
Clinical Manifestations in a Girl with NAA10-Related Syndrome and Genotype–Phenotype Correlation in Females

 ,
, {kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
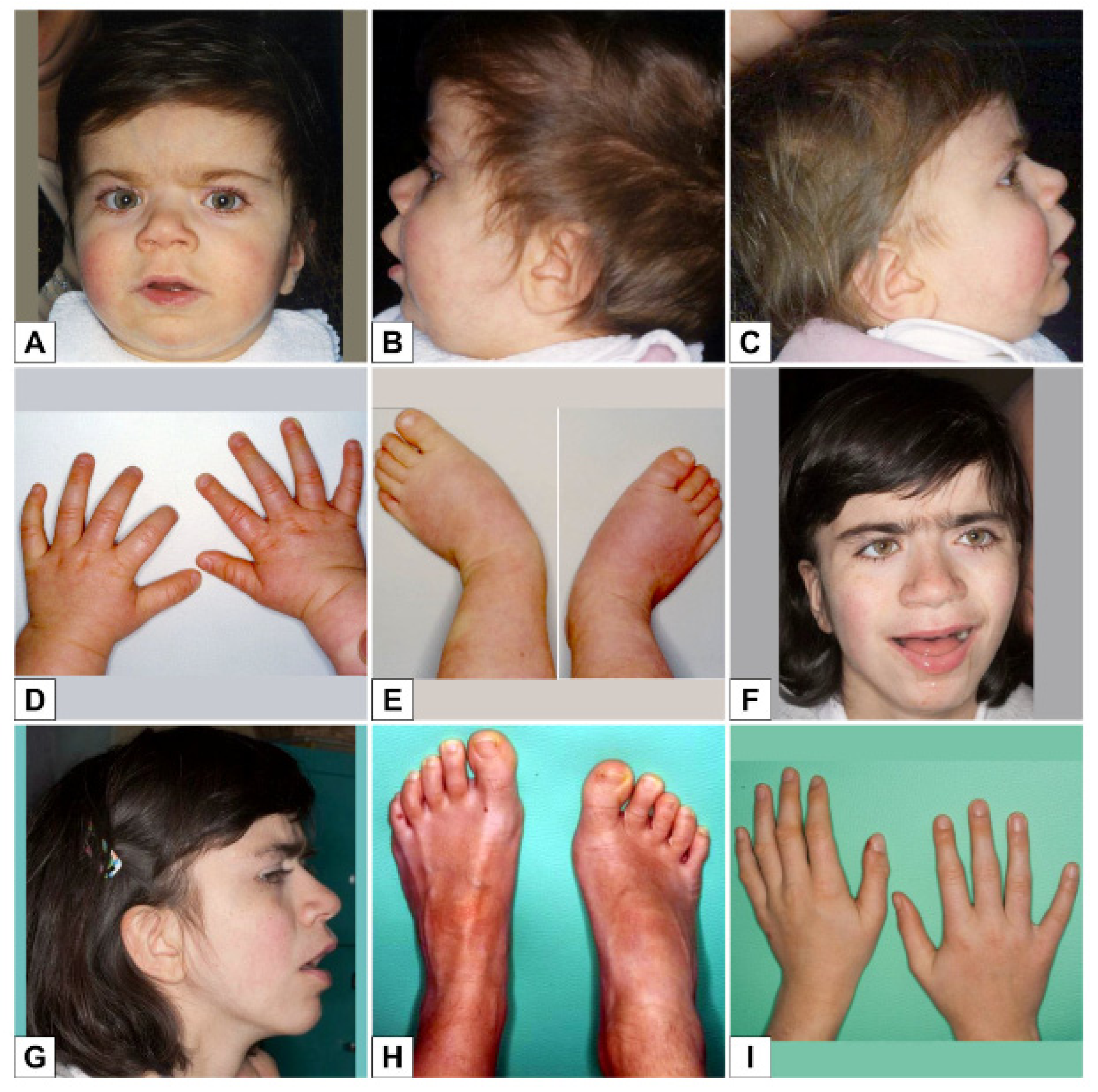
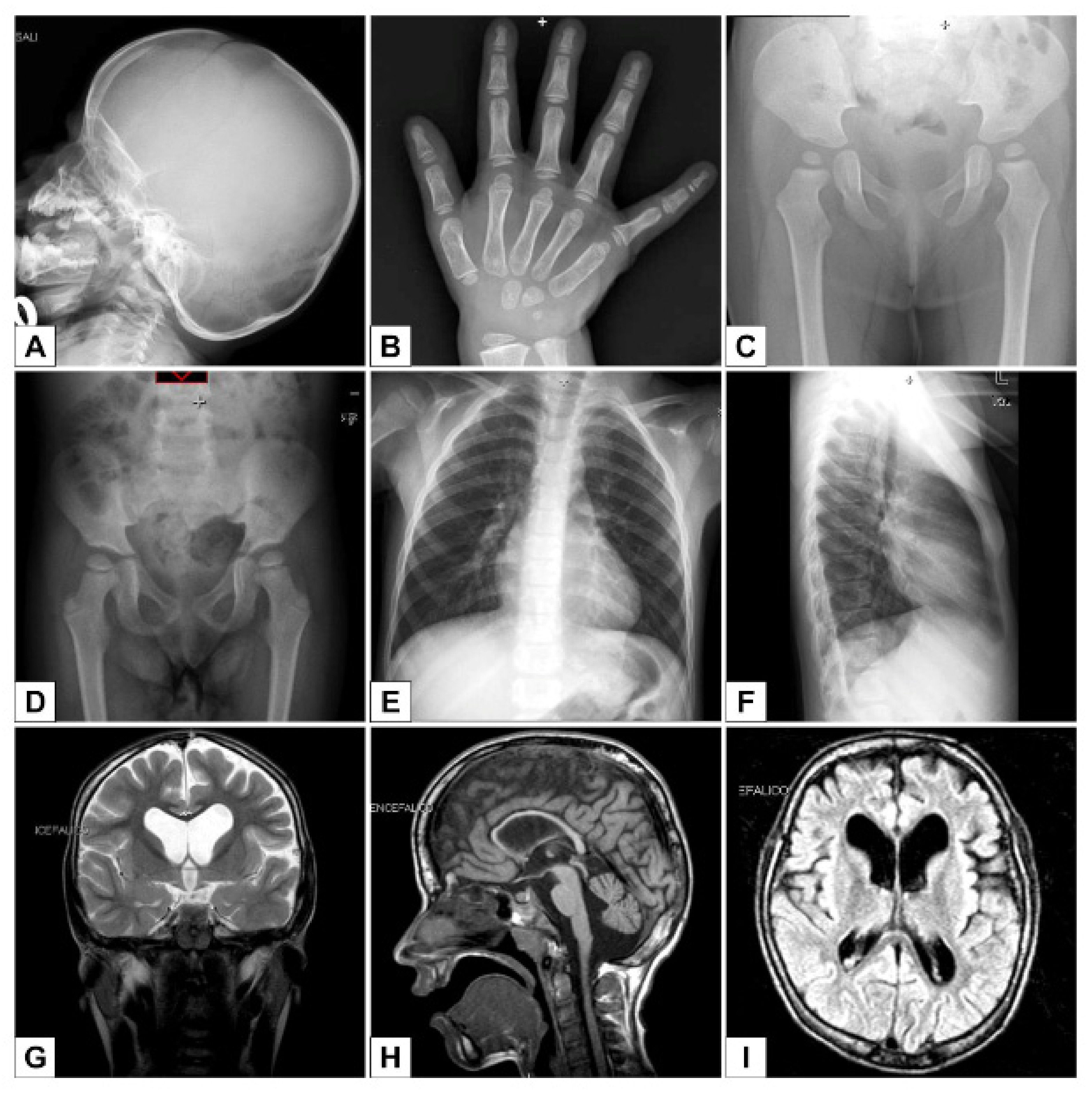
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rope, A.F.; Wang, K.; Evjenth, R.; Xing, J.; Johnston, J.J.; Swensen, J.J.; Johnson, W.E.; Moore, B.; Huff, C.D.; Bird, L.M.; et al. Using VAAST to identify an X-linked disorder resulting in lethality in male infants due to N-terminal acetyltransferase deficiency. Am. J. Hum. Genet. 2011, 89, 28–43. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Lyon, G.J. NAA10-related syndrome. Exp. Mol. Med. 2018, 50, 1–10. [Google Scholar] [CrossRef]
- Arnesen, T.; Van Damme, P.; Polevoda, B.; Helsens, K.; Evjenth, R.; Colaert, N.; Varhaug, J.E.; Vandekerckhove, J.; Lillehaug, J.R.; Sherman, F.; et al. Proteomics analyses reveal the evolutionary conservation and divergence of N-terminal acetyltransferases from yeast and humans. Proc. Natl. Acad. Sci. USA 2009, 106, 8157–8162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starheim, K.K.; Gevaert, K.; Arnesen, T. Protein N-terminal acetyltransferases: When the start matters. Trends Biochem. Sci. 2012, 37, 152–161. [Google Scholar] [CrossRef]
- Myklebust, L.M.; Van Damme, P.; Støve, S.I.; Dörfel, M.J.; Abboud, A.; Kalvik, T.V.; Grauffel, C.; Jonckheere, V.; Wu, Y.; Swensen, J.; et al. Biochemical and cellular analysis of Ogden Syndrome reveals downstream Nt-acetylation defects. Hum. Mol. Genet. 2015, 24, 1956–1976. [Google Scholar] [CrossRef] [Green Version]
- Rauch, A.; Wieczorek, D.; Graf, E.; Wieland, T.; Endele, S.; Schwarzmayr, T.; Albrecht, B.; Bartholdi, D.; Beygo, J.; Di Donato, N.; et al. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: An exome sequencing study. Lancet 2012, 380, 1674–1682. [Google Scholar] [CrossRef]
