
The Genetics of Parkinson’s Disease and Implications for Clinical Practice

Abstract
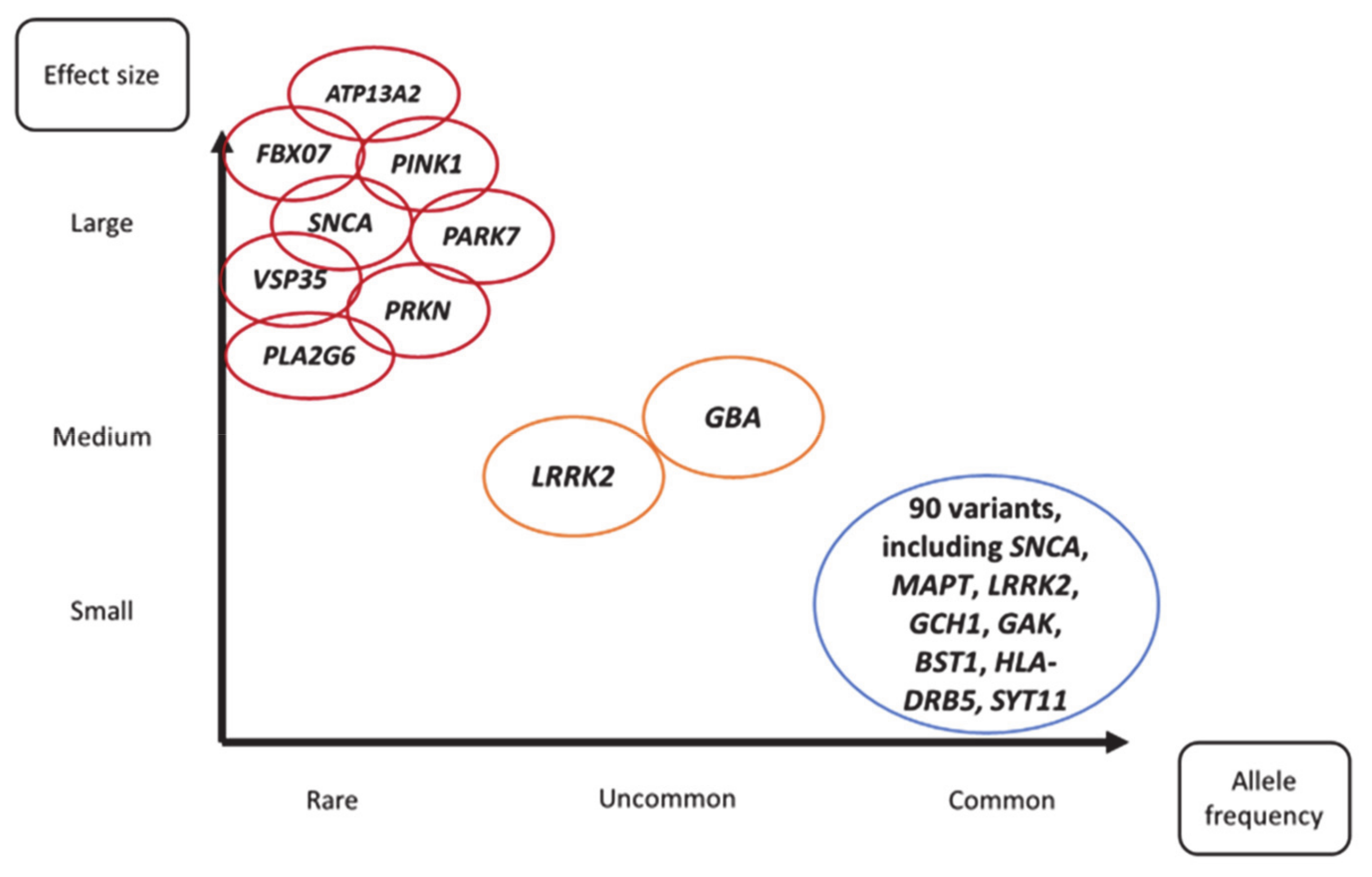
:1. Introduction
2. Monogenic Parkinson’s Disease
2.1. Single Genes with High Penetrance
2.1.1. SNCA
2.1.2. VPS35
2.1.3. PRKN/ PARK7/ PINK1
2.1.4. Others
2.2. Single Genes with Variable Penetrance
2.2.1. LRRK2
2.2.2. GBA
2.2.3. Heterozygous PRKN as a PD Genetic Risk Factor
3. Sporadic Parkinson’s Disease
4. Therapeutics
4.1. Disease-Modifying Agents for PD
4.1.1. GBA
4.1.2. LRRK2
4.1.3. SNCA
4.1.4. Mitochondria
4.2. Pharmacogenomics
5. Selecting Appropriate PD Cohorts for Clinical Trials
Prodromal PD
6. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- GBD 2016 Neurology Collaborators. Global, regional, and national burden of Parkinson’s disease, 1990-2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018, 17, 939–953. [Google Scholar] [CrossRef] [Green Version]
- Jankovic, J. Parkinson’s disease: Clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry 2008, 79, 368–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinzel, S.; Berg, D.; Gasser, T.; Chen, H.; Yao, C.; Postuma, R.B.; MDS Task Force on the Definition of Parkinson’s Disease. Update of the MDS research criteria for prodromal Parkinson’s disease. Mov. Disord. 2019, 34, 1464–1470. [Google Scholar] [CrossRef] [PubMed]
- Hughes, A.J.; Daniel, S.E.; Kilford, L.; Lees, A.J. Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: A clinico-pathological study of 100 cases. J. Neurol. Neurosurg. Psychiatry 1992, 55, 181–184. [Google Scholar] [CrossRef] [Green Version]
- Postuma, R.B.; Berg, D.; Stern, M.; Poewe, W.; Olanow, C.W.; Oertel, W.; Obeso, J.; Marek, K.; Litvan, I.; Lang, A.E.; et al. MDS clinical diagnostic criteria for Parkinson’s disease. Mov. Disord. 2015, 30, 1591–1601. [Google Scholar] [CrossRef]
- Duvoisin, R.C.; Eldridge, R.; Williams, A.; Nutt, J.; Calne, D. Twin study of Parkinson disease. Neurology 1981, 31, 77–80. [Google Scholar] [CrossRef]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [Green Version]
- Wirdefeldt, K.; Gatz, M.; Reynolds, C.A.; Prescott, C.A.; Pedersen, N.L. Heritability of Parkinson disease in Swedish twins: A longitudinal study. Neurobiol. Aging 2011, 32, 1923.e1–1923.e8. [Google Scholar] [CrossRef] [Green Version]
- Goldman, S.M.; Marek, K.; Ottman, R.; Meng, C.; Comyns, K.; Chan, P.; Ma, J.; Marras, C.; Langston, J.W.; Ross, G.W.; et al. Concordance for Parkinson’s disease in twins: A 20-year update. Ann. Neurol. 2019, 85, 600–605. [Google Scholar] [CrossRef]
- Blauwendraat, C.; Nalls, M.A.; Singleton, A.B. The genetic architecture of Parkinson’s disease. Lancet Neurol. 2020, 19, 170–178. [Google Scholar] [CrossRef]
- Noyce, A.J.; Bestwick, J.P.; Silveira-Moriyama, L.; Hawkes, C.H.; Giovannoni, G.; Lees, A.J.; Schrag, A. Meta-analysis of early nonmotor features and risk factors for Parkinson disease. Ann. Neurol. 2012, 72, 893–901. [Google Scholar] [CrossRef]
- Pang, S.Y.; Ho, P.W.; Liu, H.F.; Leung, C.T.; Li, L.; Chang, E.E.S.; Ramsden, D.B.; Ho, S.L. The interplay of aging, genetics and environmental factors in the pathogenesis of Parkinson’s disease. Transl. Neurodegener. 2019, 8, 23. [Google Scholar] [CrossRef]
- von Linstow, C.U.; DeLano-Taylor, M.; Kordower, J.H.; Brundin, P. Does Developmental Variability in the Number of Midbrain Dopamine Neurons Affect Individual Risk for Sporadic Parkinson’s Disease? J. Parkinsons Dis. 2020, 10, 405–411. [Google Scholar] [CrossRef] [Green Version]
- Ascherio, A.; Schwarzschild, M.A. The epidemiology of Parkinson’s disease: Risk factors and prevention. Lancet Neurol. 2016, 15, 1257–1272. [Google Scholar] [CrossRef]
- Breckenridge, C.B.; Berry, C.; Chang, E.T.; Sielken, R.L.; Mandel, J.S. Association between Parkinson’s Disease and Cigarette Smoking, Rural Living, Well-Water Consumption, Farming and Pesticide Use: Systematic Review and Meta-Analysis. PLoS ONE 2016, 11, e0151841. [Google Scholar] [CrossRef] [Green Version]
- Qi, H.; Li, S. Dose-response meta-analysis on coffee, tea and caffeine consumption with risk of Parkinson’s disease. Geriatr. Gerontol. Int. 2014, 14, 430–439. [Google Scholar] [CrossRef]
- Keller, M.F.; Saad, M.; Bras, J.; Bettella, F.; Nicolaou, N.; Simón-Sánchez, J.; Mittag, F.; Büchel, F.; Sharma, M.; Gibbs, J.R.; et al. Using genome-wide complex trait analysis to quantify ‘missing heritability’ in Parkinson’s disease. Hum. Mol. Genet. 2012, 21, 4996–5009. [Google Scholar] [CrossRef]
- Nalls, M.A.; Blauwendraat, C.; Vallerga, C.L.; Heilbron, K.; Bandres-Ciga, S.; Chang, D.; Tan, M.; Kia, D.A.; Noyce, A.J.; Xue, A.; et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: A meta-analysis of genome-wide association studies. Lancet Neurol. 2019, 18, 1091–1102. [Google Scholar] [CrossRef]
- Marras, C.; Lang, A. Parkinson’s disease subtypes: Lost in translation? J. Neurol. Neurosurg. Psychiatry 2013, 84, 409–415. [Google Scholar] [CrossRef]
- Correia Guedes, L.; Mestre, T.; Outeiro, T.F.; Ferreira, J.J. Are genetic and idiopathic forms of Parkinson’s disease the same disease? J. Neurochem. 2020, 152, 515–522. [Google Scholar] [CrossRef]
- Shadrina, M.I.; Slominsky, P.A.; Limborska, S.A. Molecular mechanisms of pathogenesis of Parkinson’s disease. Int. Rev. Cell Mol. Biol. 2010, 281, 229–266. [Google Scholar] [CrossRef]
- Bouchard, M.; Suchowersky, O. Tauopathies: One disease or many? Can. J. Neurol. Sci. 2011, 38, 547–556. [Google Scholar] [CrossRef]
- Marras, C.; Lang, A.; van de Warrenburg, B.P.; Sue, C.M.; Tabrizi, S.J.; Bertram, L.; Mercimek-Mahmutoglu, S.; Ebrahimi-Fakhari, D.; Warner, T.T.; Durr, A.; et al. Nomenclature of genetic movement disorders: Recommendations of the international Parkinson and movement disorder society task force. Mov. Disord. 2016, 31, 436–457. [Google Scholar] [CrossRef]
- Polymeropoulos, M.H.; Higgins, J.J.; Golbe, L.I.; Johnson, W.G.; Ide, S.E.; Di Iorio, G.; Sanges, G.; Stenroos, E.S.; Pho, L.T.; Schaffer, A.A.; et al. Mapping of a gene for Parkinson’s disease to chromosome 4q21-q23. Science 1996, 274, 1197–1199. [Google Scholar] [CrossRef] [Green Version]
- Muñoz, E.; Oliva, R.; Obach, V.; Martí, M.J.; Pastor, P.; Ballesta, F.; Tolosa, E. Identification of Spanish familial Parkinson’s disease and screening for the Ala53Thr mutation of the alpha-synuclein gene in early onset patients. Neurosci. Lett. 1997, 235, 57–60. [Google Scholar] [CrossRef]
- Vaughan, J.; Durr, A.; Tassin, J.; Bereznai, B.; Gasser, T.; Bonifati, V.; De Michele, G.; Fabrizio, E.; Volpe, G.; Bandmann, O.; et al. The alpha-synuclein Ala53Thr mutation is not a common cause of familial Parkinson’s disease: A study of 230 European cases. European Consortium on Genetic Susceptibility in Parkinson’s Disease. Ann. Neurol. 1998, 44, 270–273. [Google Scholar] [CrossRef]
- Krüger, R.; Kuhn, W.; Müller, T.; Woitalla, D.; Graeber, M.; Kösel, S.; Przuntek, H.; Epplen, J.T.; Schöls, L.; Riess, O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat. Genet. 1998, 18, 106–108. [Google Scholar] [CrossRef]
- Zarranz, J.J.; Alegre, J.; Gómez-Esteban, J.C.; Lezcano, E.; Ros, R.; Ampuero, I.; Vidal, L.; Hoenicka, J.; Rodriguez, O.; Atarés, B.; et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 2004, 55, 164–173. [Google Scholar] [CrossRef]
- Lesage, S.; Anheim, M.; Letournel, F.; Bousset, L.; Honoré, A.; Rozas, N.; Pieri, L.; Madiona, K.; Dürr, A.; Melki, R.; et al. G51D α-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann. Neurol. 2013, 73, 459–471. [Google Scholar] [CrossRef]
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. alpha-Synuclein locus triplication causes Parkinson’s disease. Science 2003, 302, 841. [Google Scholar] [CrossRef] [Green Version]
