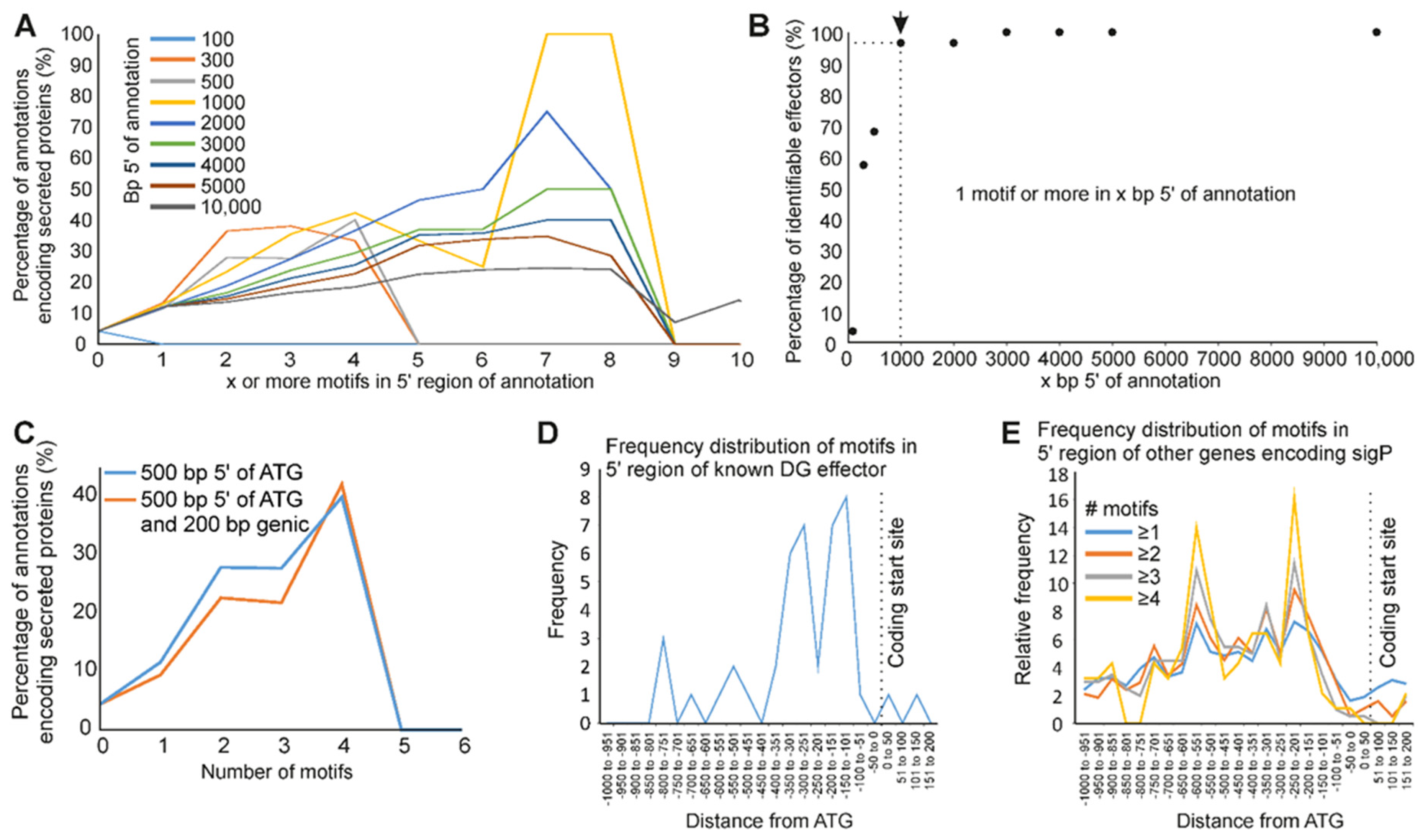
3.4. A Promoter Motif Is Associated with Dorsal Gland Effectors and Secreted Proteins
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Decraemer, W.; Hunt, D.J. Structure and classification. In Plant Nematology; Perry, R.N., Moens, M., Eds.; CABI: Wallingford, UK, 2013; pp. 3–39. ISBN 978-1-78064-151-5. [Google Scholar]
- Jones, J.T.; Haegeman, A.; Danchin, E.G.J.; Gaur, H.S.; Helder, J.; Jones, M.G.K.; Kikuchi, T.; Manzanilla-López, R.; Palomares-Rius, J.E.; Wesemael, W.M.L.; et al. Top 10 plant-parasitic nematodes in molecular plant pathology. Mol. Plant. Pathol. 2013, 14, 946–961. [Google Scholar] [CrossRef] [PubMed]
- Elling, A.A. Major emerging problems with minor Meloidogyne species. Phytopathology 2013, 103, 1092–1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goverse, A.; Smant, G. The activation and suppression of plant innate immunity by parasitic nematodes. Annu. Rev. Phytopathol. 2014, 52, 243–265. [Google Scholar] [CrossRef]
- Moens, M.; Perry, R.N.; Starr, J.L. Meloidogyne species—A diverse group of novel and important plant parasites. In Root-Knot Nematodes; Perry, R.N., Moens, M., Starr, J.L., Eds.; CABI: Wallingford, UK, 2009; pp. 1–17. ISBN 978-1-84593-492-7. [Google Scholar]
- Abad, P.; Castagnone-Sereno, P.; Rosso, M.N.; de Almeida, E.J.; Favery, B. Invasion, feeding and development. In Root-Knot Nematodes; Perry, R.N., Moens, M., Starr, J.L., Eds.; CABI: Wallingford, UK, 2009; pp. 163–181. ISBN 978-1-84593-492-7. [Google Scholar]
- Triantaphyllou, A.C.; Hirschmann, H. Post infection development of Meloidogyne incognita Chitwood 1949 (Nematoda: Heteroderidae). Ann. Inst. Phytopathol. Benaki 1960, 3, 1–11. [Google Scholar]
- Baum, T.J.; Hussey, R.S.; Davis, E.L. Root-knot and cyst nematode parasitism genes: The molecular basis of plant parasitism. Genet. Eng. 2007, 28, 17–43. [Google Scholar] [CrossRef]
- Mitchum, M.G.; Hussey, R.S.; Baum, T.J.; Wang, X.; Elling, A.A.; Wubben, M.; Davis, E.L. Nematode effector proteins: An emerging paradigm of parasitism. New Phytol. 2013, 199, 879–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, E.L.; Hussey, R.S.; Baum, T.J.; Bakker, J.; Schots, A.; Rosso, M.-N.; Abad, P. Nematode parasitism genes. Annu. Rev. Phytopathol. 2000, 38, 365–396. [Google Scholar] [CrossRef]
- Eves-van den Akker, S.; Laetsch, D.R.; Thorpe, P.; Lilley, C.J.; Danchin, E.G.J.; da Rocha, M.; Rancurel, C.; Holroyd, N.E.; Cotton, J.A.; Szitenberg, A.; et al. The genome of the yellow potato cyst nematode, Globodera rostochiensis, reveals insights into the basis of parasitism and virulence. Genome Biol. 2016, 17, 124. [Google Scholar] [CrossRef] [Green Version]