- Casey, J.P.; Støve, S.I.; McGorrian, C.; Galvin, J.; Blenski, M.; Dunne, A.; Ennis, S.; Brett, F.; King, M.D.; Arnesen, T.; et al. NAA10 mutation causing a novel intellectual disability syndrome with Long QT due to N-terminal acetyltransferase impairment. Sci. Rep. 2015, 5, 16022. [Google Scholar] [CrossRef] [PubMed]
- Popp, B.; Støve, S.I.; Endele, S.; Myklebust, L.M.; Hoyer, J.; Sticht, H.; Azzarello-Burri, S.; Rauch, A.; Arnesen, T.; Reis, A. De novo missense mutations in the NAA10 gene cause severe non-syndromic developmental delay in males and females. Eur. J. Hum. Genet. 2015, 23, 602–609. [Google Scholar] [CrossRef] [Green Version]
- Saunier, C.; Støve, S.I.; Popp, B.; Gérard, B.; Blenski, M.; Ahmew, N.; De Bie, C.; Goldenberg, P.; Isidor, B.; Keren, B.; et al. Expanding the phenotype associated with NAA10-related N-terminal acetylation deficiency. Hum. Mutat. 2016, 37, 755–764. [Google Scholar] [CrossRef] [Green Version]
- Sidhu, M.; Brady, L.; Tarnopolsky, M.; Ronen, G.M. Clinical manifestations associated with the N-terminal-acetyltransferase NAA10 gene mutation in a girl: Ogden syndrome. Pediatr. Neurol. 2017, 76, 82–85. [Google Scholar] [CrossRef]
- Støve, S.I.; Blenski, M.; Stray-Pedersen, A.; Wierenga, K.J.; Jhangiani, S.N.; Akdemir, Z.C.; Crawford, D.; McTiernan, N.; Myklebust, L.M.; Purcarin, G.; et al. A novel NAA10 variant with impaired acetyltransferase activity causes developmental delay, intellectual disability, and hypertrophic cardiomyopathy. Eur. J. Hum. Genet. 2018, 26, 1294–1305. [Google Scholar] [CrossRef] [Green Version]
- Johnston, J.J.; Williamson, K.A.; Chou, C.M.; Sapp, J.C.; Ansari, M.; Chapman, H.M.; Cooper, D.N.; Dabir, T.; Dudley, J.N.; Holt, R.J.; et al. NAA10 polyadenylation signal variants cause syndromic microphthalmia. J. Med. Genet. 2019, 56, 444–452. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.; Gottlieb, L.; Marchi, E.; Kleyner, R.; Bhardwaj, P.; Rope, A.F.; Rosenheck, S.; Moutton, S.; Philippe, C.; Eyaid, W.; et al. Phenotypic and biochemical analysis of an international cohort of individuals with variants in NAA10 and NAA15. Hum. Mol. Genet. 2019, 28, 2900–2919. [Google Scholar] [CrossRef] [PubMed]
