
Genetic Risk Factors in Early-Onset Nonalcoholic Chronic Pancreatitis: An Update


Abstract
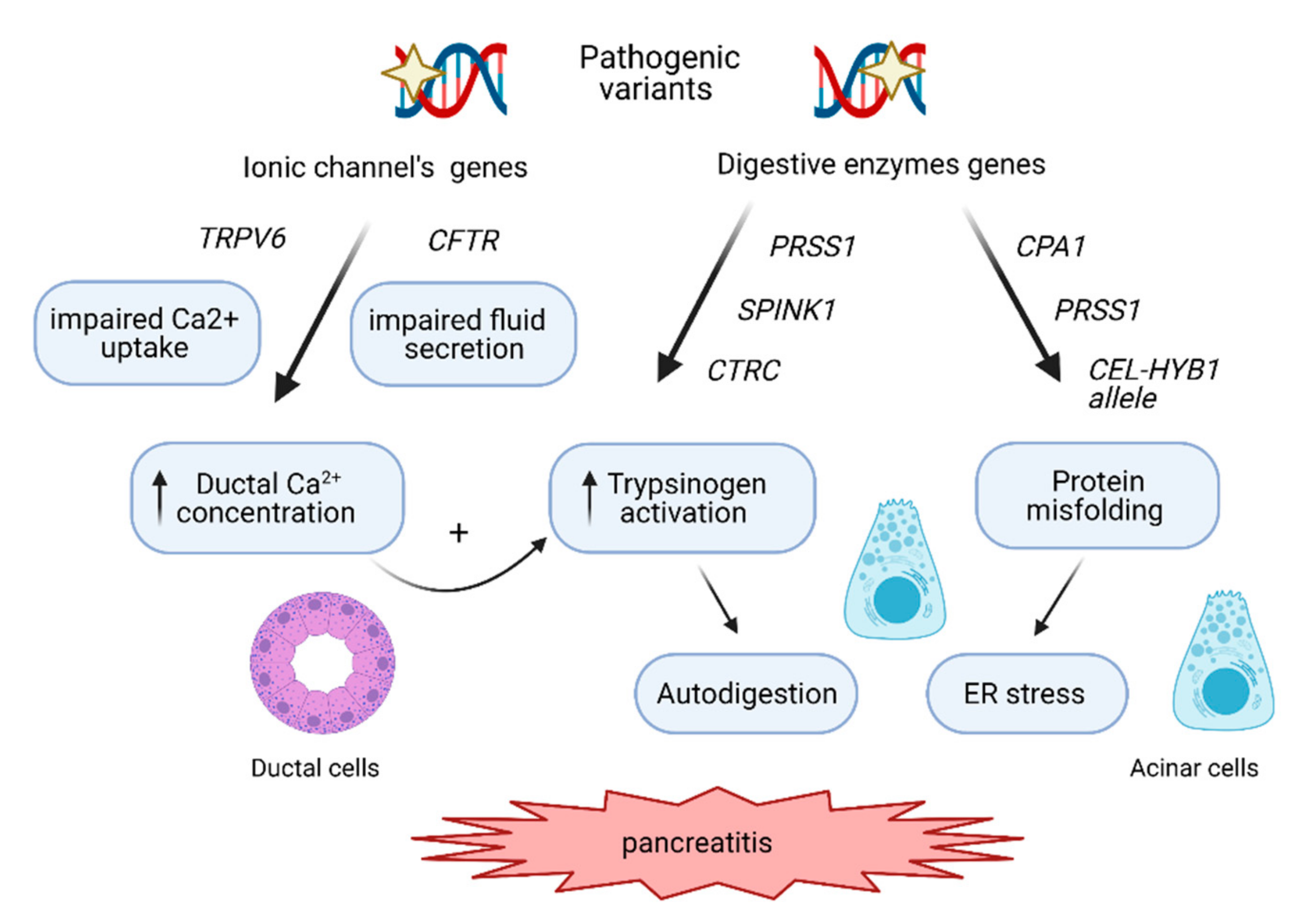
:1. Introduction
2. The Molecular Pathways Involved in CP Development
2.1. The Trypsin-Dependent Pathway
2.2. The Misfolding-Dependent Pathway
2.3. The Ductal Pathway

{kind=link}
| Gene (#OMIM) | Protein | CP Etiology | Genetic Variants # | Mechanisms | Pathway |
|---|---|---|---|---|---|
| PRSS1 (276000) | Cationic trypsinogen | HP, rarely sporadic CP | p.Asn29Ile, p.Ala16Val |
| Trypsin-dependent |
| p.Arg122His, p.Arg122Cys, p.Val39Ala |
| ||||
| Rare variants (e.g., Asp19Ala, p.Asp21Ala) |
| ||||
| Rare variants (e.g., p.Leu104Pro, p.Arg116Cys) |
| Misfolding-dependent | |||
| SPINK1 (167790) | pancreatic secretory trypsin inhibitor | ICP, | p.Asn34Ser |
| Trypsin-dependent |
| c.194+2T>C, and rare missense or nonsense variants |
| ||||
| CTRC (601405) | chymotrypisinogen C | ICP | p.Lys247_Arg254del, p.Arg254Trp, p.Val235Ile |
| Trypsin-dependent |
| CPA1 (114850) | carboxypeptidase A1 | HP and ICP | p.Asn256Lys, p.Ser282Pro, p.Arg382Trp |
| Misfolding-dependent |
| CEL-HYB1 (CEL: 114840) | Recombinant of carboxyl ester lipase and carboxyl ester lipase pseudogene | ICP | CEL-HYB1 allele (CEL and CELP recombinant) ((p.Thr488-Ile548 haplotype) |
| Misfolding-dependent |
| CFTR (602421) | cystic fibrosis transmembrane conductance regulator | ICP | p.Phe508del and other variants severely affecting CFTR expression and activity, p.Arg117His |
| Ductal |
| TRPV6 606680 | transient receptor potential cation channel, subfamily V, member 6 | ICP and familial CP | p.Glu575Lys, p.Arg345His, p.Arg483Trp |
| Ductal |
3. The Genetic Variants Associated with CP
3.1. PRSS1
3.2. SPINK1
3.3. CTRC
3.4. CPA1
3.5. CEL-HYB1 Allele
3.6. CFTR
3.7. TRPV6
4. Complex Genetic Interactions
5. Diagnostic Implications
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Braganza, J.M.; Lee, S.H.; McCloy, R.F.; McMahon, M.J. Chronic pancreatitis. Lancet 2011, 377, 1184–1197. [Google Scholar] [CrossRef]
- Löhr, J.M.; Dominguez-Munoz, E.; Rosendahl, J.; Besselink, M.; Mayerle, J.; Lerch, M.M.; Haas, S.; Akisik, F.; Kartalis, N.; Iglesias-Garcia, J.; et al. United European Gastroenterology evidence-based guidelines for the diagnosis and therapy of chronic pancreatitis (HaPanEU). United Eur. Gastroenterol J. 2017, 5, 153–199. [Google Scholar] [CrossRef] [PubMed]
- Sellers, Z.M.; MacIsaac, D.; Yu, H.; Dehghan, M.; Zhang, K.Y.; Bensen, R.; Wong, J.J.; Kin, C.; Park, K.T. Nationwide Trends in Acute and Chronic Pancreatitis Among Privately Insured Children and Non-Elderly Adults in the United States, 2007–2014. Gastroenterology 2018, 155, 469–478.e461. [Google Scholar] [CrossRef]
- Werlin, S.L.; Kugathasan, S.; Frautschy, B.C. Pancreatitis in children. J. Pediatr. Gastroenterol. Nutr. 2003, 37, 591–595. [Google Scholar] [CrossRef]
- Nydegger, A.; Couper, R.T.; Oliver, M.R. Childhood pancreatitis. J. Gastroenterol. Hepatol. 2006, 21, 499–509. [Google Scholar] [CrossRef]
- Wejnarska, K.; Kolodziejczyk, E.; Wertheim-Tysarowska, K.; Dadalski, M.; Sobczynska-Tomaszewska, A.; Kierkus, J.; Bal, J.; Rygiel, A.M.; Oracz, G. The Etiology and Clinical Course of Chronic Pancreatitis in Children with Early Onset of the Disease. J. Pediatr. Gastroenterol. Nutr. 2016, 63, 665–670. [Google Scholar] [CrossRef] [PubMed]
