
T Cell Receptor Genotype and Ubash3a Determine Susceptibility to Rat Autoimmune Diabetes

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Diabetes Induction
2.3. Vβ13a+ T Cell Depletion Studies
2.4. Generation of Vβ13 and Ubash3a Knockouts
2.5. Genotyping
2.6. Sequencing
2.7. Statistics
2.8. In Silico Modeling Procedures
3. Results
3.1. Production and Confirmation of Ubash3A and Tcrb-V13S1A1 Knockouts
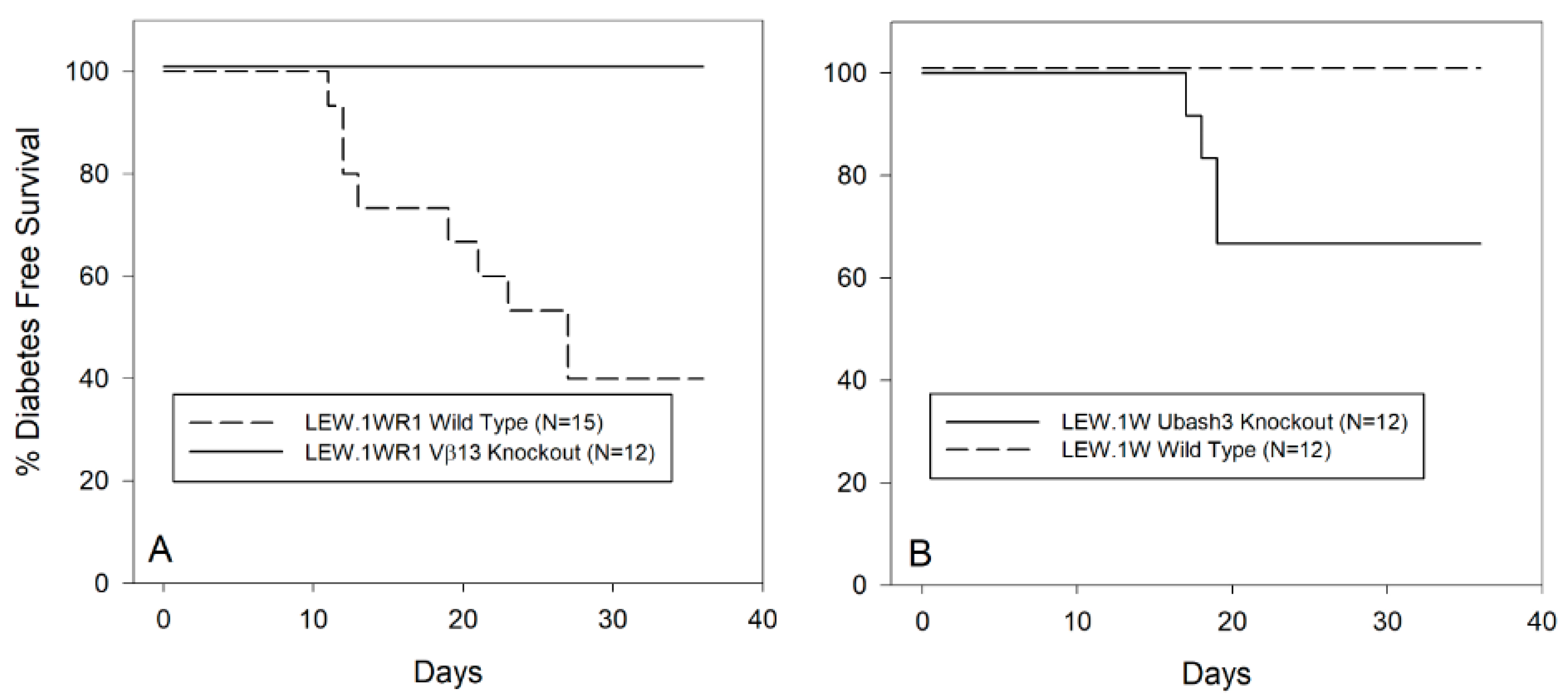
3.2. Vβ13 Knockout LEW.1WR1 Rats Are Completely Diabetes Resistant
3.3. Depletion of Vβ13a+ T Cells by Antibody Prevents T1D Only Early in Disease Progression
3.4. Ubash3A Knockout LEW.1W Rats Become Diabetes Susceptible
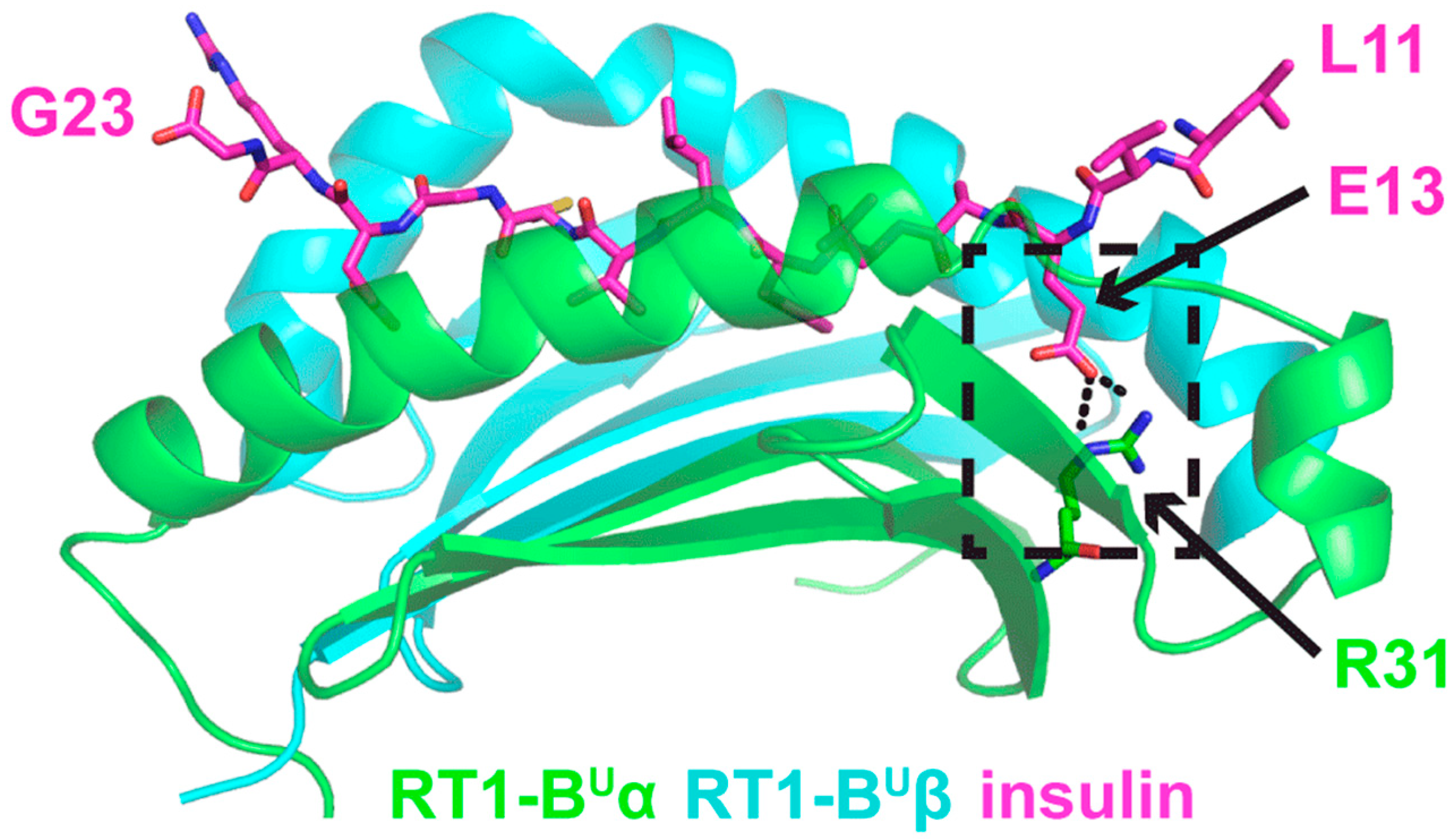
3.5. In Silico Modeling of a Vβ13 TCR and the Mechanistic Basis of Its Autoimmune Recognition
3.5.1. TCR and Insulin B:9-23 Peptide-MHC Structures
3.5.2. Model 1
3.5.3. Model 2
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Easterfield, A.J.; Bradley, J.A.; Bolton, E.M. Complementary DNA sequences encoding the rat MHC class II RT1-Bu and RT1-Du alpha and beta chains. Immunogenetics 2003, 55, 344–350. [Google Scholar] [CrossRef]
- Ellerman, K.E.; Like, A.A. Susceptibility to diabetes is widely distributed in normal class IIu haplotype rats. Diabetologia 2000, 43, 890–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mordes, J.P.; Bortell, R.; Blankenhorn, E.P.; Rossini, A.A.; Greiner, D.L. Rat models of type 1 diabetes: Genetics, environment, and autoimmunity. Ilar J. Natl. Res. Counc. Inst. Lab. Anim. Resour. 2004, 45, 278–291. [Google Scholar] [CrossRef] [Green Version]
- Yokoi, N.; Hayashi, C.; Fujiwara, Y.; Wang, H.Y.; Seino, S. Genetic reconstitution of autoimmune type 1 diabetes with two major susceptibility genes in the rat. Diabetes 2007, 56, 506–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jörns, A.; Akin, M.; Arndt, T.; Terbish, T.; Zu Vilsendorf, A.M.; Wedekind, D.; Hedrich, H.J.; Lenzen, S. Anti-TCR therapy combined with fingolimod for reversal of diabetic hyperglycemia by β cell regeneration in the LEW.1AR1-iddm rat model of type 1 diabetes. J. Mol. Med. 2014, 92, 743–755. [Google Scholar] [CrossRef]
- Wang, Z.; Xie, Z.; Lu, Q.; Chang, C.; Zhou, Z. Beyond Genetics: What Causes Type 1 Diabetes. Clin. Rev. Allergy Immunol. 2017, 52, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Bradfield, J.P.; Qu, H.Q.; Wang, K.; Zhang, H.; Sleiman, P.M.; Kim, C.E.; Mentch, F.D.; Qiu, H.; Glessner, J.T.; Thomas, K.A.; et al. A genome-wide meta-analysis of six type 1 diabetes cohorts identifies multiple associated loci. PLoS Genet. 2011, 7, e1002293. [Google Scholar] [CrossRef] [Green Version]
