
Unveiling the Pathogenesis of Psychiatric Disorders Using Network Models


Abstract
:1. Introduction
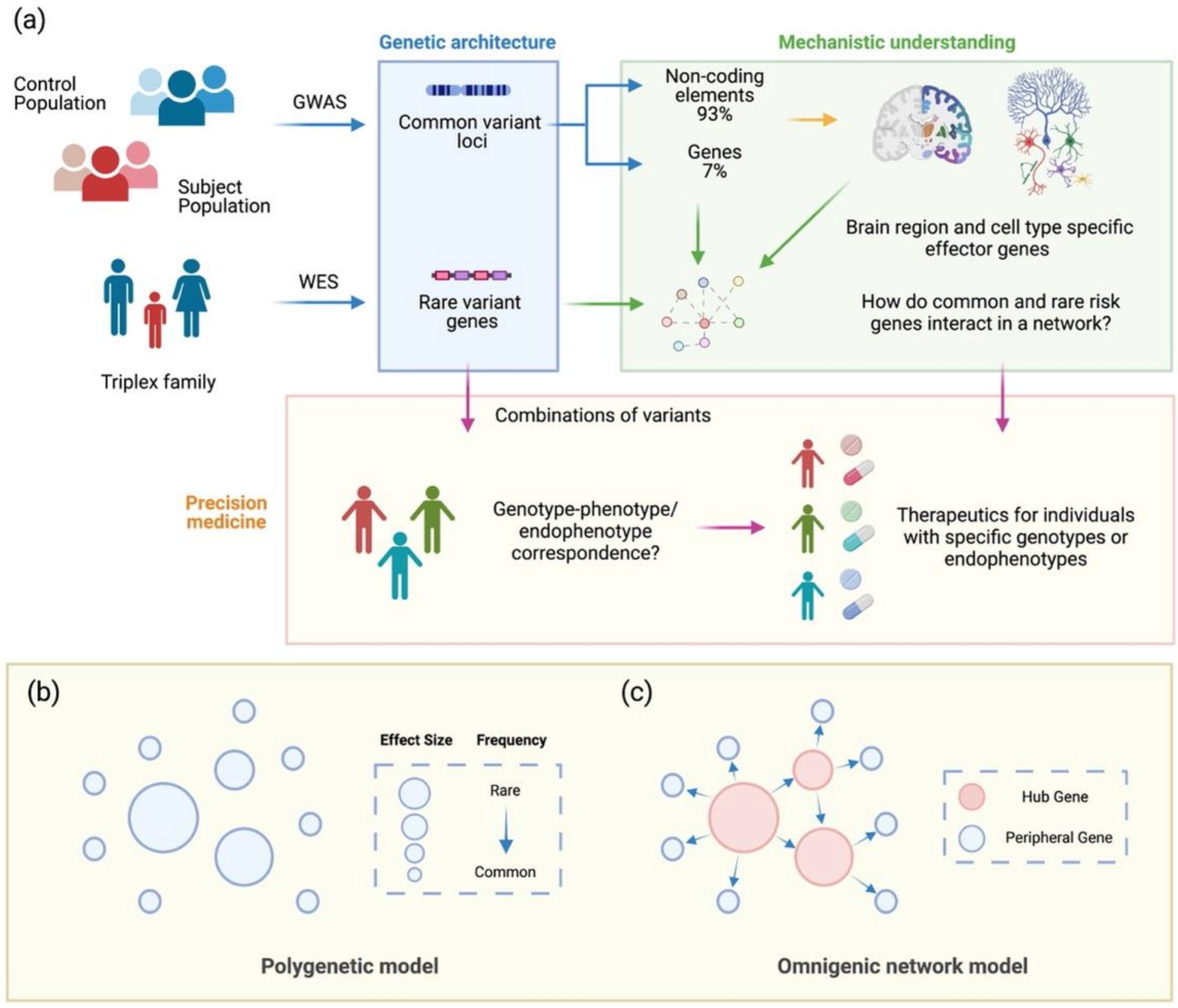
2. The Scope, Characteristics, and Genetic Architecture of Psychiatric Disorders
3. From a Polygenic Model to an Omnigenic Network Hypothesis of Psychiatric Disorders
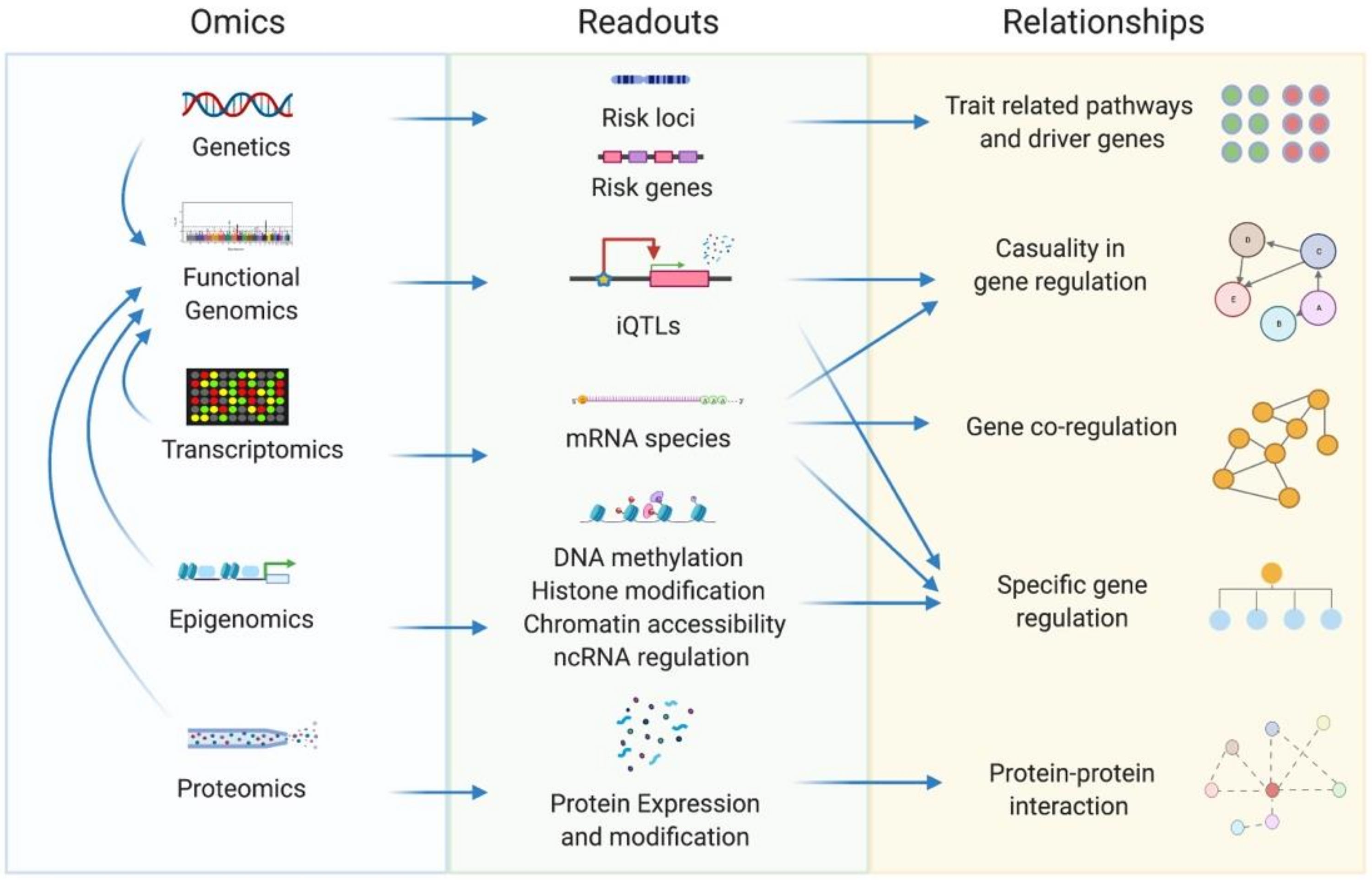
4. Connecting Disorder-Related Genetic Architecture to Network Models
5. A Survey of Current and Potential Network Methods and Applications in Psychiatric Research
5.1. Gene Regulatory Networks
5.1.1. Gene Co-expression Networks
5.1.2. Bayesian Networks (BNs)
5.1.3. Regulator-Target Pair Networks
5.2. PPI Networks
5.3. Literature-Based Networks
5.4. Hybrid Networks
5.5. Cross Disorder Network Applications
5.6. Network Applications on Treatment Response
5.7. Summary of New Insights Obtained from Network Studies of Psychiatric Disorders
6. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sullivan, P.F.; Geschwind, D.H. Defining the Genetic, Genomic, Cellular, and Diagnostic Architectures of Psychiatric Disorders. Cell 2019, 177, 162–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritchie, H.; Roser, M. Mental Health. Available online: https://ourworldindata.org/mental-health (accessed on 10 January 2021).
- Mental Illness. Available online: https://www.nimh.nih.gov/health/statistics/mental-illness.shtml (accessed on 7 January 2021).
