
Epilepsy Syndromes in the First Year of Life and Usefulness of Genetic Testing for Precision Therapy


Abstract
:1. Introduction
2. Benign and Self-Limited (Familial) Epilepsy Syndromes in Infancy
2.1. Benign (Familial) Neonatal Epilepsy
2.2. Benign (Familial) Neonatal-Infantile Epilepsy
2.3. Benign (Familial) Infantile Epilepsy
2.4. GEFS+ Spectrum (Including Febrile Seizures Plus)
2.5. Myoclonic Epilepsy in Infancy
3. Developmental and Epileptic Encephalopathies (DEEs)
3.1. Ohtahara Syndrome
3.2. Early Myoclonic Encephalopathy
3.3. Epileptic Spasms Syndrome and West Syndrome
3.4. Dravet Syndrome (DS)
3.5. Epilepsy of Infancy with Migrating Focal Seizures (EIMFS)
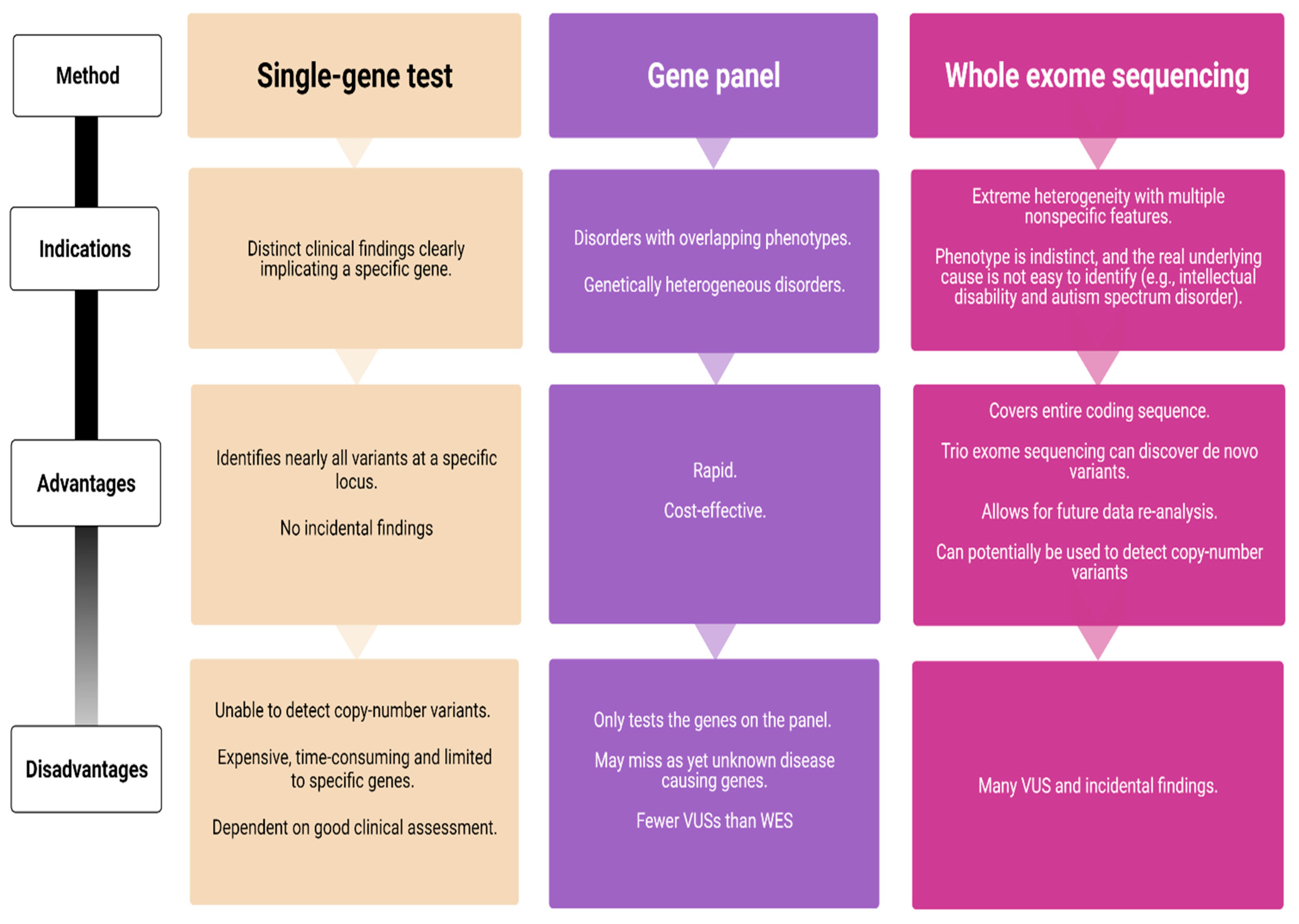
4. Advantages and Pitfalls in Genetic Testing of Subjects with Onset of Epilepsy Syndromes during the First Year of Life
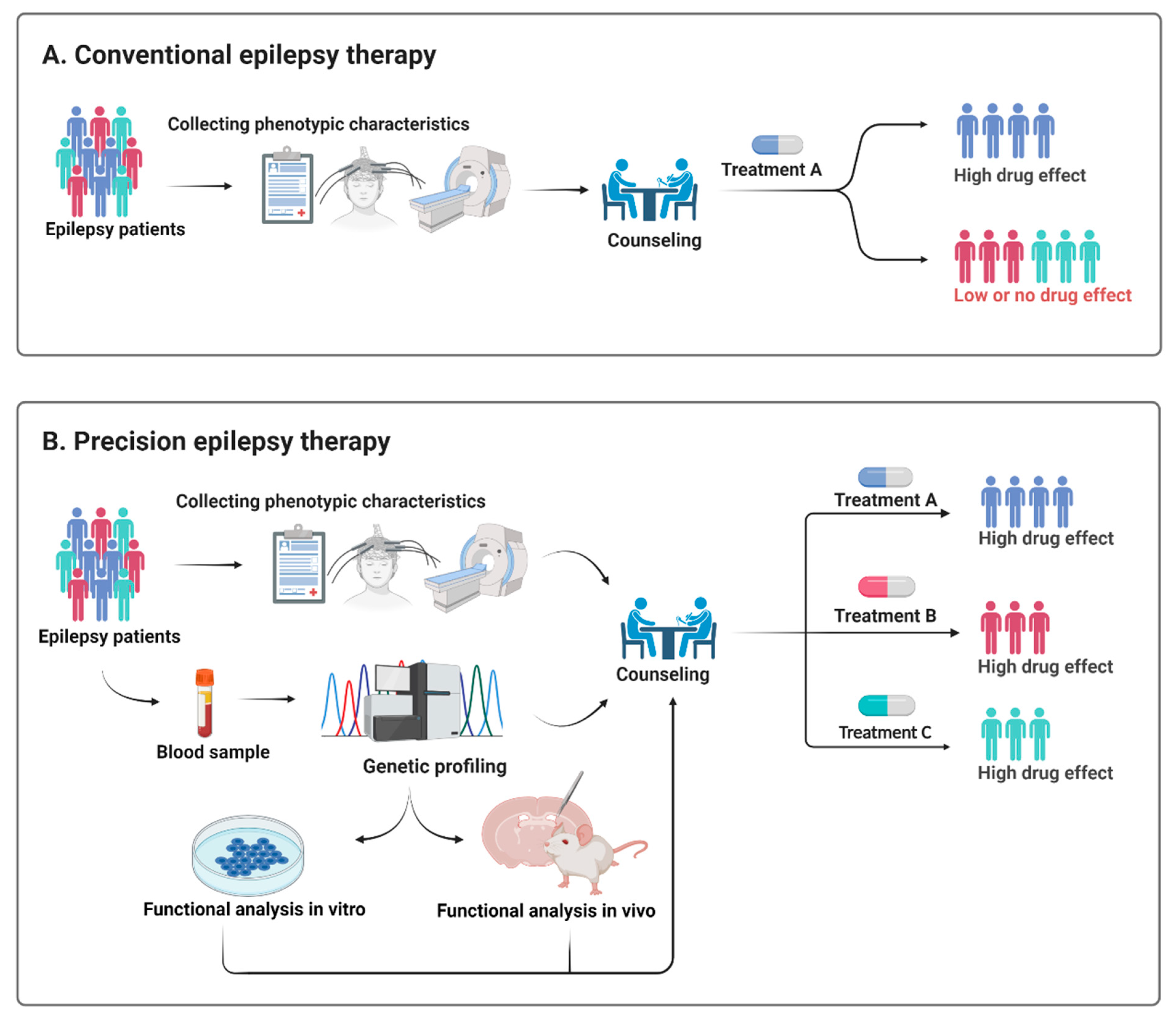
5. Utility of Early Genetic Testing for Precision Therapy Approaches
6. Genetic Testing in Self-Limiting Epilepsies
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Thomas, R.H.; Berkovic, S.F. The hidden genetics of epilepsy-a clinically important new paradigm. Nat. Rev. Neurol. 2014, 10, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Hebbar, M.; Mefford, H.C. Recent advances in epilepsy genomics and genetic testing. F1000Research 2020, 9, 185. [Google Scholar] [CrossRef] [PubMed]
- Howell, K.B.; Freeman, J.L.; Mackay, M.T.; Fahey, M.C.; Archer, J.; Berkovic, S.F.; Chan, E.; Dabscheck, G.; Eggers, S.; Hayman, M.; et al. The severe epilepsy syndromes of infancy: A population-based study. Epilepsia 2021, 62, 358–370. [Google Scholar] [CrossRef]
- Moller, R.S.; Larsen, L.H.; Johannesen, K.M.; Talvik, I.; Talvik, T.; Vaher, U.; Miranda, M.J.; Farooq, M.; Nielsen, J.E.; Svendsen, L.L.; et al. Gene Panel Testing in Epileptic Encephalopathies and Familial Epilepsies. Mol. Syndromol. 2016, 7, 210–219. [Google Scholar] [CrossRef] [Green Version]
- Hartley, T.; Lemire, G.; Kernohan, K.D.; Howley, H.E.; Adams, D.R.; Boycott, K.M. New Diagnostic Approaches for Undiagnosed Rare Genetic Diseases. Annu. Rev. Genom. Hum. Genet. 2020, 21, 351–372. [Google Scholar] [CrossRef] [Green Version]
- Kotsopoulos, I.A.; van Merode, T.; Kessels, F.G.; de Krom, M.C.; Knottnerus, J.A. Systematic review and meta-analysis of incidence studies of epilepsy and unprovoked seizures. Epilepsia 2002, 43, 1402–1409. [Google Scholar] [CrossRef]
- Lee, E.H. Epilepsy syndromes during the first year of life and the usefulness of an epilepsy gene panel. Korean J. Pediatr. 2018, 61, 101–107. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Scheffer, I.E.; Crossland, K.; Berkovic, S.F. Generalized epilepsy with febrile seizures plus: A common childhood-onset genetic epilepsy syndrome. Ann. Neurol. 1999, 45, 75–81. [Google Scholar] [CrossRef]
- Verrotti, A.; Matricardi, S.; Pavone, P.; Marino, R.; Curatolo, P. Reflex myoclonic epilepsy in infancy: A critical review. Epileptic Disord. 2013, 15, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Miceli, F.; Soldovieri, M.V.; Joshi, N.; Weckhuysen, S.; Cooper, E.; Taglialatela, M. KCNQ2-Related Disorders. In GeneReviews((R)); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Singh, N.A.; Charlier, C.; Stauffer, D.; DuPont, B.R.; Leach, R.J.; Melis, R.; Ronen, G.M.; Bjerre, I.; Quattlebaum, T.; Murphy, J.V.; et al. A novel potassium channel gene, KCNQ2, is mutated in an inherited epilepsy of newborns. Nat. Genet. 1998, 18, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Ronen, G.M.; Rosales, T.O.; Connolly, M.; Anderson, V.E.; Leppert, M. Seizure characteristics in chromosome 20 benign familial neonatal convulsions. Neurology 1993, 43, 1355–1360. [Google Scholar] [CrossRef]
- Grinton, B.E.; Heron, S.E.; Pelekanos, J.T.; Zuberi, S.M.; Kivity, S.; Afawi, Z.; Williams, T.C.; Casalaz, D.M.; Yendle, S.; Linder, I.; et al. Familial neonatal seizures in 36 families: Clinical and genetic features correlate with outcome. Epilepsia 2015, 56, 1071–1080. [Google Scholar] [CrossRef] [PubMed]
- Scalmani, P.; Rusconi, R.; Armatura, E.; Zara, F.; Avanzini, G.; Franceschetti, S.; Mantegazza, M. Effects in neocortical neurons of mutations of the Na(v)1.2 Na+ channel causing benign familial neonatal-infantile seizures. J. Neurosci. 2006, 26, 10100–10109. [Google Scholar] [CrossRef] [PubMed]