- Eves-van den Akker, S. Plant-nematode interactions. Curr. Opin. Plant. Biol. 2021, 62, 102035. [Google Scholar] [CrossRef]
- Masonbrink, R.; Maier, T.R.; Muppirala, U.; Seetharam, A.S.; Lord, E.; Juvale, P.S.; Schmutz, J.; Johnson, N.T.; Korkin, D.; Mitchum, M.G.; et al. The genome of the soybean cyst nematode (Heterodera glycines) reveals complex patterns of duplications involved in the evolution of parasitism genes. Bmc Genom. 2019, 20, 119. [Google Scholar] [CrossRef] [Green Version]
- Espada, M.; Eves-van den Akker, S.; Maier, T.; Vijayapalani, P.; Baum, T.; Mota, M.; Jones, J.T. STATAWAARS: A promoter motif associated with spatial expression in the major effector-producing tissues of the plant-parasitic nematode Bursaphelenchus xylophilus. Bmc Genom. 2018, 19, 553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vieira, P.; Maier, T.R.; Eves-van den Akker, S.; Howe, D.K.; Zasada, I.; Baum, T.J.; Eisenback, J.D.; Kamo, K. Identification of candidate effector genes of Pratylenchus penetrans. Mol. Plant. Pathol. 2018, 19, 1887–1907. [Google Scholar] [CrossRef] [Green Version]
- Blanc-Mathieu, R.; Perfus-Barbeoch, L.; Aury, J.-M.; Rocha, M.D.; Gouzy, J.; Sallet, E.; Martin-Jimenez, C.; Bailly-Bechet, M.; Castagnone-Sereno, P.; Flot, J.-F.; et al. Hybridization and polyploidy enable genomic plasticity without sex in the most devastating plant-parasitic nematodes. Plos Genet. 2017, 13, e1006777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Grynberg, P.; Togawa, R.C.; de Freitas, L.D.; Antonino, J.D.; Rancurel, C.; Costa, M.D.C.; Grossi-de-Sa, M.F.; Miller, R.N.G.; Brasileiro, A.C.M.; Guimaraes, P.M.; et al. Comparative genomics reveals novel target genes towards specific control of plant-parasitic nematodes. Genes 2020, 11, 1347. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-seq data with or without a reference genome. Bmc Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Leng, N.; Dawson, J.A.; Thomson, J.A.; Ruotti, V.; Rissman, A.I.; Smits, B.M.G.; Haag, J.D.; Gould, M.N.; Stewart, R.M.; Kendziorski, C. EBSeq: An empirical bayes hierarchical model for inference in RNA-seq experiments. Bioinformatics 2013, 29, 1035–1043. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Quevillon, E.; Silventoinen, V.; Pillai, S.; Harte, N.; Mulder, N.; Apweiler, R.; Lopez, R. InterProScan: Protein domains identifier. Nucleic Acids Res. 2005, 33, W116–W120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prüfer, K.; Muetzel, B.; Do, H.