- de la Torre-Ubieta, L.; Stein, J.L.; Won, H.; Opland, C.K.; Liang, D.; Lu, D.; Geschwind, D.H. The Dynamic Landscape of Open Chromatin during Human Cortical Neurogenesis. Cell 2018, 172, 289–304.e218. [Google Scholar] [CrossRef] [Green Version]
- Boix, C.A.; James, B.T.; Park, Y.P.; Meuleman, W.; Kellis, M. Regulatory genomic circuitry of human disease loci by integrative epigenomics. Nature 2021, 590, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Grove, J.; Ripke, S.; Als, T.D.; Mattheisen, M.; Walters, R.K.; Won, H.; Pallesen, J.; Agerbo, E.; Andreassen, O.A.; Anney, R.; et al. Identification of common genetic risk variants for autism spectrum disorder. Nat. Genet. 2019, 51, 431–444. [Google Scholar] [CrossRef] [Green Version]
- Satterstrom, F.K.; Kosmicki, J.A.; Wang, J.; Breen, M.S.; De Rubeis, S.; An, J.Y.; Peng, M.; Collins, R.; Grove, J.; Klei, L.; et al. Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell 2020, 180, 568–584.e523. [Google Scholar] [CrossRef]
- Kranzler, H.R.; Zhou, H.; Kember, R.L.; Smith, R.V.; Justice, A.C.; Damrauer, S.; Tsao, P.S.; Klarin, D.; Baras, A.; Reid, J. Genome-wide association study of alcohol consumption and use disorder in 274,424 individuals from multiple populations. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Howard, D.M.; Adams, M.J.; Clarke, T.-K.; Hafferty, J.D.; Gibson, J.; Shirali, M.; Coleman, J.R.; Hagenaars, S.P.; Ward, J.; Wigmore, E.M. Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat. Neurosci. 2019, 22, 343–352. [Google Scholar] [CrossRef] [Green Version]
- Stahl, E.A.; Breen, G.; Forstner, A.J.; McQuillin, A.; Ripke, S.; Trubetskoy, V.; Mattheisen, M.; Wang, Y.; Coleman, J.R.; Gaspar, H.A. Genome-wide association study identifies 30 loci associated with bipolar disorder. Nat. Genet. 2019, 51, 793–803. [Google Scholar] [CrossRef]
- Pardiñas, A.F.; Holmans, P.; Pocklington, A.J.; Escott-Price, V.; Ripke, S.; Carrera, N.; Legge, S.E.; Bishop, S.; Cameron, D.; Hamshere, M.L. Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong background selection. Nat. Genet. 2018, 50, 381–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nievergelt, C.M.; Maihofer, A.X.; Klengel, T.; Atkinson, E.G.; Chen, C.-Y.; Choi, K.W.; Coleman, J.R.; Dalvie, S.; Duncan, L.E.; Gelernter, J. International meta-analysis of PTSD genome-wide association studies identifies sex-and ancestry-specific genetic risk loci. Nat. Commun. 2019, 10, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amanat, S.; Requena, T.; Lopez-Escamez, J.A. A systematic review of extreme phenotype strategies to search for rare variants in genetic studies of complex disorders. Genes 2020, 11, 987. [Google Scholar] [CrossRef] [PubMed]
- Ripke, S.; Walters, J.T.; O’Donovan, M.C.; Schizophrenia Working Group of the Psychiatric Genomics Consortium. Mapping genomic loci prioritises genes and implicates synaptic biology in schizophrenia. MedRxiv 2020. [Google Scholar] [CrossRef]
- Genovese, G.; Fromer, M.; Stahl, E.A.; Ruderfer, D.M.; Chambert, K.; Landén, M.; Moran, J.L.; Purcell, S.M.; Sklar, P.; Sullivan, P.F. Increased burden of ultra-rare protein-altering variants among 4877 individuals with schizophrenia. Nat. Neurosci. 2016, 19, 1433–1441. [Google Scholar] [CrossRef] [PubMed]
- Iakoucheva, L.M.; Muotri, A.R.; Sebat, J. Getting to the cores of autism. Cell 2019, 178, 1287–1298. [Google Scholar] [CrossRef] [PubMed]
- Cai, N.; Bigdeli, T.B.; Kretzschmar, W.; Li, Y.; Liang, J.; Song, L.; Hu, J.; Li, Q.; Jin, W.; Hu, Z. Sparse whole-genome sequencing identifies two loci for major depressive disorder. Nature 2015, 523, 588–591. [Google Scholar]
- Geschwind, D.H.; Flint, J. Genetics and genomics of psychiatric disease. Science 2015, 349, 1489–1494. [Google Scholar] [CrossRef] [Green Version]
- Spencer, C.C.; Su, Z.; Donnelly, P.; Marchini, J. Designing genome-wide association studies: Sample size, power, imputation, and the choice of genotyping chip. PLoS Genet 2009, 5, e1000477. [Google Scholar] [CrossRef] [Green Version]