- Löhr, J.M.; Beuers, U.; Vujasinovic, M.; Alvaro, D.; Frøkjær, J.B.; Buttgereit, F.; Capurso, G.; Culver, E.L.; de-Madaria, E.; Della-Torre, E.; et al. European Guideline on IgG4-related digestive disease—UEG and SGF evidence-based recommendations. United Eur. Gastroenterol. J. 2020, 8, 637–666. [Google Scholar] [CrossRef] [PubMed]
- Sobczyńska-Tomaszewska, A.; Bak, D.; Oralewska, B.; Oracz, G.; Norek, A.; Czerska, K.; Mazurczak, T.; Teisseyre, M.; Socha, J.; Zagulski, M.; et al. Analysis of CFTR, SPINK1, PRSS1 and AAT mutations in children with acute or chronic pancreatitis. J. Pediatr. Gastroenterol. Nutr. 2006, 43, 299–306. [Google Scholar] [CrossRef]
- Oracz, G.; Kolodziejczyk, E.; Sobczynska-Tomaszewska, A.; Wejnarska, K.; Dadalski, M.; Grabarczyk, A.M.; Kierkus, J.; Woynarowski, M.; Wertheim-Tysarowska, K.; Ryzko, J.; et al. The clinical course of hereditary pancreatitis in children—A comprehensive analysis of 41 cases. Pancreatology 2016, 16, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Morinville, V.D.; Husain, S.Z.; Bai, H.; Barth, B.; Alhosh, R.; Durie, P.R.; Freedman, S.D.; Himes, R.; Lowe, M.E.; Pohl, J.; et al. Definitions of pediatric pancreatitis and survey of present clinical practices. J. Pediatr. Gastroenterol. Nutr. 2012, 55, 261–265. [Google Scholar] [CrossRef] [Green Version]
- Párniczky, A.; Abu-El-Haija, M.; Husain, S.; Lowe, M.; Oracz, G.; Sahin-Tóth, M.; Szabó, F.K.; Uc, A.; Wilschanski, M.; Witt, H.; et al. EPC/HPSG evidence-based guidelines for the management of pediatric pancreatitis. Pancreatology 2018, 18, 146–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wejnarska, K.; Kołodziejczyk, E.; Ryżko, J.; Oracz, G. Comparison of 72-hour fecal fat quantification and the 13C-mixed triglyceride breath test in assessing pancreatic exocrine sufficiency in children with chronic pancreatitis. Dev. Period Med. 2016, 20, 222–227. [Google Scholar] [PubMed]
- Shelton, C.; LaRusch, J.; Whitcomb, D.C. Pancreatitis Overview; University of Washington: Seattle, WA, USA, 2020. [Google Scholar]
- Whitcomb, D.C.; Gorry, M.C.; Preston, R.A.; Furey, W.; Sossenheimer, M.J.; Ulrich, C.D.; Martin, S.P.; Gates, L.K.; Amann, S.T.; Toskes, P.P.; et al. Hereditary pancreatitis is caused by a mutation in the cationic trypsinogen gene. Nat. Genet. 1996, 14, 141–145. [Google Scholar] [CrossRef] [PubMed]
- LaRusch, J.; Whitcomb, D.C. Genetics of pancreatitis. Curr. Opin. Gastroenterol. 2011, 27, 467–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witt, H.; Luck, W.; Hennies, H.C.; Classen, M.; Kage, A.; Lass, U.; Landt, O.; Becker, M. Mutations in the gene encoding the serine protease inhibitor, Kazal type 1 are associated with chronic pancreatitis. Nat. Genet. 2000, 25, 213–216. [Google Scholar] [CrossRef]
- Masson, E.; Chen, J.M.; Scotet, V.; Le Maréchal, C.; Férec, C. Association of rare chymotrypsinogen C (CTRC) gene variations in patients with idiopathic chronic pancreatitis. Hum. Genet. 2008, 123, 83–91. [Google Scholar] [CrossRef]