- Todd, J.A. Evidence that UBASH3 is a causal gene for type 1 diabetes. Eur. J. Hum. Genet. 2018, 26, 925–927. [Google Scholar] [CrossRef]
- Ge, Y.; Paisie, T.K.; Newman, J.R.B.; McIntyre, L.M.; Concannon, P. UBASH3A Mediates Risk for Type 1 Diabetes Through Inhibition of T-Cell Receptor-Induced NF-κB Signaling. Diabetes 2017, 66, 2033–2043. [Google Scholar] [CrossRef] [Green Version]
- Aly, T.A.; Baschal, E.E.; Jahromi, M.M.; Fernando, M.S.; Babu, S.R.; Fingerlin, T.E.; Kretowski, A.; Erlich, H.A.; Fain, P.R.; Rewers, M.J.; et al. Analysis of single nucleotide polymorphisms identifies major type 1A diabetes locus telomeric of the major histocompatibility complex. Diabetes 2008, 57, 770–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cort, L.; Habib, M.; Eberwine, R.A.; Hessner, M.J.; Mordes, J.P.; Blankenhorn, E.P. Diubiquitin (Ubd) is a susceptibility gene for virus-triggered autoimmune diabetes in rats. Genes Immun. 2014, 15, 168–175. [Google Scholar] [CrossRef] [Green Version]
- Mordes, J.P.; Leif, J.; Novak, S.; DeScipio, C.; Greiner, D.L.; Blankenhorn, E.P. The iddm4 locus segregates with diabetes susceptibility in congenic WF.iddm4 rats. Diabetes 2002, 51, 3254–3262. [Google Scholar] [CrossRef] [Green Version]
- Mordes, J.P.; Cort, L.; Norowski, E.; Leif, J.; Fuller, J.M.; Lernmark, A.; Greiner, D.L.; Blankenhorn, E.P. Analysis of the rat Iddm14 diabetes susceptibility locus in multiple rat strains: Identification of a susceptibility haplotype in the Tcrb-V locus. Mamm. Genome 2009, 20, 162–169. [Google Scholar] [CrossRef] [Green Version]
- Stienekemeier, M.; Hofmann, K.; Gold, R.; Herrmann, T. A polymorphism of the rat T-cell receptor beta-chain variable gene 13 (BV13S1) correlates with the frequency of BV13S1-positive CD4 cells. Immunogenetics 2000, 51, 296–305. [Google Scholar] [CrossRef]
- Liu, Z.; Cort, L.; Eberwine, R.; Herrmann, T.; Leif, J.H.; Greiner, D.L.; Yahalom, B.; Blankenhorn, E.P.; Mordes, J.P. Prevention of type 1 diabetes in the rat with an allele-specific anti-T-cell receptor antibody: Vbeta13 as a therapeutic target and biomarker. Diabetes 2012, 61, 1160–1168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Institute of Laboratory Animal Research; Committee on Care, & Use of Laboratory Animals; National Research Council. Guide for the Care and Use of Laboratory Animals; The National Academies Press: Washington, DC, USA, 1996. [Google Scholar]