- Marionneau, C.; Abriel, H. Regulation of the cardiac Na+ channel NaV1.5 by post-translational modifications. J. Mol. Cell. Cardiol. 2015, 82, 36–47. [Google Scholar] [CrossRef]
- Aksnes, H.; Drazic, A.; Arnesen, T. (Hyper)tension release by N-terminal acetylation. Trends Biochem. Sci. 2015, 40, 422–424. [Google Scholar] [CrossRef]
- Sugiura, N.; Adams, S.M.; Corriveau, R.A. An evolutionarily conserved N-terminal acetyltransferase complex associated with neuronal development. J. Biol. Chem. 2003, 278, 40113–40120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asaumi, M.; Iijima, K.; Sumioka, A.; Iijima-Ando, K.; Kirino, Y.; Nakaya, T.; Suzuki, T. Interaction of N-terminal acetyltransferase with the cytoplasmic domain of beta-amyloid precursor protein and its effect on A beta secretion. J. Biochem. 2005, 137, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Raychaudhuri, S.; Sinha, M.; Mukhopadhyay, D.; Bhattacharyya, N.P. HYPK, a Huntingtin interacting protein, reduces aggregates and apoptosis induced by N-terminal Huntingtin with 40 glutamines in Neuro2a cells and exhibits chaperone-like activity. Hum. Mol. Genet. 2007, 17, 240–255. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.-F.; Ou, S.-C.; Lee, S.-B.; Chang, L.-H.; Lin, R.-K.; Li, Y.-S.; Upadhyay, A.K.; Cheng, X.; Wang, Y.-C.; Hsu, H.-S.; et al. hNaa10p contributes to tumorigenesis by facilitating DNMT1-mediated tumor suppressor gene silencing. J. Clin. Investig. 2010, 120, 2920–2930. [Google Scholar] [CrossRef] [Green Version]
- Kalvik, T.V.; Arnesen, T. Protein N-terminal acetyltransferases in cancer. Oncogene 2013, 32, 269–276. [Google Scholar] [CrossRef] [Green Version]