- Nishino, J.; Ochi, H.; Kochi, Y.; Tsunoda, T.; Matsui, S. Sample size for successful genome-wide association study of major depressive disorder. Front. Genet. 2018, 9, 227. [Google Scholar] [CrossRef]
- Timpson, N.J.; Greenwood, C.M.; Soranzo, N.; Lawson, D.J.; Richards, J.B. Genetic architecture: The shape of the genetic contribution to human traits and disease. Nat. Rev. Genet. 2018, 19, 110. [Google Scholar] [CrossRef]
- Boyle, E.A.; Li, Y.I.; Pritchard, J.K. An expanded view of complex traits: From polygenic to omnigenic. Cell 2017, 169, 1177–1186. [Google Scholar] [CrossRef] [Green Version]
- Vuckovic, D.; Bao, E.L.; Akbari, P.; Lareau, C.A.; Mousas, A.; Jiang, T.; Chen, M.-H.; Raffield, L.M.; Tardaguila, M.; Huffman, J.E. The polygenic and monogenic basis of blood traits and diseases. MedRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Khanin, R.; Wit, E. How scale-free are biological networks. J. Comput. Biol. 2006, 13, 810–818. [Google Scholar] [CrossRef]
- Liu, X.; Li, Y.I.; Pritchard, J.K. Trans effects on gene expression can drive omnigenic inheritance. Cell 2019, 177, 1022–1034.e1026. [Google Scholar] [CrossRef]
- Sinnott-Armstrong, N.; Naqvi, S.; Rivas, M.; Pritchard, J.K. GWAS of three molecular traits highlights core genes and pathways alongside a highly polygenic background. Elife 2021, 10, e58615. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Glass, K.; Röhl, A.; Santolini, M.; Croteau-Chonka, D.C.; Weiss, S.T.; Raby, B.A.; Sharma, A. The periphery and the core properties explain the omnigenic model in the human interactome. bioRxiv 2019, 749358. [Google Scholar]
- Yang, X. Multitissue Multiomics Systems Biology to Dissect Complex Diseases. Trends Mol. Med. 2020, 26.8, 718–728. [Google Scholar] [CrossRef]
- Chen, T.; Tyagi, S. Integrative computational epigenomics to build data-driven gene regulation hypotheses. GigaScience 2020, 9, giaa064. [Google Scholar] [CrossRef] [PubMed]
- Consortium, C.T. The nature and identification of quantitative trait loci: A community’s view. Nat. Rev. Genet. 2003, 4, 911. [Google Scholar]
- Ye, Y.; Zhang, Z.; Liu, Y.; Diao, L.; Han, L. A Multi-Omics Perspective of Quantitative Trait Loci in Precision Medicine. Trends Genet. 2020, 36.8, 318–336. [Google Scholar] [CrossRef]
- Civelek, M.; Lusis, A.J. Systems genetics approaches to understand complex traits. Nat. Rev. Genet. 2014, 15, 34–48. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Wiener, M.C.; Zhang, C.; Fridman, A.; Minch, E.; Lum, P.Y.; Sachs, J.R.; Schadt, E.E. Increasing the power to detect causal associations by combining genotypic and expression data in segregating populations. PLoS Comput. Biol. 2007, 3, e69. [Google Scholar] [CrossRef]
- Zheng, J.; Erzurumluoglu, A.M.; Elsworth, B.L.; Kemp, J.P.; Howe, L.; Haycock, P.C.; Hemani, G.; Tansey, K.; Laurin, C.; Pourcain, B.S. LD Hub: A centralized database and web interface to perform LD score regression that maximizes the potential of summary level GWAS data for SNP heritability and genetic correlation analysis. Bioinformatics 2017, 33, 272–279. [Google Scholar] [CrossRef] [PubMed]
- MacArthur, J.; Bowler, E.; Cerezo, M.; Gil, L.; Hall, P.; Hastings, E.; Junkins, H.; McMahon, A.; Milano, A.; Morales, J. The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res. 2017, 45, D896–D901. [Google Scholar] [CrossRef] [PubMed]
- Consortium, G. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 2020, 369, 1318–1330. [Google Scholar] [CrossRef]
- Moore, J.E.; Purcaro, M.J.; Pratt, H.E.; Epstein, C.B.; Shoresh, N.; Adrian, J.; Kawli, T.; Davis, C.A.; Dobin, A.; Kaul, R. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 2020, 583, 699–710. [Google Scholar] [CrossRef] [PubMed]
- Ramilowski, J.A.; Yip, C.W.; Agrawal, S.; Chang, J.C.; Ciani, Y.; Kulakovskiy, I.V.; Mendez, M.; Ooi, J.L.C.; Ouyang, J.F.; Parkinson, N.; et al. Functional annotation of human long noncoding RNAs via molecular phenotyping. Genome Res 2020, 30, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Gene Expression Omnibus. Available online: https://www.ncbi.nlm.nih.gov/geo/ (accessed on 7 January 2021).