- Ross, O.A.; Braithwaite, A.T.; Skipper, L.M.; Kachergus, J.; Hulihan, M.M.; Middleton, F.A.; Nishioka, K.; Fuchs, J.; Gasser, T.; Maraganore, D.M.; et al. Genomic investigation of alpha-synuclein multiplication and parkinsonism. Ann. Neurol. 2008, 63, 743–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benitez, B.A.; Davis, A.A.; Jin, S.C.; Ibanez, L.; Ortega-Cubero, S.; Pastor, P.; Choi, J.; Cooper, B.; Perlmutter, J.S.; Cruchaga, C. Resequencing analysis of five Mendelian genes and the top genes from genome-wide association studies in Parkinson’s Disease. Mol. Neurodegener. 2016, 11, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skrahina, V.; Gaber, H.; Vollstedt, E.J.; Förster, T.M.; Usnich, T.; Curado, F.; Brüggemann, N.; Paul, J.; Bogdanovic, X.; Zülbahar, S.; et al. The Rostock International Parkinson’s Disease (ROPAD) Study: Protocol and Initial Findings. Mov. Disord. 2020. [Google Scholar] [CrossRef]
- Tan, M.M.X.; Malek, N.; Lawton, M.A.; Hubbard, L.; Pittman, A.M.; Joseph, T.; Hehir, J.; Swallow, D.M.A.; Grosset, K.A.; Marrinan, S.L.; et al. Genetic analysis of Mendelian mutations in a large UK population-based Parkinson’s disease study. Brain 2019, 142, 2828–2844. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Conway, K.A.; Harper, J.D.; Lansbury, P.T. Accelerated in vitro fibril formation by a mutant alpha-synuclein linked to early-onset Parkinson disease. Nat. Med. 1998, 4, 1318–1320. [Google Scholar] [CrossRef]
- Giasson, B.I.; Duda, J.E.; Murray, I.V.; Chen, Q.; Souza, J.M.; Hurtig, H.I.; Ischiropoulos, H.; Trojanowski, J.Q.; Lee, V.M. Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science 2000, 290, 985–989. [Google Scholar] [CrossRef]
- Xu, J.; Kao, S.Y.; Lee, F.J.; Song, W.; Jin, L.W.; Yankner, B.A. Dopamine-dependent neurotoxicity of alpha-synuclein: A mechanism for selective neurodegeneration in Parkinson disease. Nat. Med. 2002, 8, 600–606. [Google Scholar] [CrossRef]
- Peelaerts, W.; Bousset, L.; Van der Perren, A.; Moskalyuk, A.; Pulizzi, R.; Giugliano, M.; Van den Haute, C.; Melki, R.; Baekelandt, V. α-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 2015, 522, 340–344. [Google Scholar] [CrossRef]
- Perrino, G.; Wilson, C.; Santorelli, M.; di Bernardo, D. Quantitative Characterization of α-Synuclein Aggregation in Living Cells through Automated Microfluidics Feedback Control. Cell Rep. 2019, 27, 916–927.e915. [Google Scholar] [CrossRef] [Green Version]
- Narkiewicz, J.; Giachin, G.; Legname, G. In vitro aggregation assays for the characterization of α-synuclein prion-like properties. Prion 2014, 8, 19–32. [Google Scholar] [CrossRef] [Green Version]
- Neystat, M.; Lynch, T.; Przedborski, S.; Kholodilov, N.; Rzhetskaya, M.; Burke, R.E. Alpha-synuclein expression in substantia nigra and cortex in Parkinson’s disease. Mov. Disord. 1999, 14, 417–422. [Google Scholar] [CrossRef]
- Wong, Y.C.; Krainc, D. α-synuclein toxicity in neurodegeneration: Mechanism and therapeutic strategies. Nat. Med. 2017, 23, 1–13. [Google Scholar] [CrossRef]
- Vilariño-Güell, C.; Wider, C.; Ross, O.A.; Dachsel, J.C.; Kachergus, J.M.; Lincoln, S.J.; Soto-Ortolaza, A.I.; Cobb, S.A.; Wilhoite, G.J.; Bacon, J.A.; et al. VPS35 mutations in Parkinson disease. Am. J. Hum. Genet. 2011, 89, 162–167. [Google Scholar] [CrossRef] [Green Version]
- Zimprich, A.; Benet-Pagès, A.; Struhal, W.; Graf, E.; Eck, S.H.; Offman, M.N.; Haubenberger, D.; Spielberger, S.; Schulte, E.C.; Lichtner, P.; et al. A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am. J. Hum. Genet. 2011, 89, 168–175. [Google Scholar] [CrossRef] [Green Version]
- Choy, R.W.; Park, M.; Temkin, P.; Herring, B.E.; Marley, A.; Nicoll, R.A.; von Zastrow, M. Retromer mediates a discrete route of local membrane delivery to dendrites. Neuron 2014, 82, 55–62. [Google Scholar] [CrossRef] [Green Version]
- Bono, K.; Hara-Miyauchi, C.; Sumi, S.; Oka, H.; Iguchi, Y.; Okano, H.J. Endosomal dysfunction in iPSC-derived neural cells from Parkinson’s disease patients with VPS35 D620N. Mol. Brain 2020, 13, 137. [Google Scholar] [CrossRef]
- Hanss, Z.; Larsen, S.B.; Antony, P.; Mencke, P.; Massart, F.; Jarazo, J.; Schwamborn, J.C.; Barbuti, P.A.; Mellick, G.D.; Krüger, R. Mitochondrial and Clearance Impairment in p.D620N VPS35 Patient-Derived Neurons. Mov. Disord. 2020. [Google Scholar] [CrossRef]
- Wider, C.; Skipper, L.; Solida, A.; Brown, L.; Farrer, M.; Dickson, D.; Wszolek, Z.K.; Vingerhoets, F.J. Autosomal dominant dopa-responsive parkinsonism in a multigenerational Swiss family. Parkinsonism Relat. Disord. 2008, 14, 465–470. [Google Scholar] [CrossRef]
- Lesage, S.; Houot, M.; Mangone, G.; Tesson, C.; Bertrand, H.; Forlani, S.; Anheim, M.; Brefel-Courbon, C.; Broussolle, E.; Thobois, S.; et al. Genetic and Phenotypic Basis of Autosomal Dominant Parkinson’s Disease in a Large Multi-Center Cohort. Front. Neurol. 2020, 11, 682. [Google Scholar] [CrossRef]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef]
- Bonifati, V.; Rizzu, P.; van Baren, M.J.; Schaap, O.; Breedveld, G.J.; Krieger, E.; Dekker, M.C.; Squitieri, F.; Ibanez, P.; Joosse, M.; et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 2003, 299, 256–259. [Google Scholar] [CrossRef] [Green Version]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Gao, J.; Chung, K.K.; Huang, H.; Dawson, V.L.; Dawson, T.M. Parkin functions as an E2-dependent ubiquitin- protein ligase and promotes the degradation of the synaptic vesicle-associated protein, CDCrel-1. Proc. Natl. Acad. Sci. USA 2000, 97, 13354–13359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothfuss, O.; Fischer, H.; Hasegawa, T.; Maisel, M.; Leitner, P.; Miesel, F.; Sharma, M.; Bornemann, A.; Berg, D.; Gasser, T.; et al. Parkin protects mitochondrial genome integrity and supports mitochondrial DNA repair. Hum. Mol. Genet. 2009, 18, 3832–3850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizzu, P.; Hinkle, D.A.; Zhukareva, V.; Bonifati, V.; Severijnen, L.A.; Martinez, D.; Ravid, R.; Kamphorst, W.; Eberwine, J.H.; Lee, V.M.; et al. DJ-1 colocalizes with tau inclusions: A link between parkinsonism and dementia. Ann. Neurol. 2004, 55, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Sliter, D.A.; Martinez, J.; Hao, L.; Chen, X.; Sun, N.; Fischer, T.D.; Burman, J.L.; Li, Y.; Zhang, Z.; Narendra, D.P.; et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature 2018, 561, 258–262. [Google Scholar] [CrossRef]
- Xiong, H.; Wang, D.; Chen, L.; Choo, Y.S.; Ma, H.; Tang, C.; Xia, K.; Jiang, W.; Ronai, Z.; Zhuang, X.; et al. Parkin, PINK1, and DJ-1 form a ubiquitin E3 ligase complex promoting unfolded protein degradation. J. Clin. Investig. 2009, 119, 650–660. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, S.; Wakabayashi, K.; Ishikawa, A.; Nagai, H.; Saito, M.; Maruyama, M.; Takahashi, T.; Ozawa, T.; Tsuji, S.; Takahashi, H. An autopsy case of autosomal-recessive juvenile parkinsonism with a homozygous exon 4 deletion in the parkin gene. Mov. Disord. 2000, 15, 884–888. [Google Scholar] [CrossRef]
- Gouider-Khouja, N.; Larnaout, A.; Amouri, R.; Sfar, S.; Belal, S.; Ben Hamida, C.; Ben Hamida, M.; Hattori, N.; Mizuno, Y.; Hentati, F. Autosomal recessive parkinsonism linked to parkin gene in a Tunisian family. Clinical, genetic and pathological study. Parkinsonism Relat. Disord. 2003, 9, 247–251. [Google Scholar] [CrossRef]
- Mori, H.; Kondo, T.; Yokochi, M.; Matsumine, H.; Nakagawa-Hattori, Y.; Miyake, T.; Suda, K.; Mizuno, Y. Pathologic and biochemical studies of juvenile parkinsonism linked to chromosome 6q. Neurology 1998, 51, 890–892. [Google Scholar] [CrossRef]
- Miyakawa, S.; Ogino, M.; Funabe, S.; Uchino, A.; Shimo, Y.; Hattori, N.; Ichinoe, M.; Mikami, T.; Saegusa, M.; Nishiyama, K.; et al. Lewy body pathology in a patient with a homozygous parkin deletion. Mov. Disord. 2013, 28, 388–391. [Google Scholar] [CrossRef]
- Schneider, S.A.; Alcalay, R.N. Neuropathology of genetic synucleinopathies with parkinsonism: Review of the literature. Mov. Disord. 2017, 32, 1504–1523. [Google Scholar] [CrossRef]
- Ramirez, A.; Heimbach, A.; Gründemann, J.; Stiller, B.; Hampshire, D.; Cid, L.P.; Goebel, I.; Mubaidin, A.F.; Wriekat, A.L.; Roeper, J.; et al. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat. Genet. 2006, 38, 1184–1191. [Google Scholar] [CrossRef]
