
Tumor-Associated Macrophage Promotes the Survival of Cancer Cells upon Docetaxel Chemotherapy via the CSF1/CSF1R–CXCL12/CXCR4 Axis in Castration-Resistant Prostate Cancer


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. The Cell Culture, Reagents, and Generation of Genetic Engineered Cell Lines
2.2. Cell Proliferation Assay
2.3. Colony Formation Assay
2.4. Western Blot and Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
2.5. Statistical Analysis
3. Results
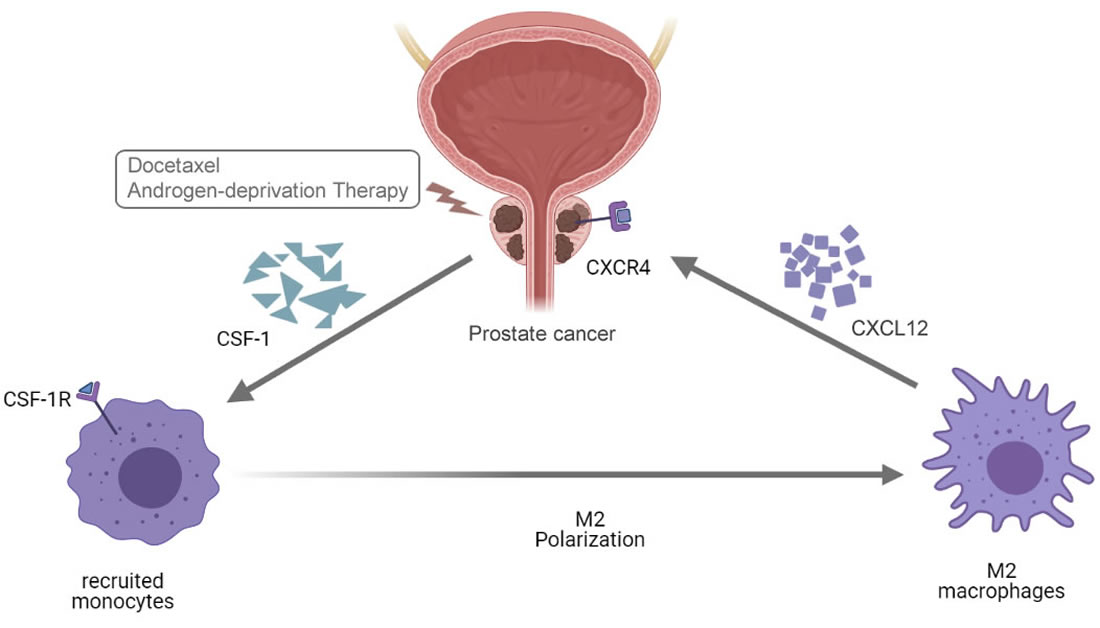
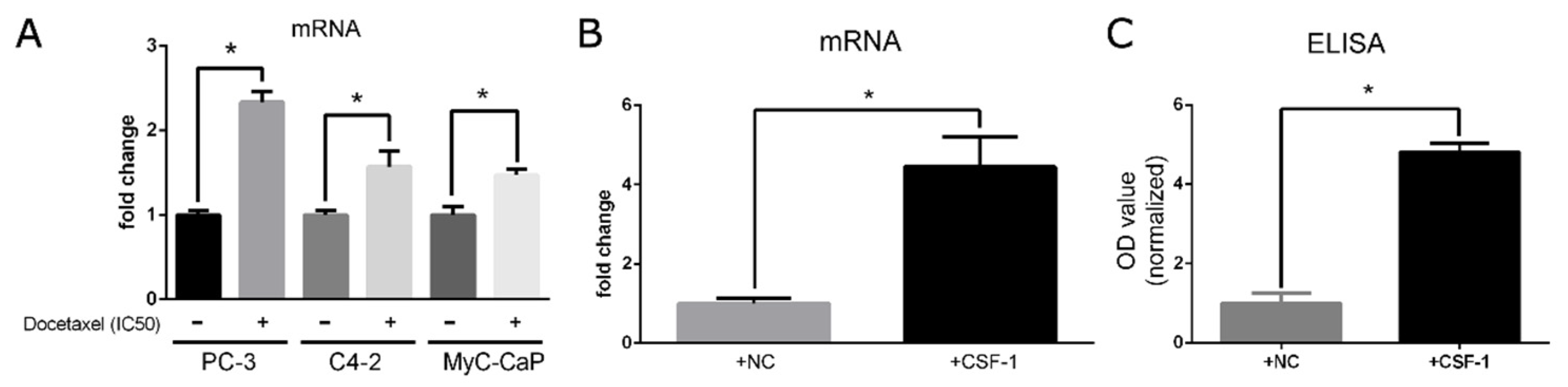
3.1. Docetaxel Treatment Induces Upregulation of CXCR4 in Prostate Cancer Cells, and Artificial Stimulation of CSF-1 Increases CXCL12 Production in Macrophages
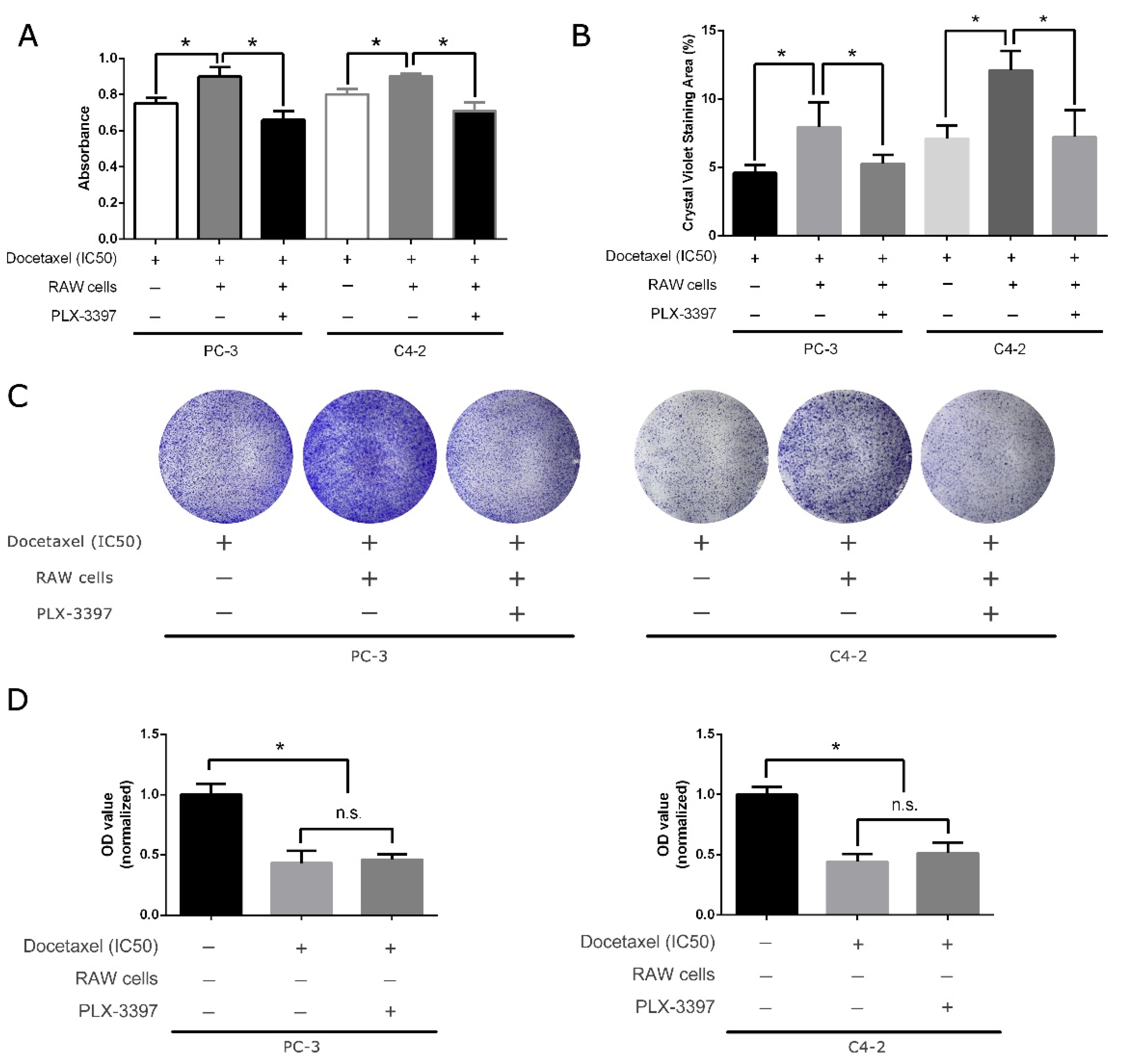
3.2. CSF-1R Inhibitor Pexidartinib Can Negate the Pro-Survival Effect of Tumor-Associated Macrophages (TAM)
3.3. Knockdown of CXCR4 Can Mimic the Effect Caused by CSF-1R Inhibition
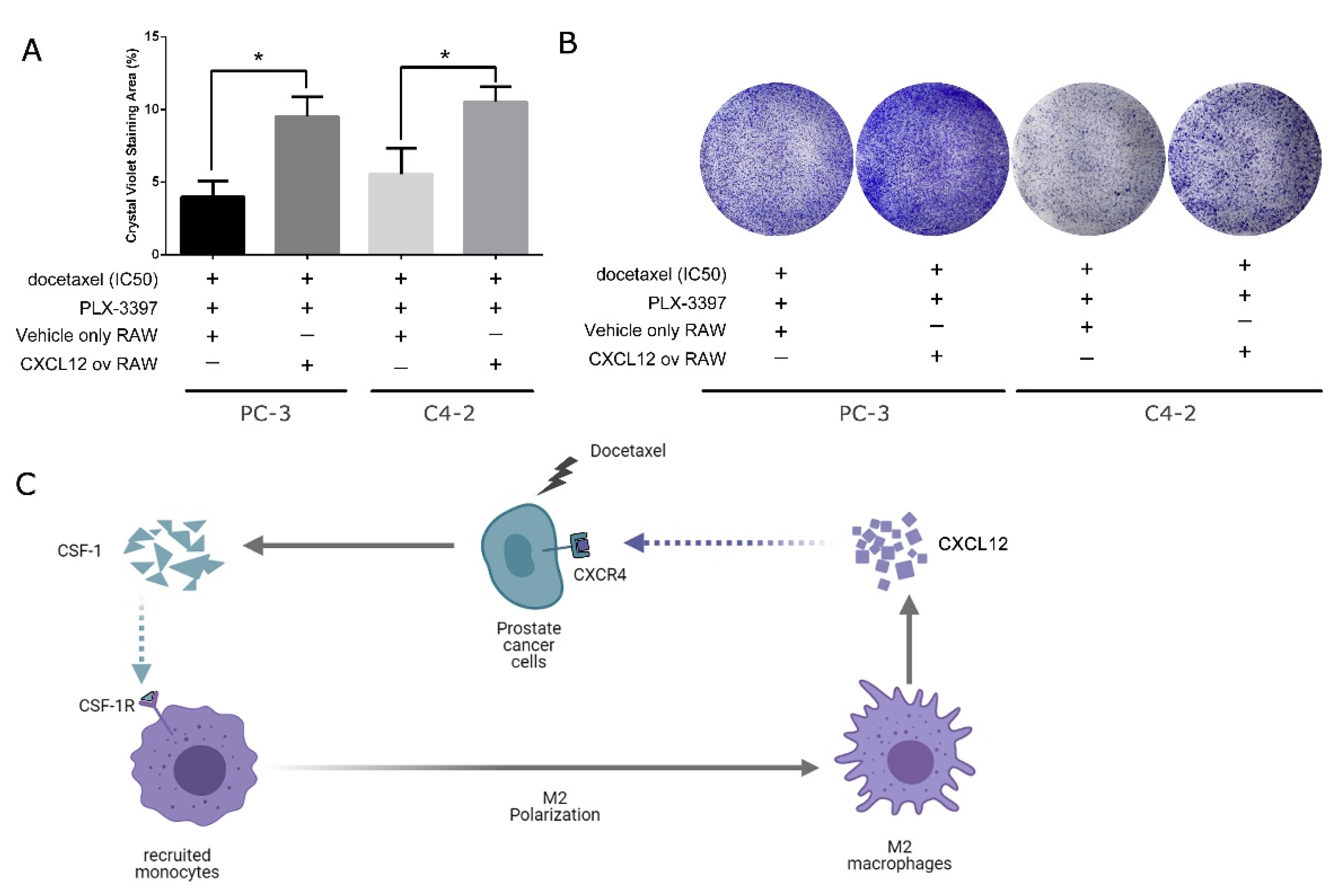
3.4. Overexpression of CXCL12 in Macrophages Can Negate the Effect Caused by CSF-1R Inhibition from Pexidartinib
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Conteduca, V.; Crabb, S.J.; Jones, R.J.; Caffo, O.; Elliott, T.; Scarpi, E.; Fabbri, P.; Derosa, L.; Massari, F.; Numico, G.; et al. Persistent Neutrophil to Lymphocyte Ratio >3 during Treatment with Enzalutamide and Clinical Outcome in Patients with Castration-Resistant Prostate Cancer. PLoS ONE 2016, 11, e0158952. [Google Scholar] [CrossRef] [PubMed]