-H.; Weiss, G.; Khaitovich, P.; Rahm, E.; Pääbo, S.; Lachmann, M.; Enard, W. FUNC: A package for detecting significant associations between gene sets and ontological annotations. Bmc Bioinform. 2007, 8, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Truong, N.M.; Nguyen, C.-N.; Abad, P.; Quentin, M.; Favery, B. Function of root-knot nematode effectors and their targets in plant parasitism. In Advances in Botanical Research; Elsevier: Amsterdam, The Netherlands, 2015; Volume 73, pp. 293–324. ISBN 978-0-12-417161-9. [Google Scholar]
- Leelarasamee, N.; Zhang, L.; Gleason, C. The root-knot nematode effector MiPFN3 disrupts plant actin filaments and promotes parasitism. Plos Pathog. 2018, 14, e1006947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwata, H.; Gotoh, O. Benchmarking spliced alignment programs including Spaln2, an extended version of Spaln that incorporates additional species-specific features. Nucleic Acids Res. 2012, 40, e161. [Google Scholar] [CrossRef] [PubMed]
- Koutsovoulos, G.D.; Poullet, M.; Elashry, A.; Kozlowski, D.K.L.; Sallet, E.; da Rocha, M.; Perfus-Barbeoch, L.; Martin-Jimenez, C.; Frey, J.E.; Ahrens, C.H.; et al. Genome assembly and annotation of Meloidogyne enterolobii, an emerging parthenogenetic root-knot nematode. Sci. Data 2020, 7, 324. [Google Scholar] [CrossRef]
- Opperman, C.H.; Bird, D.M.; Williamson, V.M.; Rokhsar, D.S.; Burke, M.; Cohn, J.; Cromer, J.; Diener, S.; Gajan, J.; Graham, S.; et al. Sequence and genetic map of Meloidogyne hapla: A compact nematode genome for plant parasitism. Proc. Natl. Acad. Sci. USA 2008, 105, 14802–14807. [Google Scholar] [CrossRef] [Green Version]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef] [Green Version]
- Heinz, S.; Benner, C.; Spann, N.; Bertolino, E.; Lin, Y.C.; Laslo, P.; Cheng, J.X.; Murre, C.; Singh, H.; Glass, C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 2010, 38, 576–589. [Google Scholar] [CrossRef] [Green Version]
- Grant, C.E.; Bailey, T.L.; Noble, W.S. FIMO: Scanning for occurrences of a given motif. Bioinformatics 2011, 27, 1017–1018. [Google Scholar] [CrossRef] [Green Version]
- Vens, C.; Rosso, M.N.; Danchin, E.G. Identifying discriminative classification-based motifs in biological sequences. Bioinformatics 2011, 27, 1231–1238. [Google Scholar] [CrossRef] [Green Version]
- Maddison, W.P.; Maddison, D.R. Mesquite: A Modular System for Evolutionary Analysis; Version 3.01. 2014. Available online: http://www.mesquiteproject.org (accessed on 13 May 2020).