- Mordes, J.P.; Guberski, D.L.; Leif, J.H.; Woda, B.A.; Flanagan, J.F.; Greiner, D.L.; Kislauskis, E.H.; Tirabassi, R.S. LEW.1WR1 rats develop autoimmune diabetes spontaneously and in response to environmental perturbation. Diabetes 2005, 54, 2727–2733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bewick, V.; Cheek, L.; Ball, J. Statistics review 12: Survival analysis. Crit. Care 2004, 8, 389–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gowthaman, R.; Pierce, B.G. TCRmodel: High resolution modeling of T cell receptors from sequence. Nucleic Acids Res. 2018, 46, W396–W401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pierce, B.G.; Weng, Z. A flexible docking approach for prediction of T cell receptor-peptide-MHC complexes. Protein Sci. 2013, 22, 35–46. [Google Scholar] [CrossRef] [Green Version]
- Eberwine, R.A.; Cort, L.; Habib, M.; Mordes, J.P.; Blankenhorn, E.P. Autoantigen-induced focusing of Vβ13+ T cells precedes onset of autoimmune diabetes in the LEW.1WR1 rat. Diabetes 2014, 63, 596–604. [Google Scholar] [CrossRef] [Green Version]
- Nakayama, M.; Castoe, T.; Sosinowski, T.; He, X.; Johnson, K.; Haskins, K.; Vignali, D.A.; Gapin, L.; Pollock, D.; Eisenbarth, G.S. Germline TRAV5D-4 T-cell receptor sequence targets a primary insulin peptide of NOD mice. Diabetes 2012, 61, 857–865. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.H.; Wucherpfennig, K.W.; Wiley, D.C. Structure of a human insulin peptide-HLA-DQ8 complex and susceptibility to type 1 diabetes. Nat. Immunol. 2001, 2, 501–507. [Google Scholar] [CrossRef]
- Pugliese, A. Autoreactive T cells in type 1 diabetes. J. Clin. Investig. 2017, 127, 2881–2891. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Jasinski, J.M.; Kobayashi, M.; Davenport, B.; Johnson, K.; Davidson, H.; Nakayama, M.; Haskins, K.; Eisenbarth, G.S. Analysis of T cell receptor beta chains that combine with dominant conserved TRAV5D-4*04 anti-insulin B:9-23 alpha chains. J. Autoimmun. 2009, 33, 42–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latek, R.R.; Suri, A.; Petzold, S.J.; Nelson, C.A.; Kanagawa, O.; Unanue, E.R.; Fremont, D.H. Structural basis of peptide binding and presentation by the type I diabetes-associated MHC class II molecule of NOD mice. Immunity 2000, 12, 699–710. [Google Scholar] [CrossRef] [Green Version]