- Rosendahl, J.; Witt, H.; Szmola, R.; Bhatia, E.; Ozsvári, B.; Landt, O.; Schulz, H.U.; Gress, T.M.; Pfützer, R.; Löhr, M.; et al. Chymotrypsin C (CTRC) variants that diminish activity or secretion are associated with chronic pancreatitis. Nat. Genet. 2008, 40, 78–82. [Google Scholar] [CrossRef] [Green Version]
- Toldi, V.; Szabó, A.; Sahin-Tóth, M. Inactivation of mesotrypsin by chymotrypsin C prevents trypsin inhibitor degradation. J. Biol. Chem. 2020, 295, 3447–3455. [Google Scholar] [CrossRef]
- Sahin-Tóth, M. Genetic risk in chronic pancreatitis: The misfolding-dependent pathway. Curr. Opin. Gastroenterol. 2017, 33, 390–395. [Google Scholar] [CrossRef] [Green Version]
- Witt, H.; Beer, S.; Rosendahl, J.; Chen, J.M.; Chandak, G.R.; Masamune, A.; Bence, M.; Szmola, R.; Oracz, G.; Macek, M.; et al. Variants in CPA1 are strongly associated with early onset chronic pancreatitis. Nat. Genet. 2013, 45, 1216–1220. [Google Scholar] [CrossRef] [Green Version]
- Kereszturi, E.; Szmola, R.; Kukor, Z.; Simon, P.; Weiss, F.U.; Lerch, M.M.; Sahin-Tóth, M. Hereditary pancreatitis caused by mutation-induced misfolding of human cationic trypsinogen: A novel disease mechanism. Hum. Mutat. 2009, 30, 575–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fjeld, K.; Weiss, F.U.; Lasher, D.; Rosendahl, J.; Chen, J.M.; Johansson, B.B.; Kirsten, H.; Ruffert, C.; Masson, E.; Steine, S.J.; et al. A recombined allele of the lipase gene CEL and its pseudogene CELP confers susceptibility to chronic pancreatitis. Nat. Genet. 2015, 47, 518–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahin-Tóth, M. Channelopathy of the Pancreas Causes Chronic Pancreatitis. Gastroenterology 2020, 158, 1538–1540. [Google Scholar] [CrossRef]
- Hegyi, E.; Sahin-Tóth, M. Genetic Risk in Chronic Pancreatitis: The Trypsin-Dependent Pathway. Dig. Dis. Sci. 2017, 62, 1692–1701. [Google Scholar] [CrossRef] [PubMed]
- Masamune, A.; Kotani, H.; Sörgel, F.L.; Chen, J.M.; Hamada, S.; Sakaguchi, R.; Masson, E.; Nakano, E.; Kakuta, Y.; Niihori, T.; et al. Variants That Affect Function of Calcium Channel TRPV6 Are Associated With Early-Onset Chronic Pancreatitis. Gastroenterology 2020, 158, 1626–1641.e1628. [Google Scholar] [CrossRef]
- Zou, W.B.; Wang, Y.C.; Ren, X.L.; Wang, L.; Deng, S.J.; Mao, X.T.; Li, Z.S.; Liao, Z. TRPV6 variants confer susceptibility to chronic pancreatitis in the Chinese population. Hum. Mutat. 2020, 41, 1351–1357. [Google Scholar] [CrossRef]
- Howes, N.; Lerch, M.M.; Greenhalf, W.; Stocken, D.D.; Ellis, I.; Simon, P.; Truninger, K.; Ammann, R.; Cavallini, G.; Charnley, R.M.; et al. Clinical and genetic characteristics of hereditary pancreatitis in Europe. Clin. Gastroenterol. Hepatol. 2004, 2, 252–261. [Google Scholar] [CrossRef]
- Keim, V.; Bauer, N.; Teich, N.; Simon, P.; Lerch, M.M.; Mössner, J. Clinical characterization of patients with hereditary pancreatitis and mutations in the cationic trypsinogen gene. Am. J. Med. 2001, 111, 622–626.e1951. [Google Scholar] [CrossRef]