- Álvarez-Ortega, S.; Brito, J.A.; Subbotin, S.A. Multigene phylogeny of root-knot nematodes and molecular characterization of Meloidogyne nataliei golden, rose & bird, 1981 (Nematoda: Tylenchida). Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Danchin, E.G.; Rosso, M.N.; Vieira, P.; de Almeida-Engler, J.; Coutinho, P.M.; Henrissat, B.; Abad, P. Multiple lateral gene transfers and duplications have promoted plant parasitism ability in nematodes. Proc. Natl. Acad. Sci. USA 2010, 107, 17651–17656. [Google Scholar] [CrossRef] [Green Version]
- Haegeman, A.; Jones, J.T.; Danchin, E.G. Horizontal gene transfer in nematodes: A catalyst for plant parasitism? Mol. Plant. Microbe Interact. 2011, 24, 879–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, G.; Gao, B.; Maier, T.; Allen, R.; Davis, E.L.; Baum, T.J.; Hussey, R.S. A profile of putative parasitism genes expressed in the esophageal gland cells of the root-knot nematode Meloidogyne incognita. Mol. Plant. Microbe Interact. 2003, 16, 376–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rutter, W.B.; Hewezi, T.; Abubucker, S.; Maier, T.R.; Huang, G.; Mitreva, M.; Hussey, R.S.; Baum, T.J. Mining novel effector proteins from the esophageal gland cells of Meloidogyne incognita. Mol. Plant. Microbe Interact. 2014, 27, 965–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Ley, I.T.; de Ley, P.; Vierstraete, A.; Karssen, G.; Moens, M.; Vanfleteren, J. Phylogenetic analyses of Meloidogyne small subunit RDNA. J. Nematol. 2002, 34, 319–327. [Google Scholar]
- Campbell, B.E.; Hofmann, A.; McCluskey, A.; Gasser, R.B. Serine/threonine phosphatases in socioeconomically important parasitic nematodes—Prospects as novel drug targets? Biotechnol. Adv. 2011, 29, 28–39. [Google Scholar] [CrossRef] [Green Version]
- Malagón, D.; Benítez, R.; Kasny, M.; Adroher, F.J. Peptidases in parasitic nematodes. A review. In Parasites: Ecology, Diseases and Management; Nova Science Publishers: Hauppauge, NY, USA, 2013; pp. 61–102. ISBN 978-1-62257-692-0. [Google Scholar]
- Hussey, R.S.; Davis, E.L.; Baum, T.J. Secrets in secretions: Genes that control nematode parasitism of plants. Braz. J. Plant Physiol. 2002, 14, 183–194. [Google Scholar] [CrossRef] [Green Version]
- Vieira, P.; Shao, J.; Vijayapalani, P.; Maier, T.R.; Pellegrin, C.; Eves-van den Akker, S.; Baum, T.J.; Nemchinov, L.G. A new Esophageal gland transcriptome reveals signatures of large scale de novo effector birth in the root lesion nematode Pratylenchus penetrans. Bmc Genom. 2020, 21, 738. [Google Scholar] [CrossRef]
- McCarter, J.P.; Mitreva, M.D.; Martin, J.; Dante, M.; Wylie, T.; Rao, U.; Pape, D.; Bowers, Y.; Theising, B.; Murphy, C.V.; et al. Analysis and functional classification of transcripts from the nematode Meloidogyne incognita. Genome Biol. 2003, 4, r26. [Google Scholar] [CrossRef] [Green Version]