- Heron, S.E.; Crossland, K.M.; Andermann, E.; Phillips, H.A.; Hall, A.J.; Bleasel, A.; Shevell, M.; Mercho, S.; Seni, M.H.; Guiot, M.C.; et al. Sodium-channel defects in benign familial neonatal-infantile seizures. Lancet 2002, 360, 851–852. [Google Scholar] [CrossRef]
- Vigevano, F.; Fusco, L.; Di Capua, M.; Ricci, S.; Sebastianelli, R.; Lucchini, P. Benign infantile familial convulsions. Eur. J. Pediatr. 1992, 151, 608–612. [Google Scholar] [CrossRef] [PubMed]
- Mulley, J.C.; Heron, S.E.; Dibbens, L.M. Proposed genetic classification of the “benign” familial neonatal and infantile epilepsies. Epilepsia 2011, 52, 649–650. [Google Scholar] [CrossRef]
- Nobile, C.; Striano, P. PRRT2: A major cause of infantile epilepsy and other paroxysmal disorders of childhood. Prog. Brain Res. 2014, 213, 141–158. [Google Scholar] [CrossRef] [PubMed]
- Ono, S.; Yoshiura, K.; Kinoshita, A.; Kikuchi, T.; Nakane, Y.; Kato, N.; Sadamatsu, M.; Konishi, T.; Nagamitsu, S.; Matsuura, M.; et al. Mutations in PRRT2 responsible for paroxysmal kinesigenic dyskinesias also cause benign familial infantile convulsions. J. Hum. Genet. 2012, 57, 338–341. [Google Scholar] [CrossRef]
- Zeng, Q.; Zhang, Y.H.; Yang, X.L.; Pu, L.H.; Zhang, J.; Liu, A.J.; Yang, Z.X.; Liu, X.Y.; Wu, X.R. Spectrum of mutations in benign familial neonatal-infantile epilepsy. Zhonghua Er Ke Za Zhi Chin. J. Pediatr. 2018, 56, 267–273. [Google Scholar] [CrossRef]
- Schubert, J.; Paravidino, R.; Becker, F.; Berger, A.; Bebek, N.; Bianchi, A.; Brockmann, K.; Capovilla, G.; Dalla Bernardina, B.; Fukuyama, Y.; et al. PRRT2 mutations are the major cause of benign familial infantile seizures. Hum. Mutat. 2012, 33, 1439–1443. [Google Scholar] [CrossRef] [PubMed]
- Heron, S.E.; Grinton, B.E.; Kivity, S.; Afawi, Z.; Zuberi, S.M.; Hughes, J.N.; Pridmore, C.; Hodgson, B.L.; Iona, X.; Sadleir, L.G.; et al. PRRT2 mutations cause benign familial infantile epilepsy and infantile convulsions with choreoathetosis syndrome. Am. J. Hum. Genet. 2012, 90, 152–160. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Zhang, Y.; Xu, X.; Wang, S.; Yang, Z.; Wu, Y.; Liu, X.; Wu, X. Phenotypes and PRRT2 mutations in Chinese families with benign familial infantile epilepsy and infantile convulsions with paroxysmal choreoathetosis. BMC Neurol. 2013, 13, 209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Zhang, Y.; Xu, X.; Yu, X.; Zhang, X.; Yang, Z.; Wang, S.; Wu, Y.; Liu, X.; Wu, X. Phenotypes and PRRT2 mutation analysis in families with benign familial infantile epilepsy. Zhonghua Er Ke Za Zhi Chin. J. Pediatr. 2014, 52, 806–811. [Google Scholar]
- Valente, P.; Castroflorio, E.; Rossi, P.; Fadda, M.; Sterlini, B.; Cervigni, R.I.; Prestigio, C.; Giovedi, S.; Onofri, F.; Mura, E.; et al. PRRT2 Is a Key Component of the Ca(2+)-Dependent Neurotransmitter Release Machinery. Cell Rep. 2016, 15, 117–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardella, E.; Moller, R.S. Phenotypic and genetic spectrum of SCN8A-related disorders, treatment options, and outcomes. Epilepsia 2019, 60 (Suppl. 3), S77–S85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheffer, I.E.; Berkovic, S.F. Generalized epilepsy with febrile seizures plus. A genetic disorder with heterogeneous clinical phenotypes. Brain 1997, 120, 479–490. [Google Scholar] [CrossRef] [Green Version]
- Myers, K.A.; Scheffer, I.E.; Berkovic, S.F.; Commission, I.G. Genetic literacy series: Genetic epilepsy with febrile seizures plus. Epileptic Disord. 2018, 20, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.H.; Burgess, R.; Malone, J.P.; Glubb, G.C.; Helbig, K.L.; Vadlamudi, L.; Kivity, S.; Afawi, Z.; Bleasel, A.; Grattan-Smith, P.; et al. Genetic epilepsy with febrile seizures plus: Refining the spectrum. Neurology 2017, 89, 1210–1219. [Google Scholar] [CrossRef]
- Moller, R.S.; Wuttke, T.V.; Helbig, I.; Marini, C.; Johannesen, K.M.; Brilstra, E.H.; Vaher, U.; Borggraefe, I.; Talvik, I.; Talvik, T.; et al. Mutations in GABRB3: From febrile seizures to epileptic encephalopathies. Neurology 2017, 88, 483–492. [Google Scholar] [CrossRef] [Green Version]
- Johannesen, K.; Marini, C.; Pfeffer, S.; Moller, R.S.; Dorn, T.; Niturad, C.E.; Gardella, E.; Weber, Y.; Sondergard, M.; Hjalgrim, H.; et al. Phenotypic spectrum of GABRA1: From generalized epilepsies to severe epileptic encephalopathies. Neurology 2016, 87, 1140–1151. [Google Scholar] [CrossRef] [PubMed]