- Santoni, M.; Bracarda, S.; Nabissi, M.; Massari, F.; Conti, A. CXC and CC chemokines as angiogenic modulators in nonhaematological tumors. BioMed Res. Int. 2014, 2014, 768758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, W.; Hu, J.; Yang, L.; Tan, P.; Tang, Z.; West, B.L.; Bollag, G.; Xu, H.; Wu, L. Inhibition of TAMs improves the response to docetaxel in castration-resistant prostate cancer. Endocr. Relat. Cancer 2019, 26, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Diao, X.-w.; Feng, J.-y.; Wang, Q.; Sun, J.; Chen, Z. SDF-1/CXCR4 axis promotes prostate cancer cell invasion and bone metastasis through p38, NF-κB and HIF-1α pathways. Int. J. Clin. Exp. Pathol. 2016, 9, 2706. [Google Scholar]
- Moughon, D.L.; He, H.; Schokrpur, S.; Jiang, Z.K.; Yaqoob, M.; David, J.; Lin, C.; Iruela-Arispe, M.L.; Dorigo, O.; Wu, L. Macrophage Blockade Using CSF1R Inhibitors Reverses the Vascular Leakage Underlying Malignant Ascites in Late-Stage Epithelial Ovarian Cancer. Cancer Res. 2015, 75, 4742–4752. [Google Scholar] [CrossRef] [Green Version]
- Yan, D.; Kowal, J.; Akkari, L.; Schuhmacher, A.J.; Huse, J.T.; West, B.L.; Joyce, J.A. Inhibition of colony stimulating factor-1 receptor abrogates microenvironment-mediated therapeutic resistance in gliomas. Oncogene 2017, 36, 6049–6058. [Google Scholar] [CrossRef]
- Mok, S.; Koya, R.C.; Tsui, C.; Xu, J.; Robert, L.; Wu, L.; Graeber, T.G.; West, B.L.; Bollag, G.; Ribas, A. Inhibition of CSF-1 Receptor Improves the Antitumor Efficacy of Adoptive Cell Transfer Immunotherapy. Cancer Res. 2014, 74, 153–161. [Google Scholar] [CrossRef] [Green Version]
- Benner, B.N. Enhancing Immunotherapy for Cancer by Targeting Suppressive Myeloid Cells. Ph.D. Thesis, The Ohio State University, Columbus, OH, USA, 2020. [Google Scholar]