- Mayerle, J.; Sendler, M.; Hegyi, E.; Beyer, G.; Lerch, M.M.; Sahin-Tóth, M. Genetics, Cell Biology, and Pathophysiology of Pancreatitis. Gastroenterology 2019, 156, 1951–1968.e1951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rebours, V.; Boutron-Ruault, M.C.; Schnee, M.; Férec, C.; Le Maréchal, C.; Hentic, O.; Maire, F.; Hammel, P.; Ruszniewski, P.; Lévy, P. The natural history of hereditary pancreatitis: A national series. Gut 2009, 58, 97–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joergensen, M.T.; Brusgaard, K.; Crüger, D.G.; Gerdes, A.M.; Schaffalitzky de Muckadell, O.B. Genetic, epidemiological, and clinical aspects of hereditary pancreatitis: A population-based cohort study in Denmark. Am. J. Gastroenterol. 2010, 105, 1876–1883. [Google Scholar] [CrossRef] [PubMed]
- Applebaum-Shapiro, S.E.; Finch, R.; Pfützer, R.H.; Hepp, L.A.; Gates, L.; Amann, S.; Martin, S.; Ulrich, C.D.; Whitcomb, D.C. Hereditary pancreatitis in North America: The Pittsburgh-Midwest Multi-Center Pancreatic Study Group Study. Pancreatology 2001, 1, 439–443. [Google Scholar] [CrossRef] [PubMed]
- Szabó, A.; Sahin-Tóth, M. Increased activation of hereditary pancreatitis-associated human cationic trypsinogen mutants in presence of chymotrypsin C. J. Biol. Chem. 2012, 287, 20701–20710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemoda, Z.; Sahin-Tóth, M. Chymotrypsin C (caldecrin) stimulates autoactivation of human cationic trypsinogen. J. Biol. Chem. 2006, 281, 11879–11886. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.M.; Kukor, Z.; Le Maréchal, C.; Tóth, M.; Tsakiris, L.; Raguénès, O.; Férec, C.; Sahin-Tóth, M. Evolution of trypsinogen activation peptides. Mol. Biol. Evol. 2003, 20, 1767–1777. [Google Scholar] [CrossRef] [PubMed]
- Geisz, A.; Hegyi, P.; Sahin-Tóth, M. Robust autoactivation, chymotrypsin C independence and diminished secretion define a subset of hereditary pancreatitis-associated cationic trypsinogen mutants. FEBS J. 2013, 280, 2888–2899. [Google Scholar] [CrossRef] [Green Version]
- Rygiel, A.M.; Beer, S.; Simon, P.; Wertheim-Tysarowska, K.; Oracz, G.; Kucharzik, T.; Tysarowski, A.; Niepokój, K.; Kierkus, J.; Jurek, M.; et al. Gene conversion between cationic trypsinogen (PRSS1) and the pseudogene trypsinogen 6 (PRSS3P2) in patients with chronic pancreatitis. Hum. Mutat. 2015, 36, 350–356. [Google Scholar] [CrossRef] [Green Version]
- Schnúr, A.; Beer, S.; Witt, H.; Hegyi, P.; Sahin-Tóth, M. Functional effects of 13 rare PRSS1 variants presumed to cause chronic pancreatitis. Gut 2014, 63, 337–343. [Google Scholar] [CrossRef] [Green Version]
- Aoun, E.; Chang, C.C.; Greer, J.B.; Papachristou, G.I.; Barmada, M.M.; Whitcomb, D.C. Pathways to injury in chronic pancreatitis: Decoding the role of the high-risk SPINK1 N34S haplotype using meta-analysis. PLoS ONE 2008, 3, e2003. [Google Scholar] [CrossRef] [Green Version]
- Gasiorowska, A.; Talar-Wojnarowska, R.; Czupryniak, L.; Smolarz, B.; Romanowicz-Makowska, H.; Kulig, A.; Malecka-Panas, E. The prevalence of cationic trypsinogen (PRSS1) and serine protease inhibitor, Kazal type 1 (SPINK1) gene mutations in Polish patients with alcoholic and idiopathic chronic pancreatitis. Dig. Dis. Sci. 2011, 56, 894–901. [Google Scholar] [CrossRef] [Green Version]