- Wolking, S.; May, P.; Mei, D.; Moller, R.S.; Balestrini, S.; Helbig, K.L.; Altuzarra, C.D.; Chatron, N.; Kaiwar, C.; Stohr, K.; et al. Clinical spectrum of STX1B-related epileptic disorders. Neurology 2019, 92, e1238–e1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldberg-Stern, H.; Aharoni, S.; Afawi, Z.; Bennett, O.; Appenzeller, S.; Pendziwiat, M.; Kuhlenbaumer, G.; Basel-Vanagaite, L.; Shuper, A.; Korczyn, A.D.; et al. Broad phenotypic heterogeneity due to a novel SCN1A mutation in a family with genetic epilepsy with febrile seizures plus. J. Child Neurol. 2014, 29, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Auvin, S.; Pandit, F.; De Bellecize, J.; Badinand, N.; Isnard, H.; Motte, J.; Villeneuve, N.; Lamblin, M.D.; Vallee, L. Benign myoclonic epilepsy in infants: Electroclinical features and long-term follow-up of 34 patients. Epilepsia 2006, 47, 387–393. [Google Scholar] [CrossRef]
- Scheffer, I.E.; Liao, J. Deciphering the concepts behind “Epileptic encephalopathy” and “Developmental and epileptic encephalopathy”. Eur. J. Paediatr. Neurol. 2020, 24, 11–14. [Google Scholar] [CrossRef]
- Scheffer, I.E.; Berkovic, S.; Capovilla, G.; Connolly, M.B.; French, J.; Guilhoto, L.; Hirsch, E.; Jain, S.; Mathern, G.W.; Moshe, S.L.; et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017, 58, 512–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheffer, I.E.; Berkovic, S.F. The genetics of human epilepsy. Trends Pharmacol. Sci. 2003, 24, 428–433. [Google Scholar] [CrossRef]
- McTague, A.; Howell, K.B.; Cross, J.H.; Kurian, M.A.; Scheffer, I.E. The genetic landscape of the epileptic encephalopathies of infancy and childhood. Lancet Neurol. 2016, 15, 304–316. [Google Scholar] [CrossRef]
- Olson, H.E.; Kelly, M.; LaCoursiere, C.M.; Pinsky, R.; Tambunan, D.; Shain, C.; Ramgopal, S.; Takeoka, M.; Libenson, M.H.; Julich, K.; et al. Genetics and genotype-phenotype correlations in early onset epileptic encephalopathy with burst suppression. Ann. Neurol. 2017, 81, 419–429. [Google Scholar] [CrossRef]
- Beal, J.C.; Cherian, K.; Moshe, S.L. Early-onset epileptic encephalopathies: Ohtahara syndrome and early myoclonic encephalopathy. Pediatr. Neurol. 2012, 47, 317–323. [Google Scholar] [CrossRef] [Green Version]
- Ohtahara, S.; Yamatogi, Y. Ohtahara syndrome: With special reference to its developmental aspects for differentiating from early myoclonic encephalopathy. Epilepsy Res. 2006, 70 (Suppl. 1), S58–S67. [Google Scholar] [CrossRef]
- Yamatogi, Y.; Ohtahara, S. Early-infantile epileptic encephalopathy with suppression-bursts, Ohtahara syndrome; its overview referring to our 16 cases. Brain Dev. 2002, 24, 13–23. [Google Scholar] [CrossRef]
- Fusco, L.; Pachatz, C.; Di Capua, M.; Vigevano, F. Video/EEG aspects of early-infantile epileptic encephalopathy with suppression-bursts (Ohtahara syndrome). Brain Dev. 2001, 23, 708–714. [Google Scholar] [CrossRef]
- Milh, M.; Villeneuve, N.; Chouchane, M.; Kaminska, A.; Laroche, C.; Barthez, M.A.; Gitiaux, C.; Bartoli, C.; Borges-Correia, A.; Cacciagli, P.; et al. Epileptic and nonepileptic features in patients with early onset epileptic encephalopathy and STXBP1 mutations. Epilepsia 2011, 52, 1828–1834. [Google Scholar] [CrossRef]
- Weckhuysen, S.; Mandelstam, S.; Suls, A.; Audenaert, D.; Deconinck, T.; Claes, L.R.; Deprez, L.; Smets, K.; Hristova, D.; Yordanova, I.; et al. KCNQ2 encephalopathy: Emerging phenotype of a neonatal epileptic encephalopathy. Ann. Neurol. 2012, 71, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Wolff, M.; Johannesen, K.M.; Hedrich, U.B.S.; Masnada, S.; Rubboli, G.; Gardella, E.; Lesca, G.; Ville, D.; Milh, M.; Villard, L.; et al. Genetic and phenotypic heterogeneity suggest therapeutic implications in SCN2A-related disorders. Brain 2017, 140, 1316–1336. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Saitoh, S.; Kamei, A.; Shiraishi, H.; Ueda, Y.; Akasaka, M.; Tohyama, J.; Akasaka, N.; Hayasaka, K. A longer polyalanine expansion mutation in the ARX gene causes early infantile epileptic encephalopathy with suppression-burst pattern (Ohtahara syndrome). Am. J. Hum. Genet. 2007, 81, 361–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molinari, F.; Kaminska, A.; Fiermonte, G.; Boddaert, N.; Raas-Rothschild, A.; Plouin, P.; Palmieri, L.; Brunelle, F.; Palmieri, F.; Dulac, O.; et al. Mutations in the mitochondrial glutamate carrier SLC25A22 in neonatal epileptic encephalopathy with suppression bursts. Clin. Genet. 2009, 76, 188–194. [Google Scholar] [CrossRef]