- van Veen, S.; Martin, S.; Van den Haute, C.; Benoy, V.; Lyons, J.; Vanhoutte, R.; Kahler, J.P.; Decuypere, J.P.; Gelders, G.; Lambie, E.; et al. ATP13A2 deficiency disrupts lysosomal polyamine export. Nature 2020, 578, 419–424. [Google Scholar] [CrossRef]
- Di Fonzo, A.; Dekker, M.C.; Montagna, P.; Baruzzi, A.; Yonova, E.H.; Correia Guedes, L.; Szczerbinska, A.; Zhao, T.; Dubbel-Hulsman, L.O.; Wouters, C.H.; et al. FBXO7 mutations cause autosomal recessive, early-onset parkinsonian-pyramidal syndrome. Neurology 2009, 72, 240–245. [Google Scholar] [CrossRef]
- Wei, L.; Ding, L.; Li, H.; Lin, Y.; Dai, Y.; Xu, X.; Dong, Q.; Long, L. Juvenile-onset parkinsonism with pyramidal signs due to compound heterozygous mutations in the F-Box only protein 7 gene. Parkinsonism Relat. Disord. 2018, 47, 76–79. [Google Scholar] [CrossRef]
- Jin, X.; An, L.; Hao, S.; Liu, Q.; Zhang, Q.; Wang, X.; Feng, X.; Zhang, C.; Cao, X.; Yan, Y.; et al. Compound heterozygous variants of the FBXO7 gene resulting in infantile-onset Parkinsonian-pyramidal syndrome in siblings of a Chinese family. J. Clin. Lab. Anal. 2020, 34, e23324. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Lear, T.B.; Verma, M.; Wang, K.Z.; Otero, P.A.; McKelvey, A.C.; Dunn, S.R.; Steer, E.; Bateman, N.W.; Wu, C.; et al. Chemical inhibition of FBXO7 reduces inflammation and confers neuroprotection by stabilizing the mitochondrial kinase PINK1. JCI Insights 2020, 5. [Google Scholar] [CrossRef]
- Morgan, N.V.; Westaway, S.K.; Morton, J.E.; Gregory, A.; Gissen, P.; Sonek, S.; Cangul, H.; Coryell, J.; Canham, N.; Nardocci, N.; et al. PLA2G6, encoding a phospholipase A2, is mutated in neurodegenerative disorders with high brain iron. Nat. Genet. 2006, 38, 752–754. [Google Scholar] [CrossRef] [Green Version]
- Paisan-Ruiz, C.; Bhatia, K.P.; Li, A.; Hernandez, D.; Davis, M.; Wood, N.W.; Hardy, J.; Houlden, H.; Singleton, A.; Schneider, S.A. Characterization of PLA2G6 as a locus for dystonia-parkinsonism. Ann. Neurol. 2009, 65, 19–23. [Google Scholar] [CrossRef]
- Chu, Y.T.; Lin, H.Y.; Chen, P.L.; Lin, C.H. Genotype-phenotype correlations of adult-onset PLA2G6-associated Neurodegeneration: Case series and literature review. BMC Neurol. 2020, 20, 101. [Google Scholar] [CrossRef] [Green Version]
- Quadri, M.; Fang, M.; Picillo, M.; Olgiati, S.; Breedveld, G.J.; Graafland, J.; Wu, B.; Xu, F.; Erro, R.; Amboni, M.; et al. Mutation in the SYNJ1 gene associated with autosomal recessive, early-onset Parkinsonism. Hum. Mutat. 2013, 34, 1208–1215. [Google Scholar] [CrossRef]
- Krebs, C.E.; Karkheiran, S.; Powell, J.C.; Cao, M.; Makarov, V.; Darvish, H.; Di Paolo, G.; Walker, R.H.; Shahidi, G.A.; Buxbaum, J.D.; et al. The Sac1 domain of SYNJ1 identified mutated in a family with early-onset progressive Parkinsonism with generalized seizures. Hum. Mutat. 2013, 34, 1200–1207. [Google Scholar] [CrossRef] [Green Version]
- Deng, H.X.; Shi, Y.; Yang, Y.; Ahmeti, K.B.; Miller, N.; Huang, C.; Cheng, L.; Zhai, H.; Deng, S.; Nuytemans, K.; et al. Identification of TMEM230 mutations in familial Parkinson’s disease. Nat. Genet. 2016, 48, 733–739. [Google Scholar] [CrossRef]
- Wang, X.; Whelan, E.; Liu, Z.; Liu, C.F.; Smith, W.W. Controversy of TMEM230 Associated with Parkinson’s Disease. Neuroscience 2021, 453, 280–286. [Google Scholar] [CrossRef]
- Saini, P.; Rudakou, U.; Yu, E.; Ruskey, J.A.; Asayesh, F.; Laurent, S.B.; Spiegelman, D.; Fahn, S.; Waters, C.; Monchi, O.; et al. Association study of DNAJC13, UCHL1, HTRA2, GIGYF2, and EIF4G1 with Parkinson’s disease. Neurobiol. Aging 2020. [Google Scholar] [CrossRef] [PubMed]
- Paisán-Ruíz, C.; Jain, S.; Evans, E.W.; Gilks, W.P.; Simón, J.; van der Brug, M.; López de Munain, A.; Aparicio, S.; Gil, A.M.; Khan, N.; et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 2004, 44, 595–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 2004, 44, 601–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marder, K.; Wang, Y.; Alcalay, R.N.; Mejia-Santana, H.; Tang, M.X.; Lee, A.; Raymond, D.; Mirelman, A.; Saunders-Pullman, R.; Clark, L.; et al. Age-specific penetrance of LRRK2 G2019S in the Michael J. Fox Ashkenazi Jewish LRRK2 Consortium. Neurology 2015, 85, 89–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, A.J.; Wang, Y.; Alcalay, R.N.; Mejia-Santana, H.; Saunders-Pullman, R.; Bressman, S.; Corvol, J.C.; Brice, A.; Lesage, S.; Mangone, G.; et al. Penetrance estimate of LRRK2 p.G2019S mutation in individuals of non-Ashkenazi Jewish ancestry. Mov. Disord. 2017, 32, 1432–1438. [Google Scholar] [CrossRef]
- Healy, D.G.; Falchi, M.; O’Sullivan, S.S.; Bonifati, V.; Durr, A.; Bressman, S.; Brice, A.; Aasly, J.; Zabetian, C.P.; Goldwurm, S.; et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: A case-control study. Lancet Neurol. 2008, 7, 583–590. [Google Scholar] [CrossRef] [Green Version]
- Shu, L.; Zhang, Y.; Pan, H.; Xu, Q.; Guo, J.; Tang, B.; Sun, Q. Clinical Heterogeneity Among. Front. Aging Neurosci. 2018, 10, 283. [Google Scholar] [CrossRef] [Green Version]
- Di Fonzo, A.; Tassorelli, C.; De Mari, M.; Chien, H.F.; Ferreira, J.; Rohé, C.F.; Riboldazzi, G.; Antonini, A.; Albani, G.; Mauro, A.; et al. Comprehensive analysis of the LRRK2 gene in sixty families with Parkinson’s disease. Eur. J. Hum. Genet. 2006, 14, 322–331. [Google Scholar] [CrossRef]
- Lüth, T.; König, I.R.; Grünewald, A.; Kasten, M.; Klein, C.; Hentati, F.; Farrer, M.; Trinh, J. Age at Onset of LRRK2 p.Gly2019Ser Is Related to Environmental and Lifestyle Factors. Mov. Disord. 2020, 35, 1854–1858. [Google Scholar] [CrossRef]
- San Luciano, M.; Tanner, C.M.; Meng, C.; Marras, C.; Goldman, S.M.; Lang, A.E.; Tolosa, E.; Schüle, B.; Langston, J.W.; Brice, A.; et al. Nonsteroidal Anti-inflammatory Use and LRRK2 Parkinson’s Disease Penetrance. Mov. Disord. 2020, 35, 1755–1764. [Google Scholar] [CrossRef]
- Heckman, M.G.; Labbé, C.; Kolicheski, A.L.; Soto-Beasley, A.I.; Walton, R.L.; Valentino, R.R.; Brennan, E.R.; Johnson, P.W.; Baheti, S.; Sarangi, V.; et al. Fine-mapping of the non-coding variation driving the Caucasian LRRK2 GWAS signal in Parkinson’s disease. Parkinsonism Relat. Disord. 2021, 83, 22–30. [Google Scholar] [CrossRef]
- Hentati, F.; Trinh, J.; Thompson, C.; Nosova, E.; Farrer, M.J.; Aasly, J.O. LRRK2 parkinsonism in Tunisia and Norway: A comparative analysis of disease penetrance. Neurology 2014, 83, 568–569. [Google Scholar] [CrossRef] [Green Version]
- Lai, D.; Alipanahi, B.; Fontanillas, P.; Schwantes-An, T.H.; Aasly, J.; Alcalay, R.N.; Beecham, G.W.; Berg, D.; Bressman, S.; Brice, A.; et al. Genomewide Association Studies of LRRK2 Modifiers of Parkinson’s Disease. Ann. Neurol. 2021. [Google Scholar] [CrossRef]
- Shu, L.; Zhang, Y.; Sun, Q.; Pan, H.; Tang, B. A Comprehensive Analysis of Population Differences in. Front. Aging Neurosci. 2019, 11, 13. [Google Scholar] [CrossRef]
- Trinh, J.; Zeldenrust, F.M.J.; Huang, J.; Kasten, M.; Schaake, S.; Petkovic, S.; Madoev, H.; Grünewald, A.; Almuammar, S.; König, I.R.; et al. Genotype-phenotype relations for the Parkinson’s disease genes SNCA, LRRK2, VPS35: MDSGene systematic review. Mov. Disord. 2018, 33, 1857–1870. [Google Scholar] [CrossRef]
- Berwick, D.C.; Heaton, G.R.; Azeggagh, S.; Harvey, K. LRRK2 Biology from structure to dysfunction: Research progresses, but the themes remain the same. Mol. Neurodegener. 2019, 14, 49. [Google Scholar] [CrossRef]
- Howlett, E.H.; Jensen, N.; Belmonte, F.; Zafar, F.; Hu, X.; Kluss, J.; Schüle, B.; Kaufman, B.A.; Greenamyre, J.T.; Sanders, L.H. LRRK2 G2019S-induced mitochondrial DNA damage is LRRK2 kinase dependent and inhibition restores mtDNA integrity in Parkinson’s disease. Hum. Mol. Genet. 2017, 26, 4340–4351. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.D.; Shin, J.H.; VanKampen, J.; Petrucelli, L.; West, A.B.; Ko, H.S.; Lee, Y.I.; Maguire-Zeiss, K.A.; Bowers, W.J.; Federoff, H.J.; et al. Inhibitors of leucine-rich repeat kinase-2 protect against models of Parkinson’s disease. Nat. Med. 2010, 16, 998–1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Maio, R.; Hoffman, E.K.; Rocha, E.M.; Keeney, M.T.; Sanders, L.H.; De Miranda, B.R.; Zharikov, A.; Van Laar, A.; Stepan, A.F.; Lanz, T.A.; et al. LRRK2 activation in idiopathic Parkinson’s disease. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, M.A.; Wegener, K.M.; Larsen, S.; Badolo, L.; Smith, G.P.; Jeggo, R.; Jensen, P.H.; Sotty, F.; Christensen, K.V.; Thougaard, A. PFE-360-induced LRRK2 inhibition induces reversible, non-adverse renal changes in rats. Toxicology 2018, 395, 15–22. [Google Scholar] [CrossRef]