- Di Leo, M.; Bianco, M.; Zuppardo, R.A.; Guslandi, M.; Calabrese, F.; Mannucci, A.; Neri, T.M.; Testoni, P.A.; Leandro, G.; Cavestro, G.M. Meta-analysis of the impact of SPINK1 p.N34S gene variation in Caucasic patients with chronic pancreatitis. An update. Dig. Liver Dis. 2017, 49, 847–853. [Google Scholar] [CrossRef] [PubMed]
- Szabó, A.; Toldi, V.; Gazda, L.D.; Demcsák, A.; Tőzsér, J.; Sahin-Tóth, M. Defective binding of SPINK1 variants is an uncommon mechanism for impaired trypsin inhibition in chronic pancreatitis. J. Biol. Chem. 2021, 100343. [Google Scholar] [CrossRef]
- Drenth, J.P.; te Morsche, R.; Jansen, J.B. Mutations in serine protease inhibitor Kazal type 1 are strongly associated with chronic pancreatitis. Gut 2002, 50, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Pfützer, R.H.; Barmada, M.M.; Brunskill, A.P.; Finch, R.; Hart, P.S.; Neoptolemos, J.; Furey, W.F.; Whitcomb, D.C. SPINK1/PSTI polymorphisms act as disease modifiers in familial and idiopathic chronic pancreatitis. Gastroenterology 2000, 119, 615–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witt, H.; Apte, M.V.; Keim, V.; Wilson, J.S. Chronic pancreatitis: Challenges and advances in pathogenesis, genetics, diagnosis, and therapy. Gastroenterology 2007, 132, 1557–1573. [Google Scholar] [CrossRef]
- Masson, E.; Chen, J.M.; Audrézet, M.P.; Cooper, D.N.; Férec, C. A conservative assessment of the major genetic causes of idiopathic chronic pancreatitis: Data from a comprehensive analysis of PRSS1, SPINK1, CTRC and CFTR genes in 253 young French patients. PLoS ONE 2013, 8, e73522. [Google Scholar] [CrossRef] [Green Version]
- Rygiel, A.M.; Wojnicka-Stolarz, M.; Niepokój, K.; Oracz, G.; Bal, J.; Wertheim-Tysarowska, K.; Gutkowski, K. Chronic Pancreatitis in a Patient With the P.Asn34ser Homozygous Spink1 Mutation--Own Experience. Dev. Period Med. 2015, 19, 347–350. [Google Scholar]
- Kume, K.; Masamune, A.; Kikuta, K.; Shimosegawa, T. [-215G>A.; IVS3+2T>C] mutation in the SPINK1 gene causes exon 3 skipping and loss of the trypsin binding site. Gut 2006, 55, 1214. [Google Scholar] [CrossRef]
- Kereszturi, E.; Király, O.; Sahin-Tóth, M. Minigene analysis of intronic variants in common SPINK1 haplotypes associated with chronic pancreatitis. Gut 2009, 58, 545–549. [Google Scholar] [CrossRef] [Green Version]
- Beer, S.; Zhou, J.; Szabó, A.; Keiles, S.; Chandak, G.R.; Witt, H.; Sahin-Tóth, M. Comprehensive functional analysis of chymotrypsin C (CTRC) variants reveals distinct loss-of-function mechanisms associated with pancreatitis risk. Gut 2013, 62, 1616–1624. [Google Scholar] [CrossRef] [Green Version]
- Szabó, A.; Ludwig, M.; Hegyi, E.; Szépeová, R.; Witt, H.; Sahin-Tóth, M. Mesotrypsin Signature Mutation in a Chymotrypsin C (CTRC) Variant Associated with Chronic Pancreatitis. J. Biol. Chem. 2015, 290, 17282–17292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosendahl, J.; Landt, O.; Bernadova, J.; Kovacs, P.; Teich, N.; Bödeker, H.; Keim, V.; Ruffert, C.; Mössner, J.; Kage, A.; et al. CFTR, SPINK1, CTRC and PRSS1 variants in chronic pancreatitis: Is the role of mutated CFTR overestimated? Gut 2013, 62, 582–592. [Google Scholar] [CrossRef] [Green Version]