- Gertler, T.; Bearden, D.; Bhattacharjee, A.; Carvill, G. KCNT1-Related Epilepsy. In GeneReviews((R)); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Borlot, F.; Abushama, A.; Morrison-Levy, N.; Jain, P.; Puthenveettil Vinayan, K.; Abukhalid, M.; Aldhalaan, H.M.; Almuzaini, H.S.; Gulati, S.; Hershkovitz, T.; et al. KCNT1-related epilepsy: An international multicenter cohort of 27 pediatric cases. Epilepsia 2020, 61, 679–692. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Ma, A.; Liu, X.; Huang, C.; Zhang, Y.; Shi, R.; Mao, S.; Geng, T.; Li, S. Infantile seizures and other epileptic phenotypes in a Chinese family with a missense mutation of KCNQ2. Eur. J. Pediatr. 2006, 165, 691–695. [Google Scholar] [CrossRef]
- Shi, X.; Yasumoto, S.; Kurahashi, H.; Nakagawa, E.; Fukasawa, T.; Uchiya, S.; Hirose, S. Clinical spectrum of SCN2A mutations. Brain Dev. 2012, 34, 541–545. [Google Scholar] [CrossRef]
- Murakami, N.; Ohtsuka, Y.; Ohtahara, S. Early infantile epileptic syndromes with suppression-bursts: Early myoclonic encephalopathy vs. Ohtahara syndrome. Jpn. J. Psychiatry Neurol. 1993, 47, 197–200. [Google Scholar] [CrossRef] [PubMed]
- Bayat, A.; Knaus, A.; Pendziwiat, M.; Afenjar, A.; Barakat, T.S.; Bosch, F.; Callewaert, B.; Calvas, P.; Ceulemans, B.; Chassaing, N.; et al. Lessons learned from 40 novel PIGA patients and a review of the literature. Epilepsia 2020, 61, 1142–1155. [Google Scholar] [CrossRef]
- Kato, M.; Saitsu, H.; Murakami, Y.; Kikuchi, K.; Watanabe, S.; Iai, M.; Miya, K.; Matsuura, R.; Takayama, R.; Ohba, C.; et al. PIGA mutations cause early-onset epileptic encephalopathies and distinctive features. Neurology 2014, 82, 1587–1596. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.; Snow, C.; Tuttle, E.; Ghoneim, D.H.; Yang, C.S.; Spencer, A.; Gunter, S.A.; Smyser, C.D.; Gurnett, C.A.; Shinawi, M.; et al. De novo mutations in SIK1 cause a spectrum of developmental epilepsies. Am. J. Hum. Genet. 2015, 96, 682–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Callaghan, F.J.; Lux, A.L.; Darke, K.; Edwards, S.W.; Hancock, E.; Johnson, A.L.; Kennedy, C.R.; Newton, R.W.; Verity, C.M.; Osborne, J.P. The effect of lead time to treatment and of age of onset on developmental outcome at 4 years in infantile spasms: Evidence from the United Kingdom Infantile Spasms Study. Epilepsia 2011, 52, 1359–1364. [Google Scholar] [CrossRef] [PubMed]
- Pavone, P.; Striano, P.; Falsaperla, R.; Pavone, L.; Ruggieri, M. Infantile spasms syndrome, West syndrome and related phenotypes: What we know in 2013. Brain Dev. 2014, 36, 739–751. [Google Scholar] [CrossRef]
- Muir, A.M.; Myers, C.T.; Nguyen, N.T.; Saykally, J.; Craiu, D.; De Jonghe, P.; Helbig, I.; Hoffman-Zacharska, D.; Guerrini, R.; Lehesjoki, A.E.; et al. Genetic heterogeneity in infantile spasms. Epilepsy Res. 2019, 156, 106181. [Google Scholar] [CrossRef]
- Michaud, J.L.; Lachance, M.; Hamdan, F.F.; Carmant, L.; Lortie, A.; Diadori, P.; Major, P.; Meijer, I.A.; Lemyre, E.; Cossette, P.; et al. The genetic landscape of infantile spasms. Hum. Mol. Genet. 2014, 23, 4846–4858. [Google Scholar] [CrossRef] [Green Version]
- Boutry-Kryza, N.; Labalme, A.; Ville, D.; de Bellescize, J.; Touraine, R.; Prieur, F.; Dimassi, S.; Poulat, A.L.; Till, M.; Rossi, M.; et al. Molecular characterization of a cohort of 73 patients with infantile spasms syndrome. Eur. J. Med. Genet. 2015, 58, 51–58. [Google Scholar] [CrossRef]
- Samanta, D. Changing Landscape of Dravet Syndrome Management: An Overview. Neuropediatrics 2020, 51, 135–145. [Google Scholar] [CrossRef]
- Dravet, C.; Bureau, M.; Oguni, H.; Fukuyama, Y.; Cokar, O. Severe myoclonic epilepsy in infancy: Dravet syndrome. Adv. Neurol. 2005, 95, 71–102. [Google Scholar] [PubMed]
- Steel, D.; Symonds, J.D.; Zuberi, S.M.; Brunklaus, A. Dravet syndrome and its mimics: Beyond SCN1A. Epilepsia 2017, 58, 1807–1816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgess, R.; Wang, S.; McTague, A.; Boysen, K.E.; Yang, X.; Zeng, Q.; Myers, K.A.; Rochtus, A.; Trivisano, M.; Gill, D.; et al. The Genetic Landscape of Epilepsy of Infancy with Migrating Focal Seizures. Ann. Neurol. 2019, 86, 821–831. [Google Scholar] [CrossRef] [PubMed]
- Koutroumanidis, M.; Arzimanoglou, A.; Caraballo, R.; Goyal, S.; Kaminska, A.; Laoprasert, P.; Oguni, H.; Rubboli, G.; Tatum, W.; Thomas, P.; et al. The role of EEG in the diagnosis and classification of the epilepsy syndromes: A tool for clinical practice by the ILAE Neurophysiology Task Force (Part 2). Epileptic Disord. 2017, 19, 385–437. [Google Scholar] [CrossRef]