- Lebovitz, C.; Wretham, N.; Osooly, M.; Milne, K.; Dash, T.; Thornton, S.; Tessier-Cloutier, B.; Sathiyaseelan, P.; Bortnik, S.; Go, N.E.; et al. Loss of Parkinson’s susceptibility gene LRRK2 promotes carcinogen-induced lung tumorigenesis. Sci. Rep. 2021, 11, 2097. [Google Scholar] [CrossRef]
- Whiffin, N.; Armean, I.M.; Kleinman, A.; Marshall, J.L.; Minikel, E.V.; Goodrich, J.K.; Quaife, N.M.; Cole, J.B.; Wang, Q.; Karczewski, K.J.; et al. The effect of LRRK2 loss-of-function variants in humans. Nat. Med. 2020, 26, 869–877. [Google Scholar] [CrossRef] [PubMed]
- Blauwendraat, C.; Reed, X.; Kia, D.A.; Gan-Or, Z.; Lesage, S.; Pihlstrøm, L.; Guerreiro, R.; Gibbs, J.R.; Sabir, M.; Ahmed, S.; et al. Frequency of Loss of Function Variants in LRRK2 in Parkinson Disease. JAMA Neurol. 2018, 75, 1416–1422. [Google Scholar] [CrossRef] [PubMed]
- Neudorfer, O.; Giladi, N.; Elstein, D.; Abrahamov, A.; Turezkite, T.; Aghai, E.; Reches, A.; Bembi, B.; Zimran, A. Occurrence of Parkinson’s syndrome in type I Gaucher disease. QJM 1996, 89, 691–694. [Google Scholar] [CrossRef] [PubMed]
- Goker-Alpan, O.; Schiffmann, R.; LaMarca, M.E.; Nussbaum, R.L.; McInerney-Leo, A.; Sidransky, E. Parkinsonism among Gaucher disease carriers. J. Med. Genet. 2004, 41, 937–940. [Google Scholar] [CrossRef] [Green Version]
- Sidransky, E.; Nalls, M.A.; Aasly, J.O.; Aharon-Peretz, J.; Annesi, G.; Barbosa, E.R.; Bar-Shira, A.; Berg, D.; Bras, J.; Brice, A.; et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N. Engl. J. Med. 2009, 361, 1651–1661. [Google Scholar] [CrossRef] [Green Version]
- Hruska, K.S.; LaMarca, M.E.; Scott, C.R.; Sidransky, E. Gaucher disease: Mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA). Hum. Mutat. 2008, 29, 567–583. [Google Scholar] [CrossRef]
- CENTOGENE. CENTOGENE. The Rare Disease Company. Introducing CentoLSD™. 2021. Available online: https://www.centogene.com/es/centolsd.html (accessed on 26 June 2021).
- Alcalay, R.N.; Dinur, T.; Quinn, T.; Sakanaka, K.; Levy, O.; Waters, C.; Fahn, S.; Dorovski, T.; Chung, W.K.; Pauciulo, M.; et al. Comparison of Parkinson risk in Ashkenazi Jewish patients with Gaucher disease and GBA heterozygotes. JAMA Neurol. 2014, 71, 752–757. [Google Scholar] [CrossRef] [Green Version]
- Anheim, M.; Elbaz, A.; Lesage, S.; Durr, A.; Condroyer, C.; Viallet, F.; Pollak, P.; Bonaïti, B.; Bonaïti-Pellié, C.; Brice, A.; et al. Penetrance of Parkinson disease in glucocerebrosidase gene mutation carriers. Neurology 2012, 78, 417–420. [Google Scholar] [CrossRef]
- Sidransky, E.; Hart, P.S. Penetrance of PD in Glucocerebrosidase Gene Mutation Carriers. Neurology 2012, 79, 106–107. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Shu, L.; Sun, Q.; Zhou, X.; Pan, H.; Guo, J.; Tang, B. Integrated Genetic Analysis of Racial Differences of Common. Front. Mol. Neurosci. 2018, 11, 43. [Google Scholar] [CrossRef]
- Gan-Or, Z.; Amshalom, I.; Kilarski, L.L.; Bar-Shira, A.; Gana-Weisz, M.; Mirelman, A.; Marder, K.; Bressman, S.; Giladi, N.; Orr-Urtreger, A. Differential effects of severe vs mild GBA mutations on Parkinson disease. Neurology 2015, 84, 880–887. [Google Scholar] [CrossRef] [Green Version]
- Mallett, V.; Ross, J.P.; Alcalay, R.N.; Ambalavanan, A.; Sidransky, E.; Dion, P.A.; Rouleau, G.A.; Gan-Or, Z. GBA p.T369M substitution in Parkinson disease: Polymorphism or association? A meta-analysis. Neurol. Genet. 2016, 2, e104. [Google Scholar] [CrossRef] [Green Version]
- Duran, R.; Mencacci, N.E.; Angeli, A.V.; Shoai, M.; Deas, E.; Houlden, H.; Mehta, A.; Hughes, D.; Cox, T.M.; Deegan, P.; et al. The glucocerobrosidase E326K variant predisposes to Parkinson’s disease, but does not cause Gaucher’s disease. Mov. Disord. 2013, 28, 232–236. [Google Scholar] [CrossRef] [Green Version]
- Park, J.K.; Tayebi, N.; Stubblefield, B.K.; LaMarca, M.E.; MacKenzie, J.J.; Stone, D.L.; Sidransky, E. The E326K mutation and Gaucher disease: Mutation or polymorphism? Clin. Genet. 2002, 61, 32–34. [Google Scholar] [CrossRef]
- Blauwendraat, C.; Reed, X.; Krohn, L.; Heilbron, K.; Bandres-Ciga, S.; Tan, M.; Gibbs, J.R.; Hernandez, D.G.; Kumaran, R.; Langston, R.; et al. Genetic modifiers of risk and age at onset in GBA associated Parkinson’s disease and Lewy body dementia. Brain 2020, 143, 234–248. [Google Scholar] [CrossRef]
- Ruskey, J.A.; Greenbaum, L.; Roncière, L.; Alam, A.; Spiegelman, D.; Liong, C.; Levy, O.A.; Waters, C.; Fahn, S.; Marder, K.S.; et al. Increased yield of full GBA sequencing in Ashkenazi Jews with Parkinson’s disease. Eur. J. Med. Genet. 2019, 62, 65–69. [Google Scholar] [CrossRef]
- Zampieri, S.; Cattarossi, S.; Bembi, B.; Dardis, A. GBA Analysis in Next-Generation Era: Pitfalls, Challenges, and Possible Solutions. J. Mol. Diagn. 2017, 19, 733–741. [Google Scholar] [CrossRef] [Green Version]
- Leija-Salazar, M.; Sedlazeck, F.J.; Toffoli, M.; Mullin, S.; Mokretar, K.; Athanasopoulou, M.; Donald, A.; Sharma, R.; Hughes, D.; Schapira, A.H.V.; et al. Evaluation of the detection of GBA missense mutations and other variants using the Oxford Nanopore MinION. Mol. Genet. Genom. Med. 2019, 7, e564. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Shu, L.; Zhou, X.; Pan, H.; Xu, Q.; Guo, J.; Tang, B.; Sun, Q. A Meta-Analysis of. Parkinsons Dis. 2018, 2018, 3136415. [Google Scholar] [CrossRef]
- Gan-Or, Z.; Giladi, N.; Rozovski, U.; Shifrin, C.; Rosner, S.; Gurevich, T.; Bar-Shira, A.; Orr-Urtreger, A. Genotype-phenotype correlations between GBA mutations and Parkinson disease risk and onset. Neurology 2008, 70, 2277–2283. [Google Scholar] [CrossRef]
- Maple-Grødem, J.; Dalen, I.; Tysnes, O.B.; Macleod, A.D.; Forsgren, L.; Counsell, C.E.; Alves, G. Association of GBA Genotype with Motor and Functional Decline in Newly Diagnosed Patients with Parkinsons Disease. Neurology 2020. [Google Scholar] [CrossRef]
- Cilia, R.; Tunesi, S.; Marotta, G.; Cereda, E.; Siri, C.; Tesei, S.; Zecchinelli, A.L.; Canesi, M.; Mariani, C.B.; Meucci, N.; et al. Survival and dementia in GBA-associated Parkinson’s disease: The mutation matters. Ann. Neurol. 2016, 80, 662–673. [Google Scholar] [CrossRef]
- Jesús, S.; Huertas, I.; Bernal-Bernal, I.; Bonilla-Toribio, M.; Cáceres-Redondo, M.T.; Vargas-González, L.; Gómez-Llamas, M.; Carrillo, F.; Calderón, E.; Carballo, M.; et al. GBA Variants Influence Motor and Non-Motor Features of Parkinson’s Disease. PLoS ONE 2016, 11, e0167749. [Google Scholar] [CrossRef]
- Iwaki, H.; Blauwendraat, C.; Leonard, H.L.; Kim, J.J.; Liu, G.; Maple-Grødem, J.; Corvol, J.C.; Pihlstrøm, L.; van Nimwegen, M.; Hutten, S.J.; et al. Genomewide association study of Parkinson’s disease clinical biomarkers in 12 longitudinal patients’ cohorts. Mov. Disord. 2019, 34, 1839–1850. [Google Scholar] [CrossRef] [PubMed]
- Iwaki, H.; Blauwendraat, C.; Leonard, H.L.; Liu, G.; Maple-Grødem, J.; Corvol, J.C.; Pihlstrøm, L.; van Nimwegen, M.; Hutten, S.J.; Nguyen, K.H.; et al. Genetic risk of Parkinson disease and progression: An analysis of 13 longitudinal cohorts. Neurol. Genet. 2019, 5, e348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gan-Or, Z.; Liong, C.; Alcalay, R.N. GBA-Associated Parkinson’s Disease and Other Synucleinopathies. Curr. Neurol. Neurosci. Rep. 2018, 18, 44. [Google Scholar] [CrossRef] [PubMed]
- Alcalay, R.N.; Levy, O.A.; Waters, C.C.; Fahn, S.; Ford, B.; Kuo, S.H.; Mazzoni, P.; Pauciulo, M.W.; Nichols, W.C.; Gan-Or, Z.; et al. Glucocerebrosidase activity in Parkinson’s disease with and without GBA mutations. Brain 2015, 138, 2648–2658. [Google Scholar] [CrossRef] [Green Version]