- Shukla, N.; Yadav, R.; Kaur, P.; Rasmussen, S.; Goel, S.; Agarwal, M.; Jagannath, A.; Gupta, R.; Kumar, A. Transcriptome analysis of root-knot nematode (Meloidogyne incognita)-infected tomato (Solanum lycopersicum) roots reveals complex gene expression profiles and metabolic networks of both host and nematode during susceptible and resistance responses. Mol. Plant. Pathol. 2018, 19, 615–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petitot, A.-S.; Dereeper, A.; da Silva, C.; Guy, J.; Fernandez, D. Analyses of the root-knot nematode (Meloidogyne graminicola) transcriptome during host infection highlight specific gene expression profiling in resistant rice plants. Pathogens 2020, 9, 644. [Google Scholar] [CrossRef] [PubMed]
- Cotton, J.A.; Lilley, C.J.; Jones, L.M.; Kikuchi, T.; Reid, A.J.; Thorpe, P.; Tsai, I.J.; Beasley, H.; Blok, V.; Cock, P.J.A.; et al. The genome and life-stage specific transcriptomes of Globodera pallida elucidate key aspects of plant parasitism by a cyst nematode. Genome Biol. 2014, 15, r43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Xu, C.-L.; Yang, S.-H.; Li, J.-Y.; Wang, H.-L.; Zhang, Z.-X.; Chen, C.; Xie, H. Life-stage specific transcriptomes of a migratory endoparasitic plant nematode, Radopholus similis elucidate a different parasitic and life strategy of plant parasitic nematodes. Sci. Rep. 2019, 9, 6277. [Google Scholar] [CrossRef] [Green Version]
- Dash, M.; Dutta, T.K.; Phani, V.; Papolu, P.K.; Shivakumara, T.N.; Rao, U. RNAi-mediated disruption of neuropeptide genes, Nlp-3 and Nlp-12, cause multiple behavioral defects in Meloidogyne incognita. Biochem. Biophys. Res. Commun. 2017, 490, 933–940. [Google Scholar] [CrossRef]
- Shivakumara, T.N.; Dutta, T.K.; Mandal, A.; Rao, U. Estimation of lipid reserves in different life stages of Meloidogyne incognita using image analysis of Nile red-stained nematodes. Nematology 2019, 21, 267–274. [Google Scholar] [CrossRef]
- Bellafiore, S.; Shen, Z.; Rosso, M.-N.; Abad, P.; Shih, P.; Briggs, S.P. Direct identification of the Meloidogyne incognita secretome reveals proteins with host cell reprogramming potential. Plos Pathog. 2008, 4, e1000192. [Google Scholar] [CrossRef] [Green Version]
- Gahoi, S.; Gautam, B. Genome-wide analysis of excretory/secretory proteins in root-knot nematode, Meloidogyne incognita provides potential targets for parasite control. Comput. Biol. Chem. 2017, 67, 225–233. [Google Scholar] [CrossRef]
- De Souza, J.D.A., Jr.; Coelho, R.R.; Lourenço, I.T.; da Rocha, R.F.; Viana, A.A.B.; de Macedo, L.L.P.; da Silva, M.C.M.; Carneiro, R.M.G.; Engler, G.; de Almeida-Engler, J.; et al. Knocking-down Meloidogyne incognita proteases by plant-delivered DsRNA has negative pleiotropic effect on nematode vigor. PLoS ONE 2013, 8, e85364. [Google Scholar] [CrossRef] [Green Version]
- Shivakumara, T.N.; Papolu, P.K.; Dutta, T.K.; Kamaraju, D.; Chaudhary, S.; Rao, U. RNAi-induced silencing of an effector confers transcriptional oscillation in another group of effectors in the root-knot nematode, Meloidogyne incognita. Nematology 2016, 18, 857–870. [Google Scholar] [CrossRef]
- Bell, C.A.; Lilley, C.J.; McCarthy, J.; Atkinson, H.J.; Urwin, P.E. Plant-parasitic nematodes respond to root exudate signals with host-specific gene expression patterns. PLoS Pathog. 2019, 15, e1007503. [Google Scholar] [CrossRef] [Green Version]