- Yoshitomi, S.; Takahashi, Y.; Imai, K.; Koshimizu, E.; Miyatake, S.; Nakashima, M.; Saitsu, H.; Matsumoto, N.; Kato, M.; Fujita, T.; et al. Different types of suppression-burst patterns in patients with epilepsy of infancy with migrating focal seizures (EIMFS). Seizure 2019, 65, 118–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonardi, C.M.; Heyne, H.O.; Fiannacca, M.; Fitzgerald, M.P.; Gardella, E.; Gunning, B.; Olofsson, K.; Lesca, G.; Verbeek, N.; Stamberger, H.; et al. KCNT1-related epilepsies and epileptic encephalopathies: Phenotypic and mutational spectrum. Brain 2021. [Google Scholar] [CrossRef]
- Lindy, A.S.; Stosser, M.B.; Butler, E.; Downtain-Pickersgill, C.; Shanmugham, A.; Retterer, K.; Brandt, T.; Richard, G.; McKnight, D.A. Diagnostic outcomes for genetic testing of 70 genes in 8565 patients with epilepsy and neurodevelopmental disorders. Epilepsia 2018, 59, 1062–1071. [Google Scholar] [CrossRef] [Green Version]
- Mercimek-Mahmutoglu, S.; Patel, J.; Cordeiro, D.; Hewson, S.; Callen, D.; Donner, E.J.; Hahn, C.D.; Kannu, P.; Kobayashi, J.; Minassian, B.A.; et al. Diagnostic yield of genetic testing in epileptic encephalopathy in childhood. Epilepsia 2015, 56, 707–716. [Google Scholar] [CrossRef]
- Ortega-Moreno, L.; Giraldez, B.G.; Soto-Insuga, V.; Losada-Del Pozo, R.; Rodrigo-Moreno, M.; Alarcon-Morcillo, C.; Sanchez-Martin, G.; Diaz-Gomez, E.; Guerrero-Lopez, R.; Serratosa, J.M.; et al. Molecular diagnosis of patients with epilepsy and developmental delay using a customized panel of epilepsy genes. PLoS ONE 2017, 12, e0188978. [Google Scholar] [CrossRef]
- Snoeijen-Schouwenaars, F.M.; van Ool, J.S.; Verhoeven, J.S.; van Mierlo, P.; Braakman, H.M.H.; Smeets, E.E.; Nicolai, J.; Schoots, J.; Teunissen, M.W.A.; Rouhl, R.P.W.; et al. Diagnostic exome sequencing in 100 consecutive patients with both epilepsy and intellectual disability. Epilepsia 2019, 60, 155–164. [Google Scholar] [CrossRef] [Green Version]
- Costain, G.; Cordeiro, D.; Matviychuk, D.; Mercimek-Andrews, S. Clinical Application of Targeted Next-Generation Sequencing Panels and Whole Exome Sequencing in Childhood Epilepsy. Neuroscience 2019, 418, 291–310. [Google Scholar] [CrossRef]
- Helbig, K.L.; Farwell Hagman, K.D.; Shinde, D.N.; Mroske, C.; Powis, Z.; Li, S.; Tang, S.; Helbig, I. Diagnostic exome sequencing provides a molecular diagnosis for a significant proportion of patients with epilepsy. Genet. Med. 2016, 18, 898–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mefford, H.C.; Yendle, S.C.; Hsu, C.; Cook, J.; Geraghty, E.; McMahon, J.M.; Eeg-Olofsson, O.; Sadleir, L.G.; Gill, D.; Ben-Zeev, B.; et al. Rare copy number variants are an important cause of epileptic encephalopathies. Ann. Neurol. 2011, 70, 974–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Kattman, B.L. ClinVar at five years: Delivering on the promise. Hum. Mutat. 2018, 39, 1623–1630. [Google Scholar] [CrossRef] [PubMed]
- Moller, R.S.; Hammer, T.B.; Rubboli, G.; Lemke, J.R.; Johannesen, K.M. From next-generation sequencing to targeted treatment of non-acquired epilepsies. Expert Rev. Mol. Diagn. 2019, 19, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Zacher, P.; Mayer, T.; Brandhoff, F.; Bartolomaeus, T.; Le Duc, D.; Finzel, M.; Heinze, A.; Horn, S.; Klockner, C.; Korber, G.; et al. The genetic landscape of intellectual disability and epilepsy in adults and the elderly: A systematic genetic work-up of 150 individuals. Genet. Med. 2021. [Google Scholar] [CrossRef]
- Johannesen, K.M.; Nikanorova, N.; Marjanovic, D.; Pavbro, A.; Larsen, L.H.G.; Rubboli, G.; Moller, R.S. Utility of genetic testing for therapeutic decision-making in adults with epilepsy. Epilepsia 2020, 61, 1234–1239. [Google Scholar] [CrossRef] [PubMed]
- Brunklaus, A.; Schorge, S.; Smith, A.D.; Ghanty, I.; Stewart, K.; Gardiner, S.; Du, J.; Perez-Palma, E.; Symonds, J.D.; Collier, A.C.; et al. SCN1A variants from bench to bedside-improved clinical prediction from functional characterization. Hum. Mutat. 2020, 41, 363–374. [Google Scholar] [CrossRef]
- Symonds, J.D.; Zuberi, S.M.; Stewart, K.; McLellan, A.; O’Regan, M.; MacLeod, S.; Jollands, A.; Joss, S.; Kirkpatrick, M.; Brunklaus, A.; et al. Incidence and phenotypes of childhood-onset genetic epilepsies: A prospective population-based national cohort. Brain 2019, 142, 2303–2318. [Google Scholar] [CrossRef] [Green Version]