- Hallett, P.J.; Huebecker, M.; Brekk, O.R.; Moloney, E.B.; Rocha, E.M.; Priestman, D.A.; Platt, F.M.; Isacson, O. Glycosphingolipid levels and glucocerebrosidase activity are altered in normal aging of the mouse brain. Neurobiol. Aging 2018, 67, 189–200. [Google Scholar] [CrossRef]
- Parnetti, L.; Chiasserini, D.; Persichetti, E.; Eusebi, P.; Varghese, S.; Qureshi, M.M.; Dardis, A.; Deganuto, M.; De Carlo, C.; Castrioto, A.; et al. Cerebrospinal fluid lysosomal enzymes and alpha-synuclein in Parkinson’s disease. Mov. Disord. 2014, 29, 1019–1027. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Clark, L.N.; Louis, E.D.; Mejia-Santana, H.; Harris, J.; Cote, L.J.; Waters, C.; Andrews, H.; Ford, B.; Frucht, S.; et al. Risk of Parkinson disease in carriers of parkin mutations: Estimation using the kin-cohort method. Arch. Neurol. 2008, 65, 467–474. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, S.A.; Scott, W.K.; Martin, E.R.; Nance, M.A.; Watts, R.L.; Hubble, J.P.; Koller, W.C.; Pahwa, R.; Stern, M.B.; Hiner, B.C.; et al. Parkin mutations and susceptibility alleles in late-onset Parkinson’s disease. Ann. Neurol. 2003, 53, 624–629. [Google Scholar] [CrossRef]
- Lücking, C.B.; Dürr, A.; Bonifati, V.; Vaughan, J.; De Michele, G.; Gasser, T.; Harhangi, B.S.; Meco, G.; Denèfle, P.; Wood, N.W.; et al. Association between early-onset Parkinson’s disease and mutations in the parkin gene. N. Engl. J. Med. 2000, 342, 1560–1567. [Google Scholar] [CrossRef]
- Yu, E.; Rudakou, U.; Krohn, L.; Mufti, K.; Ruskey, J.A.; Asayesh, F.; Estiar, M.A.; Spiegelman, D.; Surface, M.; Fahn, S.; et al. Analysis of Heterozygous PRKN Variants and Copy-Number Variations in Parkinson’s Disease. Mov. Disord. 2021, 36, 178–187. [Google Scholar] [CrossRef]
- MacArthur, J.; Bowler, E.; Cerezo, M.; Gil, L.; Hall, P.; Hastings, E.; Junkins, H.; McMahon, A.; Milano, A.; Morales, J.; et al. The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res. 2017, 45, D896–D901. [Google Scholar] [CrossRef]
- Yengo, L.; Sidorenko, J.; Kemper, K.E.; Zheng, Z.; Wood, A.R.; Weedon, M.N.; Frayling, T.M.; Hirschhorn, J.; Yang, J.; Visscher, P.M.; et al. Meta-analysis of genome-wide association studies for height and body mass index in ∼700,000 individuals of European ancestry. Hum. Mol. Genet. 2018, 27, 3641–3649. [Google Scholar] [CrossRef]
- Vujkovic, M.; Keaton, J.M.; Lynch, J.A.; Miller, D.R.; Zhou, J.; Tcheandjieu, C.; Huffman, J.E.; Assimes, T.L.; Lorenz, K.; Zhu, X.; et al. Discovery of 318 new risk loci for type 2 diabetes and related vascular outcomes among 1.4 million participants in a multi-ancestry meta-analysis. Nat. Genet. 2020, 52, 680–691. [Google Scholar] [CrossRef]
- Kao, P.Y.; Leung, K.H.; Chan, L.W.; Yip, S.P.; Yap, M.K. Pathway analysis of complex diseases for GWAS, extending to consider rare variants, multi-omics and interactions. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 335–353. [Google Scholar] [CrossRef] [Green Version]
- Nalls, M.A.; Pankratz, N.; Lill, C.M.; Do, C.B.; Hernandez, D.G.; Saad, M.; DeStefano, A.L.; Kara, E.; Bras, J.; Sharma, M.; et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat. Genet. 2014, 46, 989–993. [Google Scholar] [CrossRef]
- Escott-Price, V.; Nalls, M.A.; Morris, H.R.; Lubbe, S.; Brice, A.; Gasser, T.; Heutink, P.; Wood, N.W.; Hardy, J.; Singleton, A.B.; et al. Polygenic risk of Parkinson disease is correlated with disease age at onset. Ann. Neurol. 2015, 77, 582–591. [Google Scholar] [CrossRef] [Green Version]
- Ibanez, L.; Dube, U.; Saef, B.; Budde, J.; Black, K.; Medvedeva, A.; Del-Aguila, J.L.; Davis, A.A.; Perlmutter, J.S.; Harari, O.; et al. Parkinson disease polygenic risk score is associated with Parkinson disease status and age at onset but not with alpha-synuclein cerebrospinal fluid levels. BMC Neurol. 2017, 17, 198. [Google Scholar] [CrossRef]
- Li, W.W.; Fan, D.Y.; Shen, Y.Y.; Zhou, F.Y.; Chen, Y.; Wang, Y.R.; Yang, H.; Mei, J.; Li, L.; Xu, Z.Q.; et al. Association of the Polygenic Risk Score with the Incidence Risk of Parkinson’s Disease and Cerebrospinal Fluid α-Synuclein in a Chinese Cohort. Neurotox. Res. 2019, 36, 515–522. [Google Scholar] [CrossRef]
- Jacobs, B.M.; Belete, D.; Bestwick, J.; Blauwendraat, C.; Bandres-Ciga, S.; Heilbron, K.; Dobson, R.; Nalls, M.A.; Singleton, A.; Hardy, J.; et al. Parkinson’s disease determinants, prediction and gene-environment interactions in the UK Biobank. J. Neurol. Neurosurg. Psychiatry 2020, 91, 1046–1054. [Google Scholar] [CrossRef]
- Foo, J.N.; Chew, E.G.Y.; Chung, S.J.; Peng, R.; Blauwendraat, C.; Nalls, M.A.; Mok, K.Y.; Satake, W.; Toda, T.; Chao, Y.; et al. Identification of Risk Loci for Parkinson Disease in Asians and Comparison of Risk Between Asians and Europeans: A Genome-Wide Association Study. JAMA Neurol. 2020, 77, 746–754. [Google Scholar] [CrossRef]
- Molparia, B.; Schrader, B.N.; Cohen, E.; Wagner, J.L.; Gupta, S.R.; Gould, S.; Hwynn, N.; Spencer, E.G.; Torkamani, A. Combined accelerometer and genetic analysis to differentiate essential tremor from Parkinson’s disease. PeerJ 2018, 6, e5308. [Google Scholar] [CrossRef] [Green Version]
- Bandres-Ciga, S.; Saez-Atienzar, S.; Kim, J.J.; Makarious, M.B.; Faghri, F.; Diez-Fairen, M.; Iwaki, H.; Leonard, H.; Botia, J.; Ryten, M.; et al. Large-scale pathway specific polygenic risk and transcriptomic community network analysis identifies novel functional pathways in Parkinson disease. Acta Neuropathol. 2020, 140, 341–358. [Google Scholar] [CrossRef]
- Pankratz, N.; Beecham, G.W.; DeStefano, A.L.; Dawson, T.M.; Doheny, K.F.; Factor, S.A.; Hamza, T.H.; Hung, A.Y.; Hyman, B.T.; Ivinson, A.J.; et al. Meta-analysis of Parkinson’s disease: Identification of a novel locus, RIT2. Ann. Neurol. 2012, 71, 370–384. [Google Scholar] [CrossRef] [Green Version]
- Rudakou, U.; Yu, E.; Krohn, L.; Ruskey, J.A.; Asayesh, F.; Dauvilliers, Y.; Spiegelman, D.; Greenbaum, L.; Fahn, S.; Waters, C.H.; et al. Targeted sequencing of Parkinson’s disease loci genes highlights SYT11, FGF20 and other associations. Brain 2021, 144, 462–472. [Google Scholar] [CrossRef]
- Fung, H.C.; Scholz, S.; Matarin, M.; Simón-Sánchez, J.; Hernandez, D.; Britton, A.; Gibbs, J.R.; Langefeld, C.; Stiegert, M.L.; Schymick, J.; et al. Genome-wide genotyping in Parkinson’s disease and neurologically normal controls: First stage analysis and public release of data. Lancet Neurol. 2006, 5, 911–916. [Google Scholar] [CrossRef]
- Pankratz, N.; Wilk, J.B.; Latourelle, J.C.; DeStefano, A.L.; Halter, C.; Pugh, E.W.; Doheny, K.F.; Gusella, J.F.; Nichols, W.C.; Foroud, T.; et al. Genomewide association study for susceptibility genes contributing to familial Parkinson disease. Hum. Genet. 2009, 124, 593–605. [Google Scholar] [CrossRef] [Green Version]
- Latourelle, J.C.; Pankratz, N.; Dumitriu, A.; Wilk, J.B.; Goldwurm, S.; Pezzoli, G.; Mariani, C.B.; DeStefano, A.L.; Halter, C.; Gusella, J.F.; et al. Genomewide association study for onset age in Parkinson disease. BMC Med. Genet. 2009, 10, 98. [Google Scholar] [CrossRef] [Green Version]
- Satake, W.; Nakabayashi, Y.; Mizuta, I.; Hirota, Y.; Ito, C.; Kubo, M.; Kawaguchi, T.; Tsunoda, T.; Watanabe, M.; Takeda, A.; et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat. Genet. 2009, 41, 1303–1307. [Google Scholar] [CrossRef]
- Simón-Sánchez, J.; Schulte, C.; Bras, J.M.; Sharma, M.; Gibbs, J.R.; Berg, D.; Paisan-Ruiz, C.; Lichtner, P.; Scholz, S.W.; Hernandez, D.G.; et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat. Genet. 2009, 41, 1308–1312. [Google Scholar] [CrossRef]
- Edwards, T.L.; Scott, W.K.; Almonte, C.; Burt, A.; Powell, E.H.; Beecham, G.W.; Wang, L.; Züchner, S.; Konidari, I.; Wang, G.; et al. Genome-wide association study confirms SNPs in SNCA and the MAPT region as common risk factors for Parkinson disease. Ann. Hum. Genet. 2010, 74, 97–109. [Google Scholar] [CrossRef] [Green Version]