- Rehman, S.; Butterbach, P.; Popeijus, H.; Overmars, H.; Davis, E.L.; Jones, J.T.; Goverse, A.; Bakker, J.; Smant, G. Identification and characterization of the most abundant cellulases in stylet secretions from Globodera rostochiensis. Phytopathology 2009, 99, 194–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espada, M.; Silva, A.C.; Eves van den Akker, S.; Cock, P.J.A.; Mota, M.; Jones, J.T. Identification and characterization of parasitism genes from the pinewood nematode Bursaphelenchus xylophilus reveals a multilayered detoxification strategy. Mol. Plant. Pathol. 2016, 17, 286–295. [Google Scholar] [CrossRef] [Green Version]
- Gillet, F.-X.; Bournaud, C.; de Souza, J.D.A., Jr.; Grossi-de-Sa, M.F. Plant-parasitic nematodes: Towards understanding molecular players in stress responses. Ann. Bot. 2017, 119, 775–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vieira, P.; Kamo, K.; Eisenback, J.D. Characterization and silencing of the fatty acid- and retinol-binding Pp-Far-1 gene in Pratylenchus penetrans. Plant. Pathol. 2017, 66, 1214–1224. [Google Scholar] [CrossRef] [Green Version]
- Dubreuil, G.; Deleury, E.; Magliano, M.; Jaouannet, M.; Abad, P.; Rosso, M.-N. Peroxiredoxins from the plant parasitic root-knot nematode, Meloidogyne incognita, are required for successful development within the host. Int. J. Parasitol. 2011, 41, 385–396. [Google Scholar] [CrossRef]
- Castagnone-Sereno, P.; Deleury, E.; Danchin, E.G.; Perfus-Barbeoch, L.; Abad, P. Data-mining of the Meloidogyne incognita degradome and comparative analysis of proteases in nematodes. Genomics 2011, 97, 29–36. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, P.; Oh, B.-J.; Mani, V.; Lee, J.K.; Lee, C.-M.; Sim, J.-S.; Koo, J.C.; Hahn, B.-S. Differential metabolic profiles during the developmental stages of plant-parasitic nematode Meloidogyne incognita. Int. J. Mol. Sci. 2017, 18, 1351. [Google Scholar] [CrossRef] [Green Version]
- Abad, P.; Gouzy, J.; Aury, J.-M.; Castagnone-Sereno, P.; Danchin, E.G.J.; Deleury, E.; Perfus-Barbeoch, L.; Anthouard, V.; Artiguenave, F.; Blok, V.C.; et al. Genome sequence of the metazoan plant-parasitic nematode Meloidogyne incognita. Nat. Biotechnol. 2008, 26, 909–915. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Yang, D.; Niu, J.; Zhao, J.; Jian, H. De novo analysis of the transcriptome of Meloidogyne enterolobii to uncover potential target genes for biological control. Int. J. Mol. Sci. 2016, 17, 1442. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Cui, L.; Chen, Y.; Zhang, H.; Liu, P.; Wu, P.; Qiu, D.; Zou, J.; Yang, D.; Yang, L.; et al. Transcriptional responses of wheat and the cereal cyst nematode Heterodera avenae during their early contact stage. Sci. Rep. 2017, 7, 14471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabeh, M.; Duceppe, M.