- Grabarczyk, A.M.; Oracz, G.; Wertheim-Tysarowska, K.; Kujko, A.A.; Wejnarska, K.; Kolodziejczyk, E.; Bal, J.; Koziel, D.; Kowalik, A.; Gluszek, S.; et al. Chymotrypsinogen C Genetic Variants, Including c.180TT, Are Strongly Associated with Chronic Pancreatitis in Pediatric Patients. J. Pediatr. Gastroenterol. Nutr. 2017, 65, 652–657. [Google Scholar] [CrossRef] [PubMed]
- LaRusch, J.; Lozano-Leon, A.; Stello, K.; Moore, A.; Muddana, V.; O'Connell, M.; Diergaarde, B.; Yadav, D.; Whitcomb, D.C. The Common Chymotrypsinogen C (CTRC) Variant G60G (C.180T) Increases Risk of Chronic Pancreatitis But Not Recurrent Acute Pancreatitis in a North American Population. Clin. Transl. Gastroenterol. 2015, 6, e68. [Google Scholar] [CrossRef]
- Kujko, A.A.; Berki, D.M.; Oracz, G.; Wejnarska, K.; Antoniuk, J.; Wertheim-Tysarowska, K.; Kołodziejczyk, E.; Bal, J.; Sahin-Tóth, M.; Rygiel, A.M. A novel p.Ser282Pro. Gut 2017, 66, 1728–1730. [Google Scholar] [CrossRef]
- Németh, B.C.; Patai, Á.; Sahin-Tóth, M.; Hegyi, P. Misfolding cationic trypsinogen variant p.L104P causes hereditary pancreatitis. Gut 2017, 66, 1727–1728. [Google Scholar] [CrossRef]
- Hegyi, E.; Sahin-Tóth, M. Human. Gut 2019, 68, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Johansson, B.B.; Fjeld, K.; El Jellas, K.; Gravdal, A.; Dalva, M.; Tjora, E.; Ræder, H.; Kulkarni, R.N.; Johansson, S.; Njølstad, P.R.; et al. The role of the carboxyl ester lipase (CEL) gene in pancreatic disease. Pancreatology 2018, 18, 12–19. [Google Scholar] [CrossRef]
- Zou, W.B.; Boulling, A.; Masamune, A.; Issarapu, P.; Masson, E.; Wu, H.; Sun, X.T.; Hu, L.H.; Zhou, D.Z.; He, L.; et al. No Association Between CEL-HYB Hybrid Allele and Chronic Pancreatitis in Asian Populations. Gastroenterology 2016, 150, 1558–1560.e1555. [Google Scholar] [CrossRef] [Green Version]
- Dalva, M.; El Jellas, K.; Steine, S.J.; Johansson, B.B.; Ringdal, M.; Torsvik, J.; Immervoll, H.; Hoem, D.; Laemmerhirt, F.; Simon, P.; et al. Copy number variants and VNTR length polymorphisms of the carboxyl-ester lipase (CEL) gene as risk factors in pancreatic cancer. Pancreatology 2017, 17, 83–88. [Google Scholar] [CrossRef]
- Oracz, G.; Kujko, A.A.; Fjeld, K.; Wertheim-Tysarowska, K.; Adamus-Białek, W.; Steine, S.J.; Koziel, D.; Gluszek, S.; Molven, A.; Rygiel, A.M. The hybrid allele 1 of carboxyl-ester lipase (CEL-HYB1) in Polish pediatric patients with chronic pancreatitis. Pancreatology 2019, 19, 531–534. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, B.M.; Zino, S.; Fjeld, K.; Molven, A.; Lowe, M.E.; Xiao, X. Single nucleotide polymorphisms in CEL-HYB1 increase risk for chronic pancreatitis through proteotoxic misfolding. Hum. Mutat. 2020, 41, 1967–1978. [Google Scholar] [CrossRef] [PubMed]
- Linsdell, P. Architecture and functional properties of the CFTR channel pore. Cell. Mol. Life Sci. 2017, 74, 67–83. [Google Scholar] [CrossRef] [PubMed]
- Sharer, N.; Schwarz, M.; Malone, G.; Howarth, A.; Painter, J.; Super, M.; Braganza, J. Mutations of the cystic fibrosis gene in patients with chronic pancreatitis. N. Engl. J. Med. 1998, 339, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Castellani, C.; Cuppens, H.; Macek, M.; Cassiman, J.J.; Kerem, E.; Durie, P.; Tullis, E.; Assael, B.M.; Bombieri, C.; Brown, A.; et al. Consensus on the use and interpretation of cystic fibrosis mutation analysis in clinical practice. J. Cyst. Fibros. 2008, 7, 179–196. [Google Scholar] [CrossRef] [Green Version]