- Hamza, T.H.; Zabetian, C.P.; Tenesa, A.; Laederach, A.; Montimurro, J.; Yearout, D.; Kay, D.M.; Doheny, K.F.; Paschall, J.; Pugh, E.; et al. Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson’s disease. Nat. Genet. 2010, 42, 781–785. [Google Scholar] [CrossRef] [PubMed]
- Saad, M.; Lesage, S.; Saint-Pierre, A.; Corvol, J.C.; Zelenika, D.; Lambert, J.C.; Vidailhet, M.; Mellick, G.D.; Lohmann, E.; Durif, F.; et al. Genome-wide association study confirms BST1 and suggests a locus on 12q24 as the risk loci for Parkinson’s disease in the European population. Hum. Mol. Genet. 2011, 20, 615–627. [Google Scholar] [CrossRef] [PubMed]
- Simón-Sánchez, J.; van Hilten, J.J.; van de Warrenburg, B.; Post, B.; Berendse, H.W.; Arepalli, S.; Hernandez, D.G.; de Bie, R.M.; Velseboer, D.; Scheffer, H.; et al. Genome-wide association study confirms extant PD risk loci among the Dutch. Eur. J. Hum. Genet. 2011, 19, 655–661. [Google Scholar] [CrossRef]
- Liu, X.; Cheng, R.; Verbitsky, M.; Kisselev, S.; Browne, A.; Mejia-Sanatana, H.; Louis, E.D.; Cote, L.J.; Andrews, H.; Waters, C.; et al. Genome-wide association study identifies candidate genes for Parkinson’s disease in an Ashkenazi Jewish population. BMC Med. Genet. 2011, 12, 104. [Google Scholar] [CrossRef] [Green Version]
- Spencer, C.C.; Plagnol, V.; Strange, A.; Gardner, M.; Paisan-Ruiz, C.; Band, G.; Barker, R.A.; Bellenguez, C.; Bhatia, K.; Blackburn, H.; et al. Dissection of the genetics of Parkinson’s disease identifies an additional association 5’ of SNCA and multiple associated haplotypes at 17q21. Hum. Mol. Genet. 2011, 20, 345–353. [Google Scholar] [CrossRef] [Green Version]
- Nalls, M.A.; Plagnol, V.; Hernandez, D.G.; Sharma, M.; Sheerin, U.M.; Saad, M.; Simón-Sánchez, J.; Schulte, C.; Lesage, S.; Sveinbjörnsdóttir, S.; et al. Imputation of sequence variants for identification of genetic risks for Parkinson’s disease: A meta-analysis of genome-wide association studies. Lancet 2011, 377, 641–649. [Google Scholar] [CrossRef] [Green Version]
- Do, C.B.; Tung, J.Y.; Dorfman, E.; Kiefer, A.K.; Drabant, E.M.; Francke, U.; Mountain, J.L.; Goldman, S.M.; Tanner, C.M.; Langston, J.W.; et al. Web-based genome-wide association study identifies two novel loci and a substantial genetic component for Parkinson’s disease. PLoS Genet. 2011, 7, e1002141. [Google Scholar] [CrossRef] [Green Version]
- Chung, S.J.; Armasu, S.M.; Biernacka, J.M.; Anderson, K.J.; Lesnick, T.G.; Rider, D.N.; Cunningham, J.M.; Eric Ahlskog, J.; Frigerio, R.; Maraganore, D.M. Genomic determinants of motor and cognitive outcomes in Parkinson’s disease. Parkinsonism Relat. Disord. 2012, 18, 881–886. [Google Scholar] [CrossRef] [Green Version]
- Hernandez, D.G.; Nalls, M.A.; Ylikotila, P.; Keller, M.; Hardy, J.A.; Majamaa, K.; Singleton, A.B. Genome wide assessment of young onset Parkinson’s disease from Finland. PLoS ONE 2012, 7, e41859. [Google Scholar] [CrossRef] [Green Version]
- Davis, M.F.; Cummings, A.C.; D’Aoust, L.N.; Jiang, L.; Velez Edwards, D.R.; Laux, R.; Reinhart-Mercer, L.; Fuzzell, D.; Scott, W.K.; Pericak-Vance, M.A.; et al. Parkinson disease loci in the mid-western Amish. Hum. Genet. 2013, 132, 1213–1221. [Google Scholar] [CrossRef] [Green Version]
- Hill-Burns, E.M.; Wissemann, W.T.; Hamza, T.H.; Factor, S.A.; Zabetian, C.P.; Payami, H. Identification of a novel Parkinson’s disease locus via stratified genome-wide association study. BMC Genom. 2014, 15, 118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Deng, L.; Zhang, J.; Fang, X.; Mei, P.; Cao, X.; Lin, J.; Wei, Y.; Zhang, X.; Xu, R. A Pooling Genome-Wide Association Study Combining a Pathway Analysis for Typical Sporadic Parkinson’s Disease in the Han Population of Chinese Mainland. Mol. Neurobiol. 2016, 53, 4302–4318. [Google Scholar] [CrossRef] [PubMed]
- Hill-Burns, E.M.; Ross, O.A.; Wissemann, W.T.; Soto-Ortolaza, A.I.; Zareparsi, S.; Siuda, J.; Lynch, T.; Wszolek, Z.K.; Silburn, P.A.; Mellick, G.D.; et al. Identification of genetic modifiers of age-at-onset for familial Parkinson’s disease. Hum. Mol. Genet. 2016, 25, 3849–3862. [Google Scholar] [CrossRef] [Green Version]
- Siitonen, A.; Nalls, M.A.; Hernández, D.; Gibbs, J.R.; Ding, J.; Ylikotila, P.; Edsall, C.; Singleton, A.; Majamaa, K. Genetics of early-onset Parkinson’s disease in Finland: Exome sequencing and genome-wide association study. Neurobiol. Aging 2017, 53, 195.e7–195.e10. [Google Scholar] [CrossRef] [Green Version]
- Foo, J.N.; Tan, L.C.; Irwan, I.D.; Au, W.L.; Low, H.Q.; Prakash, K.M.; Ahmad-Annuar, A.; Bei, J.; Chan, A.Y.; Chen, C.M.; et al. Genome-wide association study of Parkinson’s disease in East Asians. Hum. Mol. Genet. 2017, 26, 226–232. [Google Scholar] [CrossRef] [Green Version]
- Chang, D.; Nalls, M.A.; Hallgrímsdóttir, I.B.; Hunkapiller, J.; van der Brug, M.; Cai, F.; Kerchner, G.A.; Ayalon, G.; Bingol, B.; Sheng, M.; et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat. Genet. 2017, 49, 1511–1516. [Google Scholar] [CrossRef]
- Wallen, Z.D.; Chen, H.; Hill-Burns, E.M.; Factor, S.A.; Zabetian, C.P.; Payami, H. Plasticity-related gene 3. Neurol. Genet. 2018, 4, e271. [Google Scholar] [CrossRef] [Green Version]
- Blauwendraat, C.; Heilbron, K.; Vallerga, C.L.; Bandres-Ciga, S.; von Coelln, R.; Pihlstrøm, L.; Simón-Sánchez, J.; Schulte, C.; Sharma, M.; Krohn, L.; et al. Parkinson’s disease age at onset genome-wide association study: Defining heritability, genetic loci, and α-synuclein mechanisms. Mov. Disord. 2019, 34, 866–875. [Google Scholar] [CrossRef] [Green Version]
- Bandres-Ciga, S.; Ahmed, S.; Sabir, M.S.; Blauwendraat, C.; Adarmes-Gómez, A.D.; Bernal-Bernal, I.; Bonilla-Toribio, M.; Buiza-Rueda, D.; Carrillo, F.; Carrión-Claro, M.; et al. The Genetic Architecture of Parkinson Disease in Spain: Characterizing Population-Specific Risk, Differential Haplotype Structures, and Providing Etiologic Insight. Mov. Disord. 2019, 34, 1851–1863. [Google Scholar] [CrossRef]
- Ryu, H.S.; Park, K.W.; Choi, N.; Kim, J.; Park, Y.M.; Jo, S.; Kim, M.J.; Kim, Y.J.; Kim, K.; Koh, S.B.; et al. Genomic Analysis Identifies New Loci Associated With Motor Complications in Parkinson’s Disease. Front. Neurol. 2020, 11, 570. [Google Scholar] [CrossRef]
- Cha, P.C.; Satake, W.; Ando-Kanagawa, Y.; Yamamoto, K.; Murata, M.; Toda, T. Genome-wide association study identifies zonisamide responsive gene in Parkinson’s disease patients. J. Hum. Genet. 2020, 65, 693–704. [Google Scholar] [CrossRef]
- Tan, M.M.X.; Lawton, M.A.; Jabbari, E.; Reynolds, R.H.; Iwaki, H.; Blauwendraat, C.; Kanavou, S.; Pollard, M.I.; Hubbard, L.; Malek, N.; et al. Genome-Wide Association Studies of Cognitive and Motor Progression in Parkinson’s Disease. Mov. Disord. 2020. [Google Scholar] [CrossRef]
- Toffoli, M.; Vieira, S.R.L.; Schapira, A.H.V. Genetic causes of PD: A pathway to disease modification. Neuropharmacology 2020, 170, 108022. [Google Scholar] [CrossRef]
- Mullin, S.; Smith, L.; Lee, K.; D’Souza, G.; Woodgate, P.; Elflein, J.; Hällqvist, J.; Toffoli, M.; Streeter, A.; Hosking, J.; et al. Ambroxol for the Treatment of Patients With Parkinson Disease With and Without Glucocerebrosidase Gene Mutations: A Nonrandomized, Noncontrolled Trial. JAMA Neurol. 2020, 77, 427–434. [Google Scholar] [CrossRef] [Green Version]
- Silveira, C.R.A.; MacKinley, J.; Coleman, K.; Li, Z.; Finger, E.; Bartha, R.; Morrow, S.A.; Wells, J.; Borrie, M.; Tirona, R.G.; et al. Ambroxol as a novel disease-modifying treatment for Parkinson’s disease dementia: Protocol for a single-centre, randomized, double-blind, placebo-controlled trial. BMC Neurol. 2019, 19, 20. [Google Scholar] [CrossRef] [Green Version]
- Sardi, S.P.; Viel, C.; Clarke, J.; Treleaven, C.M.; Richards, A.M.; Park, H.; Olszewski, M.A.; Dodge, J.C.; Marshall, J.; Makino, E.; et al. Glucosylceramide synthase inhibition alleviates aberrations in synucleinopathy models. Proc. Natl. Acad. Sci. USA 2017, 114, 2699–2704. [Google Scholar] [CrossRef] [Green Version]
- Sanofi. PRESS RELEASES. 5 February 2021. Available online: https://www.sanofi.com/en/media-room/press-releases/2021/2021-02-05-07-30-00 (accessed on 26 June 2021).