- Kim, J.; Takeuchi, H.; Lam, S.T.; Turner, R.R.; Wang, H.J.; Kuo, C.; Foshag, L.; Bilchik, A.J.; Hoon, D.S. Chemokine receptor CXCR4 expression in colorectal cancer patients increases the risk for recurrence and for poor survival. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2005, 23, 2744–2753. [Google Scholar] [CrossRef]
- Longo-Imedio, M.I.; Longo, N.; Treviño, I.; Lázaro, P.; Sánchez-Mateos, P. Clinical significance of CXCR3 and CXCR4 expression in primary melanoma. Int. J. Cancer J. Int. Cancer 2005, 117, 861–865. [Google Scholar] [CrossRef]
- Scala, S.; Ottaiano, A.; Ascierto, P.A.; Cavalli, M.; Simeone, E.; Giuliano, P.; Napolitano, M.; Franco, R.; Botti, G.; Castello, G. Expression of CXCR4 predicts poor prognosis in patients with malignant melanoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2005, 11, 1835–1841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, M.; Kitayama, J.; Kazama, S.; Nagawa, H. Expression pattern of CXC chemokine receptor-4 is correlated with lymph node metastasis in human invasive ductal carcinoma. Breast Cancer Res. BCR 2003, 5, R144–R150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.X.; Fang, M.; Wang, J.; Cooper, C.R.; Pienta, K.J.; Taichman, R.S. Expression and activation of alpha v beta 3 integrins by SDF-1/CXC12 increases the aggressiveness of prostate cancer cells. Prostate 2007, 67, 61–73. [Google Scholar] [CrossRef] [Green Version]