- Helbig, I.; Ellis, C.A. Personalized medicine in genetic epilepsies—Possibilities, challenges, and new frontiers. Neuropharmacology 2020, 172, 107970. [Google Scholar] [CrossRef] [PubMed]
- Sanders, S.J.; Campbell, A.J.; Cottrell, J.R.; Moller, R.S.; Wagner, F.F.; Auldridge, A.L.; Bernier, R.A.; Catterall, W.A.; Chung, W.K.; Empfield, J.R.; et al. Progress in Understanding and Treating SCN2A-Mediated Disorders. Trends Neurosci. 2018, 41, 442–456. [Google Scholar] [CrossRef] [PubMed]
- Millichap, J.J.; Park, K.L.; Tsuchida, T.; Ben-Zeev, B.; Carmant, L.; Flamini, R.; Joshi, N.; Levisohn, P.M.; Marsh, E.; Nangia, S.; et al. KCNQ2 encephalopathy: Features, mutational hot spots, and ezogabine treatment of 11 patients. Neurol. Genet. 2016, 2, e96. [Google Scholar] [CrossRef] [Green Version]
- Bialer, M.; Johannessen, S.I.; Koepp, M.J.; Levy, R.H.; Perucca, E.; Perucca, P.; Tomson, T.; White, H.S. Progress report on new antiepileptic drugs: A summary of the Fifteenth Eilat Conference on New Antiepileptic Drugs and Devices (EILAT XV). II. Drugs in more advanced clinical development. Epilepsia 2020, 61, 2365–2385. [Google Scholar] [CrossRef]
- Cornet, M.C.; Morabito, V.; Lederer, D.; Glass, H.C.; Ferrao Santos, S.; Numis, A.L.; Ferriero, D.M.; Sands, T.T.; Cilio, M.R. Neonatal presentation of genetic epilepsies: Early differentiation from acute provoked seizures. Epilepsia 2021. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Chen, C.; Christiansen, A.; Ji, S.; Lin, Q.; Anumonwo, C.; Liu, C.; Leiser, S.C.; Aznarez, I.; Liau, G.; et al. Antisense oligonucleotides increase Scn1a expression and reduce seizures and SUDEP incidence in a mouse model of Dravet syndrome. Sci. Transl. Med. 2020, 12, eaaz6100. [Google Scholar] [CrossRef]
- Lenk, G.M.; Jafar-Nejad, P.; Hill, S.F.; Huffman, L.D.; Smolen, C.E.; Wagnon, J.L.; Petit, H.; Yu, W.; Ziobro, J.; Bhatia, K.; et al. Scn8a Antisense Oligonucleotide Is Protective in Mouse Models of SCN8A Encephalopathy and Dravet Syndrome. Ann. Neurol. 2020, 87, 339–346. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Epilepsy Syndrome | Gene Involved | Protein Product | Mode of Inheritance | |
|---|---|---|---|---|
| Benign familial epilepsy syndromes | BFNE | KCNQ2 | Subunit of voltage-gated K+ channel | AD |
| KCNQ3 | Subunit of voltage-gated K+ channel | AD | ||
| BFNIE | SCN2A | Subunit of voltage-gated Na+ channel | AD | |
| BFIE | PRRT2 | Protein-rich transmembrane protein 2 | AD | |
| SCN2A | Subunit of voltage-gated Na+ channel | AD | ||
| SCN8A | Subunit of voltage-gated Na+ channel | AD | ||
| GEFS+ | SCN1A | Subunit of voltage-gated Na+ channel | AD | |
| SCN1B | Subunit of voltage-gated Na+ channel | AD, AR | ||
| GABRG2 | Subunit of GABAa receptor | AD | ||
| STX1B | Syntaxin 1B | AD | ||
| Myoclonic epilepsy in infancy | Unknown | - | - | |
| Developmental and epileptic encephalopathies | Ohtahara syndrome | STXBP1 (in ~30%) | Syntaxin binding protein 1 | AD |
| KCNQ2 (in ~20%) | Subunit of voltage-gated K+ channel | AD | ||
| SCN2A (in ~10%) | Subunit of voltage-gated Na+ channel | AD | ||
| GNAO1 | Guanine nucleotide-binding protein, α-activating activity polypeptide O | AD | ||
| KCNT1 | Subunit of voltage-gated K+ channel | AD | ||
| SNC8A | Subunit of voltage-gated NA+ channel | AD | ||
| SIK1 | Salt-inducable kinase 1 | AD | ||
| AARS | Alanyl-tRNA synthetase 1 | AR | ||
| BRAT1 | BRACT1-associated atm activator 1 | AR | ||
| CACNA2D2 | Calcium channel, voltage-dependent, α-2/delta subunit 2 | AR | ||
| NECAP1 | NECAP endocytosis-associated protein 1 | AR | ||
| PIGQ | Phosphatidylinositol glycan anchor biosynthesis class Q protein | AR | ||
| SLC25A22 | Solute carrier family 25, member 22 | AR | ||
| ARX | Aristaless-related homeobox | XR | ||
| PIGA | Phosphatidylinositol glycan anchor biosynthesis class A protein | XR | ||
| Early myoclonic encephalopathy | SETBP1 | SET-binding protein 1 | AD | |
| SIK1 | Salt-inducable kinase 1 | AD | ||
| SLC25A22 | Solute carrier family 25, member 22 | AR | ||
| PIGA | Phosphatidylinositol glycan anchor biosynthesis class A protein | XR | ||
| Infantile spasms syndrome (including WEST syndrome) | STXBP1 (in ~2%) | Syntaxin binding protein 1 | AD | |
| CHD2 | Chromodomain helicase DNA-binding protein 2 | AD | ||