- Sheehan, P.; Heckman, L.D.; Fenn, T.; Wong, L.C.; Nelson, S.; Garimalla, S.; Haller, J.; Daily, J.; Politi, J.; Dai, Y.; et al. PR001 gene therapy improved phenotypes in models of Parkinson’s disease with GBA1 mutation. Alzheimer’s Dement. 2020, 16. [Google Scholar] [CrossRef]
- Jeon, J.Y.; Sparreboom, A.; Baker, S.D. Kinase Inhibitors: The Reality Behind the Success. Clin. Pharm. 2017, 102, 726–730. [Google Scholar] [CrossRef]
- Jennings, D.; Wetering de Rooij, J.; Vissers, M.; Heuberger, J.; Groeneveld, G.; Maciuca, R.; Kay, A.; Borin, M.; Wong, B.; Daryani, V.; et al. P54: LRRK2 Inhibition by BIIB122 / DNL151 in Double-Blind, Placebo-Controlled Phase 1 Healthy Volunteer and Phase 1B. In Proceedings of the XXVI World Congress on Parkinson’s Disease and Related Disorders, Prague, Czech Republic, 1–4 May 2021. [Google Scholar]
- Zhao, H.T.; John, N.; Delic, V.; Ikeda-Lee, K.; Kim, A.; Weihofen, A.; Swayze, E.E.; Kordasiewicz, H.B.; West, A.B.; Volpicelli-Daley, L.A. LRRK2 Antisense Oligonucleotides Ameliorate α-Synuclein Inclusion Formation in a Parkinson’s Disease Mouse Model. Mol. Nucleic Acids 2017, 8, 508–519. [Google Scholar] [CrossRef] [Green Version]
- Volc, D.; Poewe, W.; Kutzelnigg, A.; Lührs, P.; Thun-Hohenstein, C.; Schneeberger, A.; Galabova, G.; Majbour, N.; Vaikath, N.; El-Agnaf, O.; et al. Safety and immunogenicity of the α-synuclein active immunotherapeutic PD01A in patients with Parkinson’s disease: A randomised, single-blinded, phase 1 trial. Lancet Neurol. 2020, 19, 591–600. [Google Scholar] [CrossRef]
- Simuni, T.; Fiske, B.; Merchant, K.; Coffey, C.S.; Klingner, E.; Caspell-Garcia, C.; Lafontant, D.E.; Matthews, H.; Wyse, R.K.; Brundin, P.; et al. Efficacy of Nilotinib in Patients With Moderately Advanced Parkinson Disease: A Randomized Clinical Trial. JAMA Neurol. 2020. [Google Scholar] [CrossRef]
- Payne, T.; Sassani, M.; Buckley, E.; Moll, S.; Anton, A.; Appleby, M.; Maru, S.; Taylor, R.; McNeill, A.; Hoggard, N.; et al. Ursodeoxycholic acid as a novel disease-modifying treatment for Parkinson’s disease: Protocol for a two-centre, randomised, double-blind, placebo-controlled trial, The ‘UP’ study. BMJ Open 2020, 10, e038911. [Google Scholar] [CrossRef] [PubMed]
- De Franco, E.; Flanagan, S.E.; Houghton, J.A.; Lango Allen, H.; Mackay, D.J.; Temple, I.K.; Ellard, S.; Hattersley, A.T. The effect of early, comprehensive genomic testing on clinical care in neonatal diabetes: An international cohort study. Lancet 2015, 386, 957–963. [Google Scholar] [CrossRef] [Green Version]
- Bowman, P.; Sulen, Å.; Barbetti, F.; Beltrand, J.; Svalastoga, P.; Codner, E.; Tessmann, E.H.; Juliusson, P.B.; Skrivarhaug, T.; Pearson, E.R.; et al. Effectiveness and safety of long-term treatment with sulfonylureas in patients with neonatal diabetes due to KCNJ11 mutations: An international cohort study. Lancet Diabetes Endocrinol. 2018, 6, 637–646. [Google Scholar] [CrossRef]
- Bowman, P.; Mathews, F.; Barbetti, F.; Shepherd, M.H.; Sanchez, J.; Piccini, B.; Beltrand, J.; Letourneau-Freiberg, L.R.; Polak, M.; Greeley, S.A.W.; et al. Long-term Follow-up of Glycemic and Neurological Outcomes in an International Series of Patients with Sulfonylurea-Treated. Diabetes Care 2021, 44, 35–42. [Google Scholar] [CrossRef]
- Bacon, S.; Kyithar, M.P.; Rizvi, S.R.; Donnelly, E.; McCarthy, A.; Burke, M.; Colclough, K.; Ellard, S.; Byrne, M.M. Successful maintenance on sulphonylurea therapy and low diabetes complication rates in a HNF1A-MODY cohort. Diabet. Med. 2016, 33, 976–984. [Google Scholar] [CrossRef]
- Shepherd, M.; Shields, B.; Ellard, S.; Rubio-Cabezas, O.; Hattersley, A.T. A genetic diagnosis of HNF1A diabetes alters treatment and improves glycaemic control in the majority of insulin-treated patients. Diabet. Med. 2009, 26, 437–441. [Google Scholar] [CrossRef]
- Stride, A.; Shields, B.; Gill-Carey, O.; Chakera, A.J.; Colclough, K.; Ellard, S.; Hattersley, A.T. Cross-sectional and longitudinal studies suggest pharmacological treatment used in patients with glucokinase mutations does not alter glycaemia. Diabetologia 2014, 57, 54–56. [Google Scholar] [CrossRef] [Green Version]
- Lynam, A.; McDonald, T.; Hill, A.; Dennis, J.; Oram, R.; Pearson, E.; Weedon, M.; Hattersley, A.; Owen, K.; Shields, B.; et al. Development and validation of multivariable clinical diagnostic models to identify type 1 diabetes requiring rapid insulin therapy in adults aged 18-50 years. BMJ Open 2019, 9, e031586. [Google Scholar] [CrossRef] [Green Version]
- Redenšek, S.; Dolžan, V. The role of pharmacogenomics in the personalization of Parkinson’s disease treatment. Pharmacogenomics 2020, 21, 1033–1043. [Google Scholar] [CrossRef] [PubMed]
- Contin, M.; Martinelli, P.; Mochi, M.; Riva, R.; Albani, F.; Baruzzi, A. Genetic polymorphism of catechol-O-methyltransferase and levodopa pharmacokinetic-pharmacodynamic pattern in patients with Parkinson’s disease. Mov. Disord. 2005, 20, 734–739. [Google Scholar] [CrossRef]
- Corvol, J.C.; Bonnet, C.; Charbonnier-Beaupel, F.; Bonnet, A.M.; Fiévet, M.H.; Bellanger, A.; Roze, E.; Meliksetyan, G.; Ben Djebara, M.; Hartmann, A.; et al. The COMT Val158Met polymorphism affects the response to entacapone in Parkinson’s disease: A randomized crossover clinical trial. Ann. Neurol. 2011, 69, 111–118. [Google Scholar] [CrossRef]
- Kraemmer, J.; Smith, K.; Weintraub, D.; Guillemot, V.; Nalls, M.A.; Cormier-Dequaire, F.; Moszer, I.; Brice, A.; Singleton, A.B.; Corvol, J.C. Clinical-genetic model predicts incident impulse control disorders in Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 2016, 87, 1106–1111. [Google Scholar] [CrossRef] [Green Version]
- Redenšek, S.; Jenko Bizjan, B.; Trošt, M.; Dolžan, V. Clinical-Pharmacogenetic Predictive Models for Time to Occurrence of Levodopa Related Motor Complications in Parkinson’s Disease. Front. Genet. 2019, 10, 461. [Google Scholar] [CrossRef]
- Over, L.; Brüggemann, N.; Lohmann, K. Therapies for Genetic Forms of Parkinson’s Disease: Systematic Literature Review. J. Neuromuscul. Dis. 2021. [Google Scholar] [CrossRef]
- Lythe, V.; Athauda, D.; Foley, J.; Mencacci, N.E.; Jahanshahi, M.; Cipolotti, L.; Hyam, J.; Zrinzo, L.; Hariz, M.; Hardy, J.; et al. GBA-Associated Parkinson’s Disease: Progression in a Deep Brain Stimulation Cohort. J. Parkinsons Dis. 2017, 7, 635–644. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Parkinson Disease and DBS: Cognitive Effects in GBA Mutation Carriers. Available online: https://www.clinicaltrials.gov/ct2/show/NCT03234478 (accessed on 26 June 2021).
- ClinicalTrials.gov Identifier: NCT03234478. Available online: https://clinicaltrials.gov/ct2/show/NCT03234478 (accessed on 26 June 2021).