- Dehghani, M.; Kianpour, S.; Zangeneh, A.; Mostafavi-Pour, Z. CXCL12 Modulates Prostate Cancer Cell Adhesion by Altering the Levels or Activities of β1-Containing Integrins. Int. J. Cell Biol. 2014, 2014, 981750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaday, G.G.; Hua, S.B.; Peehl, D.M.; Pauling, M.H.; Lin, Y.H.; Zhu, L.; Lawrence, D.M.; Foda, H.D.; Zucker, S. CXCR4 and CXCL12 (SDF-1) in prostate cancer: Inhibitory effects of human single chain Fv antibodies. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2004, 10, 5630–5639. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Guan, E.; Roderiquez, G.; Calvert, V.; Alvarez, R.; Norcross, M.A. Role of tyrosine phosphorylation in ligand-independent sequestration of CXCR4 in human primary monocytes-macrophages. J. Biol. Chem. 2001, 276, 49236–49243. [Google Scholar] [CrossRef] [Green Version]
- Moriuchi, M.; Moriuchi, H.; Turner, W.; Fauci, A.S. Cloning and analysis of the promoter region of CXCR4, a coreceptor for HIV-1 entry. J. Immunol. 1997, 159, 4322–4329. [Google Scholar]
- Jourdan, P.; Vendrell, J.P.; Huguet, M.F.; Segondy, M.; Bousquet, J.; Pène, J.; Yssel, H. Cytokines and cell surface molecules independently induce CXCR4 expression on CD4+ CCR7+ human memory T cells. J. Immunol. 2000, 165, 716–724. [Google Scholar] [CrossRef] [Green Version]
- Jung, Y.; Wang, J.; Schneider, A.; Sun, Y.X.; Koh-Paige, A.J.; Osman, N.I.; McCauley, L.K.; Taichman, R.S. Regulation of SDF-1 (CXCL12) production by osteoblasts; a possible mechanism for stem cell homing. Bone 2006, 38, 497–508. [Google Scholar] [CrossRef]
- Domanska, U.M.; Timmer-Bosscha, H.; Nagengast, W.B.; Oude Munnink, T.H.; Kruizinga, R.C.; Ananias, H.J.K.; Kliphuis, N.M.; Huls, G.; De Vries, E.G.E.; de Jong, I.J.; et al. CXCR4 inhibition with AMD3100 sensitizes prostate cancer to docetaxel chemotherapy. Neoplasia 2012, 14, 709–718. [Google Scholar] [CrossRef] [Green Version]