| DNM1 | Dynamin 1 | AD | ||
| FOXG1 (duplications) | Forkhead box G1 | AD | ||
| GABRA1 | Subunit of GABAa receptor | AD | ||
| GABRB3 | Subunit of GABAa receptor | AD | ||
| GABRG2 | Subunit of GABAa receptor | AD | ||
| GNAO1 | Guanine nucleotide-binding protein, α-activating activity polypeptide O | AD | ||
| GRIN1 | Glutamate receptor, ionotropic, n-methyl-d-aspartate, subunit 1 | AD, AR | ||
| GRIN2A, | Glutamate receptor, ionotropic, n-methyl-d-aspartate, subunit 2A | AD | ||
| GRIN2B | Glutamate receptor, ionotropic, n-methyl-d-aspartate, subunit 2B | AD | ||
| HCN1 | Hyperpolarization activated cyclic nucleotide gated potassium channel 1 | AD | ||
| KCNA2 | Subunit of voltage-gated K+ channel | AD | ||
| KCNT1 | Subunit of voltage-gated K+ channel | AD | ||
| MAGI2 | Membrane-associated guanylate kinase, WW and PDZ domains-containing 2 | AD | ||
| MEF2C | MADS BOX transcription enhancer factor 2, polypeptide C | AD | ||
| NDP | Norrin cystine knot growth factor NDP | AD | ||
| PTEN | Phosphatase and tensin homolog | AD | ||
| SCA2 | Spinocerebellar ataxia 2 | AD | ||
| SPTAN1 | Spectrin, α, non-erythrocytic 1 | AD | ||
| SETBP1 | SET-binding protein 1 | AD | ||
| SIK1 | Salt-inducable kinase 1 | AD | ||
| SCN1A | Subunit of voltage-gated Na+ channel | AD | ||
| SCN1B | Subunit of voltage-gated Na+ channel | AD, AR | ||
| SCN2A | Subunit of voltage-gated Na+ channel | AD | ||
| SCN8A | Subunit of voltage-gated Na+ channel | AD | ||
| SCN9A | Subunit of voltage-gated Na+ channel | AD | ||
| STXBP1 | Syntaxin binding protein 1 | AD | ||
| TCF4 | Transcription factor 4 | AD | ||
| TSC1 | Tuberous sclerosis complex 1 | AD | ||
| TSC2 | Tuberous sclerosis complex 2 | AD | ||
| DOCK7 | Dedicator of cytokinesis 7 | AR | ||
| NRXN1 | Neurexin 1 | AR | ||
| PIGN | Phosphatidylinositol glycan anchor biosynthesis class N protein | AR | ||
| PIGP | Phosphatidylinositol glycan anchor biosynthesis class P protein | AR | ||
| PIGQ | Phosphatidylinositol glycan anchor biosynthesis class Q protein | AR | ||
| PIGS | Phosphatidylinositol glycan anchor biosynthesis class S protein | AR | ||
| PLCB1 | Phospholipase C, β-1 | AR | ||
| SLC25A22 | Solute carrier family 25, member 22 | AR | ||
| ST3GAL3 | ST3 β-galactoside α-2,3-sialyltransferase 3 | AR | ||
| TBC1D24 | TBC1 domain family, member 24 | AR | ||
| WWOX | WW domain-containing oxidoreductase | AR | ||
| CDKL5 | Cyclin-dependent kinase-like 5 | XD | ||
| ARX | Aristaless-related homeobox | XR | ||
| PIGA | Phosphatidylinositol glycan anchor biosynthesis class A protein | XR | ||
| ALG13 | ALG13 UDP-N-acetylglucosaminyltransferase subunit | XL | ||
| PCDH19 | Protocadherin subclass of the cadherin superfamily | XL | ||
| Dravet/dravet-like phenotypes | SCN1A (in ~90%) | Subunit of voltage-gated Na+ channel | AD | |
| CHD2 | Chromodomain helicase DNA-binding protein 2 | AD | ||
| HCN1 * | Hyperpolarization activated cyclic nucleotide gated potassium channel 1 | AD | ||
| GABRA1 | Subunit of GABAa receptor | AD | ||
| GABRB3 | Subunit of GABAa receptor | AD | ||
| GABRG2 | Subunit of GABAa receptor | AD | ||
| KCNA2 | Subunit of voltage-gated K+ channel | AD | ||
| SCN1B | Subunit of voltage-gated Na+ channel | AD, AR | ||
| SCN2A | Subunit of voltage-gated Na+ channel | AD | ||
| SCN8A | Subunit of voltage-gated Na+ channel | AD | ||
| SCN9A | Subunit of voltage-gated Na+ channel | AD | ||
| STXBP1 | Syntaxin binding protein 1 | AD | ||
| PCDH19 * | Protocadherin subclass of the cadherin superfamily | XL | ||
| EIMFS | KCNT1 (in ~27%) | Subunit of voltage-gated K+ channel | AD | |
| SCN2A (in ~7%) | Subunit of voltage-gated Na+ channel | AD | ||
| SCN1A | Subunit of voltage-gated Na+ channel | AD | ||
| GABRA1 | Subunit of GABAa receptor | AD | ||
| GABRB1 | Subunit of GABAa receptor | AD | ||
| GABRB3 | Subunit of GABAa receptor | AD | ||
| HCN1 | Hyperpolarization-activated cyclic nucleotide-gated potassium channel 1 | AD | ||
| KCNQ2 | Subunit of voltage-gated K+ channel | AD | ||
| SCN8A | Subunit of voltage-gated Na+ channel | AD | ||
| ATP1A3 | ATPase, Na+/K+ transporting, α-3 polypeptide | AD | ||
| AIMP1 | Aminoacyl-tRNA synthetase complex-interacting multifunctional protein 1 | AR | ||
| BRAT1 | BRCA1-associated ATM activator 1 | AR | ||
| ITPA | Inosine triphosphatase | AR | ||
| KARS | Lysyl-tRNA synthetase 1 | AR | ||
| PLCB1 | Phospholipase C, β-1 | AR | ||