-O.; St-Arnaud, M.; Mimee, B. Transcriptome-wide selection of a reliable set of reference genes for gene expression studies in potato cyst nematodes (Globodera spp.). PLoS ONE 2018, 13, e0193840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Wafula, E.K.; Honaas, L.A.; Zhang, H.; Das, M.; Fernandez-Aparicio, M.; Huang, K.; Bandaranayake, P.C.G.; Wu, B.; Der, J.P.; et al. Comparative transcriptome analyses reveal core parasitism genes and suggest gene duplication and repurposing as sources of structural novelty. Mol. Biol. Evol. 2015, 32, 767–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunt, V.L.; Hino, A.; Yoshida, A.; Kikuchi, T. Comparative transcriptomics gives insights into the evolution of parasitism in Strongyloides nematodes at the genus, subclade and species level. Sci. Rep. 2018, 8, 5192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunt, V.L.; Tsai, I.J.; Coghlan, A.; Reid, A.J.; Holroyd, N.; Foth, B.J.; Tracey, A.; Cotton, J.A.; Stanley, E.J.; Beasley, H.; et al. The genomic basis of parasitism in the Strongyloides clade of nematodes. Nat. Genet. 2016, 48, 299–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rai, K.M.; Balasubramanian, V.K.; Welker, C.M.; Pang, M.; Hii, M.M.; Mendu, V. Genome wide comprehensive analysis and web resource development on cell wall degrading enzymes from phyto-parasitic nematodes. Bmc Plant. Biol. 2015, 15, 187. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, S.E.; Dayi, M.; Maeda, Y.; Tsai, I.J.; Tanaka, R.; Bligh, M.; Takeuchi-Kaneko, Y.; Fukuda, K.; Kanzaki, N.; Kikuchi, T. Stage-specific transcriptome of Bursaphelenchus xylophilus reveals temporal regulation of effector genes and roles of the dauer-like stages in the lifecycle. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- International Helminth Genomes Consortium. Comparative genomics of the major parasitic worms. Nat. Genet. 2019, 51, 163–174. [Google Scholar] [CrossRef] [Green Version]
- Davis, E.L.; Hussey, R.S.; Mitchum, M.G.; Baum, T.J. Parasitism proteins in nematode—Plant interactions. Curr. Opin. Plant. Biol. 2008, 11, 360–366. [Google Scholar] [CrossRef]
- Jones, J.T.; Furlanetto, C.; Bakker, E.; Banks, B.; Blok, V.; Chen, Q.; Phillips, M.; Prior, A. Characterization of a chorismate mutase from the potato cyst nematode Globodera pallida. Mol. Plant. Pathol. 2003, 4, 43–50. [Google Scholar] [CrossRef]
- Nguyen, C.-N.; Perfus-Barbeoch, L.; Quentin, M.; Zhao, J.; Magliano, M.; Marteu, N.; Rocha, M.D.; Nottet, N.; Abad, P.; Favery, B. A root-knot nematode small glycine and cysteine-rich secreted effector, MiSGCR1, is involved in plant parasitism. New Phytol. 2018, 217, 687–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardoso, J.M.S.; Fonseca, L.; Abrantes, I. α-l-fucosidases from Bursaphelenchus xylophilus secretome—Molecular characterization and their possible role in breaking down plant cell walls. Forests 2020, 11, 265. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.; Ma, R.; Zhu, N.; Guo, K.; Guo, Y.; Bai, L.; Yu, H.; Hu, J.; Zhang, X. Bxy-fuca encoding α-L-fucosidase plays crucial roles in development and reproduction of the pathogenic pinewood nematode, Bursaphelenchus xylophilus. Pest. Manag. Sci. 2020, 76, 205–214. [Google Scholar] [CrossRef]
- Kranse, O.; Beasley, H.; Adams, S.; da Silva, A.P.; Bell, C.; Lilley, C.; Urwin, P.; Bird, D.; Miska, E.; Smant, G.; et al. Towards genetic modification of plant-parasitic nematodes: Delivery of macromolecules to adults and expression of exogenous MRNA in second stage juveniles. bioRxiv 2020. [Google Scholar] [CrossRef]