- Bhardwaj, A.; Srivastava, S.K.; Singh, S.; Arora, S.; Tyagi, N.; Andrews, J.; McClellan, S.; Carter, J.E.; Singh, A.P. CXCL12/CXCR4 signaling counteracts docetaxel-induced microtubule stabilization via p21-activated kinase 4-dependent activation of LIM domain kinase 1. Oncotarget 2014, 5, 11490–11500. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guan, W.; Li, F.; Zhao, Z.; Zhang, Z.; Hu, J.; Zhang, Y. Tumor-Associated Macrophage Promotes the Survival of Cancer Cells upon Docetaxel Chemotherapy via the CSF1/CSF1R–CXCL12/CXCR4 Axis in Castration-Resistant Prostate Cancer. Genes 2021, 12, 773. https://doi.org/10.3390/genes12050773
Guan W, Li F, Zhao Z, Zhang Z, Hu J, Zhang Y. Tumor-Associated Macrophage Promotes the Survival of Cancer Cells upon Docetaxel Chemotherapy via the CSF1/CSF1R–CXCL12/CXCR4 Axis in Castration-Resistant Prostate Cancer. Genes. 2021; 12(5):773. https://doi.org/10.3390/genes12050773
Chicago/Turabian StyleGuan, Wei, Fan Li, Zhenyu Zhao, Zongbiao Zhang, Junhui Hu, and Yan Zhang. 2021. "Tumor-Associated Macrophage Promotes the Survival of Cancer Cells upon Docetaxel Chemotherapy via the CSF1/CSF1R–CXCL12/CXCR4 Axis in Castration-Resistant Prostate Cancer" Genes 12, no. 5: 773. https://doi.org/10.3390/genes12050773
APA StyleGuan, W., Li, F., Zhao, Z., Zhang, Z., Hu, J., & Zhang, Y. (2021). Tumor-Associated Macrophage Promotes the Survival of Cancer Cells upon Docetaxel Chemotherapy via the CSF1/CSF1R–CXCL12/CXCR4 Axis in Castration-Resistant Prostate Cancer. Genes, 12(5), 773. https://doi.org/10.3390/genes12050773