- Esmailpour, T.; Riazifar, H.; Liu, L.; Donkervoort, S.; Huang, V.H.; Madaan, S.; Shoucri, B.M.; Busch, A.; Wu, J.; Towbin, A.; et al. A splice donor mutation in NAA10 results in the dysregulation of the retinoic acid signalling pathway and causes Lenz microphthalmia syndrome. J. Med. Genet. 2014, 51, 185–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ree, R.; Geithus, A.S.; Tørring, P.M.; Sørensen, K.P.; Damkjær, M.; Lynch, S.A.; Arnesen, T. A novel NAA10 p.(R83H) variant with impaired acetyltransferase activity identified in two boys with ID and microcephaly. BMC Med. Genet. 2019, 20, 101. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Muzny, D.M.; Xia, F.; Niu, Z.; Person, R.; Ding, Y.; Ward, P.; Braxton, A.; Wang, M.; Buhay, C.; et al. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA 2014, 312, 1870–1879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thevenon, J.; Duffourd, Y.; Masurel-Paulet, A.; Lefebvre, M.; Feillet, F.; El Chehadeh-Djebbar, S.; St-Onge, J.; Steinmetz, A.; Huet, F.; Chouchane, M.; et al. Diagnostic odyssey in severe neurodevelopmental disorders: Toward clinical whole-exome sequencing as a first-line diagnostic test. Clin. Genet. 2016, 89, 700–707. [Google Scholar] [CrossRef]
- McTiernan, N.; Støve, S.I.; Aukrust, I.; Mårli, M.T.; Myklebust, L.M.; Houge, G.; Arnesen, T. NAA10 dysfunction with normal NatA-complex activity in a girl with non-syndromic ID and a de novo NAA10 p.(V111G) variant—A case report. BMC Med. Genet. 2018, 19, 47. [Google Scholar] [CrossRef] [Green Version]
- Bader, I.; McTiernan, N.; Darbakk, C.; Boltshauser, E.; Ree, R.; Ebner, S.; Mayr, J.A.; Arnesen, T. Severe syndromic ID and skewed X-inactivation in a girl with NAA10 dysfunction and a novel heterozygous de novo NAA10 p.(His16Pro) variant—A case report. BMC Med. Genet. 2020, 21, 1–9. [Google Scholar] [CrossRef]