- Arroyo-Velez, N.; González-Fuente, M.; Peeters, N.; Lauber, E.; Noël, L.D. From effectors to effectomes: Are functional studies of individual effectors enough to decipher plant pathogen infectious strategies? PLoS Pathog. 2020, 16, e1009059. [Google Scholar] [CrossRef]
- Iqbal, S.; Fosu-Nyarko, J.; Jones, M.G.K. Attempt to silence genes of the RNAi pathways of the root-knot nematode, Meloidogyne incognita results in diverse responses including increase and no change in expression of some genes. Front. Plant. Sci. 2020, 11. [Google Scholar] [CrossRef] [Green Version]
- Lilley, C.J.; Davies, L.J.; Urwin, P.E. RNA interference in plant parasitic nematodes: A summary of the current status. Parasitology 2012, 139, 630–640. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.A.B.; John, E.; Rybak, K.; Phan, H.T.T.; Singh, K.B.; Lin, S.-Y.; Solomon, P.S.; Oliver, R.P.; Tan, K.-C. A specific fungal transcription factor controls effector gene expression and orchestrates the establishment of the necrotrophic pathogen lifestyle on wheat. Sci. Rep. 2019, 9, 15884. [Google Scholar] [CrossRef] [PubMed]
- Eves-van den Akker, S.; Birch, P.R.J. Opening the effector protein toolbox for plant—Parasitic cyst nematode interactions. Mol. Plant. 2016, 9, 1451–1453. [Google Scholar] [CrossRef] [Green Version]
- Vieira, P.; Gleason, C. Plant-parasitic nematode effectors—Insights into their diversity and new tools for their identification. Curr. Opin. Plant. Biol. 2019, 50, 37–43. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Da Rocha, M.; Bournaud, C.; Dazenière, J.; Thorpe, P.; Bailly-Bechet, M.; Pellegrin, C.; Péré, A.; Grynberg, P.; Perfus-Barbeoch, L.; Eves-van den Akker, S.; et al. Genome Expression Dynamics Reveal the Parasitism Regulatory Landscape of the Root-Knot Nematode Meloidogyne incognita and a Promoter Motif Associated with Effector Genes. Genes 2021, 12, 771. https://doi.org/10.3390/genes12050771
Da Rocha M, Bournaud C, Dazenière J, Thorpe P, Bailly-Bechet M, Pellegrin C, Péré A, Grynberg P, Perfus-Barbeoch L, Eves-van den Akker S, et al. Genome Expression Dynamics Reveal the Parasitism Regulatory Landscape of the Root-Knot Nematode Meloidogyne incognita and a Promoter Motif Associated with Effector Genes. Genes. 2021; 12(5):771. https://doi.org/10.3390/genes12050771
Chicago/Turabian StyleDa Rocha, Martine, Caroline Bournaud, Julie Dazenière, Peter Thorpe, Marc Bailly-Bechet, Clément Pellegrin, Arthur Péré, Priscila Grynberg, Laetitia Perfus-Barbeoch, Sebastian Eves-van den Akker, and et al. 2021. "Genome Expression Dynamics Reveal the Parasitism Regulatory Landscape of the Root-Knot Nematode Meloidogyne incognita and a Promoter Motif Associated with Effector Genes" Genes 12, no. 5: 771. https://doi.org/10.3390/genes12050771
APA StyleDa Rocha, M., Bournaud, C., Dazenière, J., Thorpe, P., Bailly-Bechet, M., Pellegrin, C., Péré, A., Grynberg, P., Perfus-Barbeoch, L., Eves-van den Akker, S., & Danchin, E. G. J. (2021). Genome Expression Dynamics Reveal the Parasitism Regulatory Landscape of the Root-Knot Nematode Meloidogyne incognita and a Promoter Motif Associated with Effector Genes. Genes, 12(5), 771. https://doi.org/10.3390/genes12050771