- Sosinowski, T.; Eisenbarth, G.S. Type 1 diabetes: Primary antigen/peptide/register/trimolecular complex. Immunol. Res. 2013, 55, 270–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landry, L.G.; Anderson, A.M.; Russ, H.A.; Yu, L.; Kent, S.C.; Atkinson, M.A.; Mathews, C.E.; Michels, A.W.; Nakayama, M. Proinsulin-Reactive CD4 T Cells in the Islets of Type 1 Diabetes Organ Donors. Front. Endocrinol. 2021, 12, 622647. [Google Scholar] [CrossRef]
- Levisetti, M.G.; Suri, A.; Petzold, S.J.; Unanue, E.R. The insulin-specific T cells of nonobese diabetic mice recognize a weak MHC-binding segment in more than one form. J. Immunol. 2007, 178, 6051–6057. [Google Scholar] [CrossRef] [Green Version]
- Stadinski, B.D.; Zhang, L.; Crawford, F.; Marrack, P.; Eisenbarth, G.S.; Kappler, J.W. Diabetogenic T cells recognize insulin bound to IAg7 in an unexpected, weakly binding register. Proc. Natl. Acad. Sci.USA 2010, 107, 10978–10983. [Google Scholar] [CrossRef] [Green Version]
- Eswar, N.; Webb, B.; Marti-Renom, M.A.; Madhusudhan, M.S.; Eramian, D.; Shen, M.Y.; Pieper, U.; Sali, A. Comparative protein structure modeling using Modeller. Curr. Protoc. Bioinform. 2006, 15, 5–6. [Google Scholar] [CrossRef] [Green Version]
- Pierce, B.; Weng, Z. A combination of rescoring and refinement significantly improves protein docking performance. Proteins 2008, 72, 270–279. [Google Scholar] [CrossRef] [Green Version]
- Leaver-Fay, A.; Tyka, M.; Lewis, S.M.; Lange, O.F.; Thompson, J.; Jacak, R.; Kaufman, K.; Renfrew, P.D.; Smith, C.A.; Sheffler, W.; et al. ROSETTA3: An object-oriented software suite for the simulation and design of macromolecules. Methods Enzym. 2011, 487, 545–574. [Google Scholar]
- Chao, N.J.; Timmerman, L.; McDevitt, H.O.; Jacob, C.O. Molecular characterization of MHC class II antigens (beta 1 domain) in the BB diabetes-prone and -resistant rat. Immunogenetics 1989, 29, 231–234. [Google Scholar] [CrossRef]
- Rudolph, M.G.; Stanfield, R.L.; Wilson, I.A. How TCRs bind MHCs, peptides, and coreceptors. Annu. Rev. Immunol. 2006, 24, 419–466. [Google Scholar] [CrossRef] [PubMed]
- Gowthaman, R.; Pierce, B.G. TCR3d: The T cell receptor structural repertoire database. Bioinformatics 2019, 35, 5323–5325. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Li, Y.; Mariuzza, R.A. Structural basis for self-recognition by autoimmune T-cell receptors. Immunol. Rev. 2012, 250, 32–48. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sosinowski, T.; Novikov, A.; Crawford, F.; White, J.; Jin, N.; Liu, Z.; Zou, J.; Neau, D.; Davidson, H.W.; et al. How C-terminal additions to insulin B-chain fragments create superagonists for T cells in mouse and human type 1 diabetes. Sci. Immunol. 2019, 4. [Google Scholar] [CrossRef]