- Schneider, A.; Larusch, J.; Sun, X.; Aloe, A.; Lamb, J.; Hawes, R.; Cotton, P.; Brand, R.E.; Anderson, M.A.; Money, M.E.; et al. Combined bicarbonate conductance-impairing variants in CFTR and SPINK1 variants are associated with chronic pancreatitis in patients without cystic fibrosis. Gastroenterology 2011, 140, 162–171. [Google Scholar] [CrossRef] [Green Version]
- Freeman, A.J.; Ooi, C.Y. Pancreatitis and pancreatic cystosis in Cystic Fibrosis. J. Cyst. Fibros. 2017, 16, S79–S86.e2226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pallagi, P.; Venglovecz, V.; Rakonczay, Z.; Borka, K.; Korompay, A.; Ozsvári, B.; Judák, L.; Sahin-Tóth, M.; Geisz, A.; Schnúr, A.; et al. Trypsin reduces pancreatic ductal bicarbonate secretion by inhibiting CFTR Cl⁻ channels and luminal anion exchangers. Gastroenterology 2011, 141, 2228–2239.e2226. [Google Scholar] [CrossRef] [Green Version]
- De Boeck, K.; Zolin, A.; Cuppens, H.; Olesen, H.V.; Viviani, L. The relative frequency of CFTR mutation classes in European patients with cystic fibrosis. J. Cyst. Fibros. 2014, 13, 403–409. [Google Scholar] [CrossRef] [Green Version]
- Hegyi, P.; Wilschanski, M.; Muallem, S.; Lukacs, G.L.; Sahin-Tóth, M.; Uc, A.; Gray, M.A.; Rakonczay, Z.; Maléth, J. CFTR: A New Horizon in the Pathomechanism and Treatment of Pancreatitis. Rev. Physiol. Biochem. Pharmacol. 2016, 170, 37–66. [Google Scholar] [CrossRef] [Green Version]
- LaRusch, J.; Jung, J.; General, I.J.; Lewis, M.D.; Park, H.W.; Brand, R.E.; Gelrud, A.; Anderson, M.A.; Banks, P.A.; Conwell, D.; et al. Mechanisms of CFTR functional variants that impair regulated bicarbonate permeation and increase risk for pancreatitis but not for cystic fibrosis. PLoS Genet. 2014, 10, e1004376. [Google Scholar] [CrossRef] [Green Version]
- Saotome, K.; Singh, A.K.; Yelshanskaya, M.V.; Sobolevsky, A.I. Crystal structure of the epithelial calcium channel TRPV6. Nature 2016, 534, 506–511. [Google Scholar] [CrossRef] [Green Version]
- Segerstolpe, Å.; Palasantza, A.; Eliasson, P.; Andersson, E.M.; Andréasson, A.C.; Sun, X.; Picelli, S.; Sabirsh, A.; Clausen, M.; Bjursell, M.K.; et al. Single-Cell Transcriptome Profiling of Human Pancreatic Islets in Health and Type 2 Diabetes. Cell Metab. 2016, 24, 593–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oracz, G.; Zarod, M.; Gambin, T.; Drozak, A.; Kwiatkowski, S.; Wertheim-Tysarowska, K.; Kolodziejczyk, E.; Sawicka, J.; Jackiewicz, M.; Kosińska, J.; et al. TRPV6-defective variants are associated with chronic pancreatitis in Polish pediatric patients. In Proceedings of the 52nd Meeting of the European Pancreatic Club Combined with the International Association of Pancreatology, Paris, France, 1–3 July 2020; pp. S1–S194. [Google Scholar]
- Whitcomb, D.C. Genetic risk factors for pancreatic disorders. Gastroenterology 2013, 144, 1292–1302. [Google Scholar] [CrossRef] [Green Version]