- Paul, K.C.; Schulz, J.; Bronstein, J.M.; Lill, C.M.; Ritz, B.R. Association of Polygenic Risk Score With Cognitive Decline and Motor Progression in Parkinson Disease. JAMA Neurol. 2018, 75, 360–366. [Google Scholar] [CrossRef]
- Latourelle, J.C.; Beste, M.T.; Hadzi, T.C.; Miller, R.E.; Oppenheim, J.N.; Valko, M.P.; Wuest, D.M.; Church, B.W.; Khalil, I.G.; Hayete, B.; et al. Large-scale identification of clinical and genetic predictors of motor progression in patients with newly diagnosed Parkinson’s disease: A longitudinal cohort study and validation. Lancet Neurol. 2017, 16, 908–916. [Google Scholar] [CrossRef]
- Leonard, H.; Blauwendraat, C.; Krohn, L.; Faghri, F.; Iwaki, H.; Ferguson, G.; Day-Williams, A.G.; Stone, D.J.; Singleton, A.B.; Nalls, M.A.; et al. Genetic variability and potential effects on clinical trial outcomes: Perspectives in Parkinson’s disease. J. Med. Genet. 2020, 57, 331–338. [Google Scholar] [CrossRef]
- Lai, D.; Alipanahi, B.; Fontanillas, P.; Schwantes-An, T.-H.; Aasly, J.; Alcalay, R.N.; Beecham, G.W.; Berg, D.; Bressman, S.; Brice, A.; et al. Genome-wide association studies of LRRK2 modifiers of Parkinson’s disease. medRxiv 2020. [Google Scholar] [CrossRef]
- Liu, G.; Boot, B.; Locascio, J.J.; Jansen, I.E.; Winder-Rhodes, S.; Eberly, S.; Elbaz, A.; Brice, A.; Ravina, B.; van Hilten, J.J.; et al. Specifically neuropathic Gaucher’s mutations accelerate cognitive decline in Parkinson’s. Ann. Neurol. 2016, 80, 674–685. [Google Scholar] [CrossRef] [PubMed]
- Mahlknecht, P.; Gasperi, A.; Willeit, P.; Kiechl, S.; Stockner, H.; Willeit, J.; Rungger, G.; Sawires, M.; Nocker, M.; Rastner, V.; et al. Prodromal Parkinson’s disease as defined per MDS research criteria in the general elderly community. Mov. Disord. 2016, 31, 1405–1408. [Google Scholar] [CrossRef] [PubMed]
- Postuma, R.B.; Gagnon, J.F.; Bertrand, J.A.; Génier Marchand, D.; Montplaisir, J.Y. Parkinson risk in idiopathic REM sleep behavior disorder: Preparing for neuroprotective trials. Neurology 2015, 84, 1104–1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullin, S.; Beavan, M.; Bestwick, J.; McNeill, A.; Proukakis, C.; Cox, T.; Hughes, D.; Mehta, A.; Zetterberg, H.; Schapira, A.H.V. Evolution and clustering of prodromal parkinsonian features in GBA1 carriers. Mov. Disord. 2019, 34, 1365–1373. [Google Scholar] [CrossRef] [Green Version]
- Mirelman, A.; Saunders-Pullman, R.; Alcalay, R.N.; Shustak, S.; Thaler, A.; Gurevich, T.; Raymond, D.; Mejia-Santana, H.; Orbe Reilly, M.; Ozelius, L.; et al. Application of the Movement Disorder Society prodromal criteria in healthy G2019S-LRRK2 carriers. Mov. Disord. 2018, 33, 966–973. [Google Scholar] [CrossRef]
- Noyce, A.J.; R’Bibo, L.; Peress, L.; Bestwick, J.P.; Adams-Carr, K.L.; Mencacci, N.E.; Hawkes, C.H.; Masters, J.M.; Wood, N.; Hardy, J.; et al. PREDICT-PD: An online approach to prospectively identify risk indicators of Parkinson’s disease. Mov. Disord. 2017, 32, 219–226. [Google Scholar] [CrossRef]
- Griffanti, L.; Klein, J.C.; Szewczyk-Krolikowski, K.; Menke, R.A.L.; Rolinski, M.; Barber, T.R.; Lawton, M.; Evetts, S.G.; Begeti, F.; Crabbe, M.; et al. Cohort profile: The Oxford Parkinson’s Disease Centre Discovery Cohort MRI substudy (OPDC-MRI). BMJ Open 2020, 10, e034110. [Google Scholar] [CrossRef]
- Jennings, D.; Siderowf, A.; Stern, M.; Seibyl, J.; Eberly, S.; Oakes, D.; Marek, K.; Investigators, P. Conversion to Parkinson Disease in the PARS Hyposmic and Dopamine Transporter-Deficit Prodromal Cohort. JAMA Neurol. 2017, 74, 933–940. [Google Scholar] [CrossRef] [Green Version]
- Krohn, L.; Ruskey, J.A.; Rudakou, U.; Leveille, E.; Asayesh, F.; Hu, M.T.M.; Arnulf, I.; Dauvilliers, Y.; Högl, B.; Stefani, A.; et al. Variants in REM sleep behavior disorder: A multicenter study. Neurology 2020, 95, e1008–e1016. [Google Scholar] [CrossRef]
- Artzi, M.; Even-Sapir, E.; Lerman Shacham, H.; Thaler, A.; Urterger, A.O.; Bressman, S.; Marder, K.; Hendler, T.; Giladi, N.; Ben Bashat, D.; et al. DaT-SPECT assessment depicts dopamine depletion among asymptomatic G2019S LRRK2 mutation carriers. PLoS ONE 2017, 12, e0175424. [Google Scholar] [CrossRef]
- Salat, D.; Noyce, A.J.; Schrag, A.; Tolosa, E. Challenges of modifying disease progression in prediagnostic Parkinson’s disease. Lancet Neurol. 2016, 15, 637–648. [Google Scholar] [CrossRef]
- Tolosa, E.; Vila, M.; Klein, C.; Rascol, O. LRRK2 in Parkinson disease: Challenges of clinical trials. Nat. Rev. Neurol. 2020, 16, 97–107. [Google Scholar] [CrossRef]
- Klein, C.; Hattori, N.; Marras, C. MDSGene: Closing Data Gaps in Genotype-Phenotype Correlations of Monogenic Parkinson’s Disease. J. Parkinsons Dis. 2018, 8, S25–S30. [Google Scholar] [CrossRef] [Green Version]
- Grenn, F.P.; Kim, J.J.; Makarious, M.B.; Iwaki, H.; Illarionova, A.; Brolin, K.; Kluss, J.H.; Schumacher-Schuh, A.F.; Leonard, H.; Faghri, F.; et al. The Parkinson’s Disease Genome-Wide Association Study Locus Browser. Mov. Disord. 2020, 35, 2056–2067. [Google Scholar] [CrossRef]
- Program GPsG. GP2: The Global Parkinson’s Genetics Program. Mov. Disord. 2021. [CrossRef]
- Rapsodi. PD Frontline. Rapsodi Study. 2021. Available online: https://pdfrontline.com/en (accessed on 26 June 2021).
- Parkinson’s Foundation. PD GENEration:Mapping the Future of Parkinson’s Disease: Parkinson’s Foundation. 2021. Available online: https://www.parkinson.org/PDGENEration (accessed on 26 June 2021).


{kind=link}
| Gene (HGNC Approved Name) | Alternative Gene Names | Inheritance | Pathogenicity | PD Phenotype | Function | |
|---|---|---|---|---|---|---|
| High penetrance | SNCA | PARK1, PARK4, NCAP | AD | Pathogenic | Early-onset | Uncertain (encodes α-synuclein) |
| VPS35 | PARK17, MEM3 | AD | Pathogenic | Typical | Retromer and endosomal trafficking | |
| PINK1 | PARK6 | AR | Pathogenic | Early-onset | Mitochondrial | |
| PARK7 | DJ-1 | AR | Pathogenic | Early-onset | ||
| PRKN | PARK2, PARKIN | AR | Pathogenic | Early-onset | ||
| PLA2G6 | PARK14, IPLA2 | AR | Pathogenic | Early-onset, atypical | Cell membrane | |
| ATP13A2 | PARK9 | AR | Pathogenic | Early-onset, atypical | Lysosomal | |
| FBXO7 | PARK15, FBX7 | AR | Pathogenic | Early-onset, atypical | Mitochondrial | |
| POLG | POLG1, POLGA | AD | Pathogenic | Early-onset, atypical | Mitochondrial DNA maintenance | |
| DNAJC6 | PARK19, DJC6 | AR | Likely pathogenic | Early-onset | Synaptic vesicle formation and trafficking | |
| DNAJC13 | PARK21, RME8 | AD | Conflicting reports | Typical | ||
| TMEM230 | C20ORF30 | AD | Conflicting reports | Typical | ||
| SYNJ1 | PARK20 | AR | Pathogenic | Early-onset, atypical | ||
| VPS13C | PARK23 | AR | Pathogenic | Early-onset | Mitochondrial | |
| CHCHD2 | - | AD | Pathogenic | Typical | Uncertain | |
| DCTN1 | - | AD | Pathogenic | Atypical | Microtubule | |
| Variable penetrance | LRRK2 | PARK8, DARDARIN | AD | Pathogenic | Typical | Lysosomal, mitochondrial, microtubule |
| GBA | GBA1 | AD | Pathogenic | Typical | Lysosomal | |
| Associated with PD but unlikely to be pathogenic | HTRA2 | - | AD | Uncertain/likely benign | - | Mitochondrial |
| UCHL1 | PARK5 | AD | Uncertain/likely benign | - | Ubiquitin-proteasome | |
| GIGYF2 | PARK11 | AD | Uncertain/likely benign | - | Uncertain | |
| EIF4G1 | - | AD | Benign | - | mRNA translation | |
| LRP10 | LRP9 | AD 1 | Uncertain | - | Uncertain |
| Study | Year | Cohort Size (Cases: Controls) | Trait | Ethnicity | Number of Genome-Wide Significant Loci |
|---|---|---|---|---|---|
| Fung et al. [147] | 2006 | 267:270 | PD | European | 0 |
| Pankratz et al. [148] | 2009 | 857:867 | Familial PD | European | 0 |
| Latourelle et al. [149] | 2009 | 1604:440 | PD age of onset | European | 0 |
| Satake et al. [150] | 2009 | 1921:18274 | PD | East Asian | 4 |
| Simón-Sánchez et al. [151] | 2009 | 5074:8551 | PD | European | 3 |
| Edwards et al. [152] | 2010 | 1752:1745 | PD | European | 2 |
| Hamza et al. [153] | 2010 | 2000:1986 | PD | European | 4 |
| Saad et al. [154] | 2011 | 4271:9048 | PD | European | 2 |
| Simón-Sánchez et al. [155] | 2011 | 772:2024 | PD | European | 0 |
| Liu et al. [156] | 2011 | 2050:1836 | PD | European | 0 |
| Spencer et al. [157] | 2011 | 2744:7159 | PD | European | 3 |
| Nalls et al. [158] | 2011 | 12386:21026 | PD | European | 11 |
| Do et al. [159] | 2011 | 3426:29624 | PD | European | 8 |
| Chung et al. [160] | 2012 | 443:0 | PD motor and cognitive outcomes | European | 0 |
| Hernandez et al. [161] | 2012 | 387:496 | Early-onset PD | Finnish | 0 |
| Pankratz et al. [145] | 2012 | 7976:6350 | PD | European | 5 |
| Davis et al. [162] | 2013 | 31:767 | PD | European | 0 |
| Hill-Burns et al. [163] | 2014 | 4235:2782 | Familial and sporadic PD | European | 4 |
| Nalls et al. [137] | 2014 | 19061:100833 | PD | European | 24 |
| Hu et al. [164] | 2016 | 250:250 | PD | East Asian | 0 |
| Hill-Burns et al. [165] | 2016 | 1168:0 | Familial PD age of onset | European | 2 |
| Siitonen et al. [166] | 2017 | 403:1650 | Early-onset PD | Finnish | 13 |
| Foo et al. [167] | 2017 | 5904:30831 | PD | East Asian | 3 |
| Chang et al. [168] | 2017 | 26035:403190 | PD | European | 41 |
| Wallen et al. [169] | 2018 | 2676:0 | PD age of onset | European | 1 |
| Blauwendraat et al. [170] | 2019 | 28568:0 | PD age of onset | European | 2 |
| Bandres-Ciga et al. [171] | 2019 | 4783:3066 | PD and PD age of onset | European | 5 |
| Nalls et al. [18] | 2019 | 37688(plus 18618 proxy cases):1417791 | PD | European | 90 |
| Ryu et al. [172] | 2020 | 741:0 | Motor complications of PD | East Asian | 1 |
| Cha et al. [173] | 2020 | 200:0 | PD motor response to zonisamide | East Asian | 1 |
| Tan et al. [174] | 2020 | 2755:0 | PD progression | European | 1 |
| Foo et al. [142] | 2020 | 65257:1896188 | PD | East Asian and European | 10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Day, J.O.; Mullin, S. The Genetics of Parkinson’s Disease and Implications for Clinical Practice. Genes 2021, 12, 1006. https://doi.org/10.3390/genes12071006
Day JO, Mullin S. The Genetics of Parkinson’s Disease and Implications for Clinical Practice. Genes. 2021; 12(7):1006. https://doi.org/10.3390/genes12071006
Chicago/Turabian StyleDay, Jacob Oliver, and Stephen Mullin. 2021. "The Genetics of Parkinson’s Disease and Implications for Clinical Practice" Genes 12, no. 7: 1006. https://doi.org/10.3390/genes12071006
APA StyleDay, J. O., & Mullin, S. (2021). The Genetics of Parkinson’s Disease and Implications for Clinical Practice. Genes, 12(7), 1006. https://doi.org/10.3390/genes12071006