- Martin, A.M.; Blankenhorn, E.P.; Maxson, M.N.; Zhao, M.; Leif, J.; Mordes, J.P.; Greiner, D.L. Non-major histocompatibility complex-linked diabetes susceptibility loci on chromosomes 4 and 13 in a backcross of the DP-BB/Wor rat to the WF rat. Diabetes 1999, 48, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Concannon, P.; Onengut-Gumuscu, S.; Todd, J.A.; Smyth, D.J.; Pociot, F.; Bergholdt, R.; Akolkar, B.; Erlich, H.A.; Hilner, J.E.; Julier, C.; et al. Type 1 Diabetes Genetics, C., A human type 1 diabetes susceptibility locus maps to chromosome 21q22.3. Diabetes 2008, 57, 2858–2861. [Google Scholar] [CrossRef] [Green Version]
- Tsygankov, A.Y. TULA-family proteins: A new class of cellular regulators. J. Cell Physiol. 2013, 228, 43–49. [Google Scholar] [CrossRef] [PubMed]
- San Luis, B.; Sondgeroth, B.; Nassar, N.; Carpino, N. Sts-2 is a phosphatase that negatively regulates zeta-associated protein (ZAP)-70 and T cell receptor signaling pathways. J. Biol. Chem. 2011, 286, 15943–15954. [Google Scholar] [CrossRef] [Green Version]
- Wucherpfennig, K.W. The first structures of T cell receptors bound to peptide-MHC. J. Immunol. 2010, 185, 6391–6393. [Google Scholar] [CrossRef] [PubMed]
- Sim, B.C.; Zerva, L.; Greene, M.I.; Gascoigne, N.R. Control of MHC restriction by TCR Valpha CDR1 and CDR2. Science 1996, 273, 963–966. [Google Scholar] [CrossRef]
- Broughton, S.E.; Petersen, J.; Theodossis, A.; Scally, S.W.; Loh, K.L.; Thompson, A.; van Bergen, J.; Kooy-Winkelaar, Y.; Henderson, K.N.; Beddoe, T.; et al. Biased T cell receptor usage directed against human leukocyte antigen DQ8-restricted gliadin peptides is associated with celiac disease. Immunity 2012, 37, 611–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, J.J.; Narayanan, S.; Liu, B.; Birnbaum, M.E.; Kruse, A.C.; Bowerman, N.A.; Chen, W.; Levin, A.M.; Connolly, J.M.; Zhu, C.; et al. T cell receptor signaling is limited by docking geometry to peptide-major histocompatibility complex. Immunity 2011, 35, 681–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sethi, D.K.; Gordo, S.; Schubert, D.A.; Wucherpfennig, K.W. Crossreactivity of a human autoimmune TCR is dominated by a single TCR loop. Nat. Commun. 2013, 4, 2623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahn, M.; Nicholson, M.J.; Pyrdol, J.; Wucherpfennig, K.W. Unconventional topology of self peptide-major histocompatibility complex binding by a human autoimmune T cell receptor. Nat. Immunol. 2005, 6, 490–496. [Google Scholar] [CrossRef]