| QARS | Glutaminyl-tRNA synthetase 1 | AR | ||
| SLC12A5 | Solute carrier family 12 | AR | ||
| SLC25A22 | Solute carrier family 25, member 22 | AR | ||
| TBC1D24 | TBC1 domain family, member 24 | AR | ||
| WWOX | WW domain-containing oxidoreductase | AR | ||
| CDKL5 | Cyclin-dependent kinase-like 5 | XD | ||
| SMC1A | Structural maintenance of chromosomes 1A | XD | ||
| PIGA | Phosphatidylinositol glycan anchor biosynthesis class A protein | XR | ||
| Gene | Associated Epilepsy Syndromes | Associated with Structural Brain Anomalies/Lesions | Potential Therapeutic Approaches |
|---|---|---|---|
| ARX | Ohtahara syndrome | Yes | Currently none available |
| Epileptic spasms syndrome | |||
| Myoclonic epilepsy | |||
| Nonsyndromic intellectual disability with or without epilepsy | |||
| Developmental and epileptic encephalopathy 1 | |||
| CDKL5 | Epileptic spasms syndrome | Yes | Ganaxolone (PMID 33165915) |
| Developmental and epileptic encephalopathy 2 | |||
| KCNA2 | Developmental and epileptic encephalopathy 32 | Yes | GoF: 4-Aminopyridine Sodium channel blockers (PMID 33515866) |
| ESES | |||
| KCNB1 | Developmental and epileptic encephalopathy 26 | Yes | Currently none available |
| Epileptic spasms syndrome | |||
| ESES | |||
| KCNQ2 | Benign familial neonatal epilepsy | Yes | LoF: Sodium channel blockers (PMID 24371303 and 25880994) Retigabine (PMID 27602407) |
| Ohtahara syndrome | |||
| Neonatal epileptic encephalopathy | |||
| EIMFS | |||
| Developmental and epileptic encephalopathy 7 | |||
| KCNQ3 | Benign familial neonatal epilepsy | Yes | LoF: Sodium channel blockers (PMID 27888506) Retigabine (PMID 27602407) |
| KCNT1 | Autosomal dominant sleep-related hypermotor epilepsy | Yes | GoF: Quinidine (PMID 31054119) |
| EIMFS | |||
| Developmental and epileptic encephalopathy 14 | |||
| PRRT2 | Benign familial infantile epilepsy | Yes | Carbamazepine (PMID 32413583) |
| Infantile convulsion and choreoathetosis syndrome | |||
| Paroxysmal kinesigenic dyskinesia | |||
| SCN1A | Dravet syndrome | Yes | Lof: Stiripentol (+ valproate + clobazam) Fenfluramine Cannabidiol Avoid sodium channel blockers (PMID 32413583) |
| Genetic epilepsy with febrile seizure plus | |||
| EIMFS | |||
| Developmental and epileptic encephalopathy 6 | |||
| SCN2A | Benign familial neonatal infantile epilepsy | Yes | GoF: Sodium channel blockers (PMID 32413583) LoF: Avoid sodium channel blockers (PMID 32413583) |
| Genetic epilepsy with febrile seizures plus | |||
| Epileptic spasms syndrome | |||
| EIMFS | |||
| Ohtahara syndrome | |||
| Developmental and epileptic encephalopathy 11 | |||
| SCN8A | Benign familial neonatal infantile epilepsy | Yes | GoF: Sodium channel blockers (PMID 32413583) LoF: Avoid sodium channel blockers (PMID 32413583) |
| Developmental and epileptic encephalopathy 13 | |||
| STXBP1 | Ohtahara syndrome. | Yes | Levetiracetam may have superior effect on seizures and movement disorder (PMID 29896790). |
| Epileptic spasms syndrome | |||
| Nonsyndromic intellectual disability with or without epilepsy | |||
| Developmental and epileptic encephalopathy 4 | |||
| SLC25A22 | Ohtahara syndrome | Yes | Currently none available |
| Early myoclonic epilepsy | |||
| EIMFS | |||
| Developmental and epileptic encephalopathy 3 | |||
| TSC1 | Epileptic spasms syndrome | Yes | Everolimus and other mTOR inhibitors (PMID 27351628) |
| TSC2 | Epileptic spasms syndrome | Yes | Everolimus and other mTOR inhibitors (PMID 27351628) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bayat, A.; Bayat, M.; Rubboli, G.; Møller, R.S. Epilepsy Syndromes in the First Year of Life and Usefulness of Genetic Testing for Precision Therapy. Genes 2021, 12, 1051. https://doi.org/10.3390/genes12071051
Bayat A, Bayat M, Rubboli G, Møller RS. Epilepsy Syndromes in the First Year of Life and Usefulness of Genetic Testing for Precision Therapy. Genes. 2021; 12(7):1051. https://doi.org/10.3390/genes12071051
Chicago/Turabian StyleBayat, Allan, Michael Bayat, Guido Rubboli, and Rikke S. Møller. 2021. "Epilepsy Syndromes in the First Year of Life and Usefulness of Genetic Testing for Precision Therapy" Genes 12, no. 7: 1051. https://doi.org/10.3390/genes12071051
APA StyleBayat, A., Bayat, M., Rubboli, G., & Møller, R. S. (2021). Epilepsy Syndromes in the First Year of Life and Usefulness of Genetic Testing for Precision Therapy. Genes, 12(7), 1051. https://doi.org/10.3390/genes12071051