- Ivanovski, I.; Caraffi, S.G.; Magnani, E.; Rosato, S.; Pollazzon, M.; Matalonga, L.; Piana, S.; Nicoli, D.; Baldo, C.; Bernasconi, S.; et al. Alazami syndrome: The first case of papillary thyroid carcinoma. J. Hum. Genet. 2019, 65, 133–141. [Google Scholar] [CrossRef]
- Thompson, D.J.; Genovese, G.; Halvardson, J.; Ulirsch, J.C.; Wright, D.J.; Terao, C.; Davidsson, O.B.; Day, F.R.; Sulem, P.; Jiang, Y.; et al. Genetic predisposition to mosaic Y chromosome loss in blood. Nature 2019, 575, 652–657. [Google Scholar] [CrossRef]
- Loh, P.R.; Genovese, G.; McCarroll, S.A. Monogenic and polygenic inheritance become instruments for clonal selection. Nature 2020, 584, 136–141. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maini, I.; Caraffi, S.G.; Peluso, F.; Valeri, L.; Nicoli, D.; Laurie, S.; Baldo, C.; Zuffardi, O.; Garavelli, L. Clinical Manifestations in a Girl with NAA10-Related Syndrome and Genotype–Phenotype Correlation in Females. Genes 2021, 12, 900. https://doi.org/10.3390/genes12060900
Maini I, Caraffi SG, Peluso F, Valeri L, Nicoli D, Laurie S, Baldo C, Zuffardi O, Garavelli L. Clinical Manifestations in a Girl with NAA10-Related Syndrome and Genotype–Phenotype Correlation in Females. Genes. 2021; 12(6):900. https://doi.org/10.3390/genes12060900
Chicago/Turabian StyleMaini, Ilenia, Stefano G. Caraffi, Francesca Peluso, Lara Valeri, Davide Nicoli, Steven Laurie, Chiara Baldo, Orsetta Zuffardi, and Livia Garavelli. 2021. "Clinical Manifestations in a Girl with NAA10-Related Syndrome and Genotype–Phenotype Correlation in Females" Genes 12, no. 6: 900. https://doi.org/10.3390/genes12060900
APA StyleMaini, I., Caraffi, S. G., Peluso, F., Valeri, L., Nicoli, D., Laurie, S., Baldo, C., Zuffardi, O., & Garavelli, L. (2021). Clinical Manifestations in a Girl with NAA10-Related Syndrome and Genotype–Phenotype Correlation in Females. Genes, 12(6), 900. https://doi.org/10.3390/genes12060900