- Noone, P.G.; Zhou, Z.; Silverman, L.M.; Jowell, P.S.; Knowles, M.R.; Cohn, J.A. Cystic fibrosis gene mutations and pancreatitis risk: Relation to epithelial ion transport and trypsin inhibitor gene mutations. Gastroenterology 2001, 121, 1310–1319. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.M.; Férec, C. Chronic pancreatitis: Genetics and pathogenesis. Annu. Rev. Genomics Hum. Genet. 2009, 10, 63–87. [Google Scholar] [CrossRef] [PubMed]
- Rebours, V.; Boutron-Ruault, M.C.; Schnee, M.; Férec, C.; Maire, F.; Hammel, P.; Ruszniewski, P.; Lévy, P. Risk of pancreatic adenocarcinoma in patients with hereditary pancreatitis: A national exhaustive series. Am. J. Gastroenterol. 2008, 103, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer statistics, 2013. CA Cancer J. Clin. 2013, 63, 11–30. [Google Scholar] [CrossRef] [Green Version]
- Chhoda, A.; Lu, L.; Clerkin, B.M.; Risch, H.; Farrell, J.J. Current Approaches to Pancreatic Cancer Screening. Am. J. Pathol. 2019, 189, 22–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadaj-Lipka, R.; Lipiński, M.; Adrych, K.; Durlik, M.; Gąsiorowska, A.; Jarosz, M.; Jurkowska, G.; Małecka-Panas, E.; Oracz, G.; Rosołowski, M.; et al. Diagnostic and therapeutic recommendations for chronic pancreatitis. Recommendations of the Working Group of the Polish Society of Gastroenterology and the Polish Pancreas Club. Prz. Gastroenterol. 2018, 13, 167–181. [Google Scholar] [CrossRef] [Green Version]
- Whitcomb, D.C.; Shimosegawa, T.; Chari, S.T.; Forsmark, C.E.; Frulloni, L.; Garg, P.; Hegyi, P.; Hirooka, Y.; Irisawa, A.; Ishikawa, T.; et al. International consensus statements on early chronic Pancreatitis. Recommendations from the working group for the international consensus guidelines for chronic pancreatitis in collaboration with The International Association of Pancreatology, American Pancreatic Association, Japan Pancreas Society, PancreasFest Working Group and European Pancreatic Club. Pancreatology 2018, 18, 516–527. [Google Scholar] [CrossRef] [PubMed]
- Girodon, E.; Rebours, V.; Chen, J.M.; Pagin, A.; Levy, P.; Ferec, C.; Bienvenu, T. Clinical interpretation of SPINK1 and CTRC variants in pancreatitis. Pancreatology 2020, 20, 1354–1367. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Minowa, K.; Nakano, S.; Isayama, H.; Shimizu, T. Genetic Abnormalities in Pancreatitis: An Update on Diagnosis, Clinical Features, and Treatment. Diagnostics 2020, 11, 31. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wertheim-Tysarowska, K.; Oracz, G.; Rygiel, A.M. Genetic Risk Factors in Early-Onset Nonalcoholic Chronic Pancreatitis: An Update. Genes 2021, 12, 785. https://doi.org/10.3390/genes12050785
Wertheim-Tysarowska K, Oracz G, Rygiel AM. Genetic Risk Factors in Early-Onset Nonalcoholic Chronic Pancreatitis: An Update. Genes. 2021; 12(5):785. https://doi.org/10.3390/genes12050785
Chicago/Turabian StyleWertheim-Tysarowska, Katarzyna, Grzegorz Oracz, and Agnieszka Magdalena Rygiel. 2021. "Genetic Risk Factors in Early-Onset Nonalcoholic Chronic Pancreatitis: An Update" Genes 12, no. 5: 785. https://doi.org/10.3390/genes12050785
APA StyleWertheim-Tysarowska, K., Oracz, G., & Rygiel, A. M. (2021). Genetic Risk Factors in Early-Onset Nonalcoholic Chronic Pancreatitis: An Update. Genes, 12(5), 785. https://doi.org/10.3390/genes12050785