- Deng, L.; Langley, R.J.; Brown, P.H.; Xu, G.; Teng, L.; Wang, Q.; Gonzales, M.I.; Callender, G.G.; Nishimura, M.I.; Topalian, S.L.; et al. Structural basis for the recognition of mutant self by a tumor-specific, MHC class II-restricted T cell receptor. Nat. Immunol. 2007, 8, 398–408. [Google Scholar] [CrossRef]
- Deng, L.; Langley, R.J.; Wang, Q.; Topalian, S.L.; Mariuzza, R.A. Structural insights into the editing of germ-line-encoded interactions between T-cell receptor and MHC class II by Valpha CDR3. Proc. Natl. Acad. Sci. USA 2012, 109, 14960–14965. [Google Scholar] [CrossRef] [Green Version]
- Petersen, J.; Montserrat, V.; Mujico, J.R.; Loh, K.L.; Beringer, D.X.; van Lummel, M.; Thompson, A.; Mearin, M.L.; Schweizer, J.; Kooy-Winkelaar, Y.; et al. T-cell receptor recognition of HLA-DQ2-gliadin complexes associated with celiac disease. Nat. Struct. Mol. Biol. 2014, 21, 480–488. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Start of Anti-Vβ13 mAb Relative to Poly I:C | Day −5 or +5 | Day +10 |
|---|---|---|
| Diabetic | 1 | 6 |
| Non-diabetic | 9 | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mordes, J.P.; Cort, L.; Liu, Z.; Eberwine, R.; Blankenhorn, E.P.; Pierce, B.G. T Cell Receptor Genotype and Ubash3a Determine Susceptibility to Rat Autoimmune Diabetes. Genes 2021, 12, 852. https://doi.org/10.3390/genes12060852
Mordes JP, Cort L, Liu Z, Eberwine R, Blankenhorn EP, Pierce BG. T Cell Receptor Genotype and Ubash3a Determine Susceptibility to Rat Autoimmune Diabetes. Genes. 2021; 12(6):852. https://doi.org/10.3390/genes12060852
Chicago/Turabian StyleMordes, John P., Laura Cort, Zhijun Liu, Ryan Eberwine, Elizabeth P. Blankenhorn, and Brian G. Pierce. 2021. "T Cell Receptor Genotype and Ubash3a Determine Susceptibility to Rat Autoimmune Diabetes" Genes 12, no. 6: 852. https://doi.org/10.3390/genes12060852
APA StyleMordes, J. P., Cort, L., Liu, Z., Eberwine, R., Blankenhorn, E. P., & Pierce, B. G. (2021). T Cell Receptor Genotype and Ubash3a Determine Susceptibility to Rat Autoimmune Diabetes. Genes, 12(6), 852. https://doi.org/10.3390/genes12060852