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Blencowe, M.; Karunanayake, T.; Wier, J.; Hsu, N.; Yang, X. Network modeling approaches and applications to unravelling non-alcoholic fatty liver disease. Genes 2019, 10, 966. [Google Scholar] [CrossRef] [Green Version]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Song, W.-M.; Zhang, B. Multiscale embedded gene co-expression network analysis. PLoS Comput. Biol. 2015, 11, e1004574. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Zhang, B.; Smith, E.N.; Drees, B.; Brem, R.B.; Kruglyak, L.; Bumgarner, R.E.; Schadt, E.E. Integrating large-scale functional genomic data to dissect the complexity of yeast regulatory networks. Nat. Genet. 2008, 40, 854–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margolin, A.A.; Nemenman, I.; Basso, K.; Wiggins, C.; Stolovitzky, G.; Dalla Favera, R.; Califano, A. ARACNE: An algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context. BMC Bioinform. 2006, 7.1, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, V.; Bell, G.W.; Nam, J.-W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. elife 2015, 4, e05005. [Google Scholar] [CrossRef] [PubMed]
- Gilman, S.R.; Iossifov, I.; Levy, D.; Ronemus, M.; Wigler, M.; Vitkup, D. Rare de novo variants associated with autism implicate a large functional network of genes involved in formation and function of synapses. Neuron 2011, 70, 898–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, J.; Tunbridge, E.M.; Sandor, C.; Lyall, L.M.; Ferguson, A.; Strawbridge, R.J.; Lyall, D.M.; Cullen, B.; Graham, N.; Johnston, K.J. The genomic basis of mood instability: Identification of 46 loci in 363,705 UK Biobank participants, genetic correlation with psychiatric disorders, and association with gene expression and function. Mol. Psychiatry 2020, 25, 3091–3099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.K.; Carlin, D.E.; Yu, M.K.; Zhang, W.; Kreisberg, J.F.; Tamayo, P.; Ideker, T. Systematic evaluation of molecular networks for discovery of disease genes. Cell Syst. 2018, 6, 484–495.e485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnan, K.C.; Kurt, Z.; Barrere-Cain, R.; Sabir, S.; Das, A.; Floyd, R.; Vergnes, L.; Zhao, Y.; Che, N.; Charugundla, S. Integration of multi-omics data from mouse diversity panel highlights mitochondrial dysfunction in non-alcoholic fatty liver disease. Cell Syst. 2018, 6, 103–115.e107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurt, Z.; Barrere-Cain, R.; LaGuardia, J.; Mehrabian, M.; Pan, C.; Hui, S.T.; Norheim, F.; Zhou, Z.; Hasin, Y.; Lusis, A.J. Tissue-specific pathways and networks underlying sexual dimorphism in non-alcoholic fatty liver disease. Biol. Sex Differ. 2018, 9, 1–14. [Google Scholar] [CrossRef]
- Kapoor, M.; Wang, J.-C.; Farris, S.P.; Liu, Y.; McClintick, J.; Gupta, I.; Meyers, J.L.; Bertelsen, S.; Chao, M.; Nurnberger, J. Analysis of whole genome-transcriptomic organization in brain to identify genes associated with alcoholism. Transl. Psychiatry 2019, 9, 1–11. [Google Scholar] [CrossRef]
- Parikshak, N.N.; Luo, R.; Zhang, A.; Won, H.; Lowe, J.K.; Chandran, V.; Horvath, S.; Geschwind, D.H. Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell 2013, 155, 1008–1021. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.J.; Kawasawa, Y.I.; Cheng, F.; Zhu, Y.; Xu, X.; Li, M.; Sousa, A.M.; Pletikos, M.; Meyer, K.A.; Sedmak, G. Spatio-temporal transcriptome of the human brain. Nature 2011, 478, 483–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parikshak, N.N.; Swarup, V.; Belgard, T.G.; Irimia, M.; Ramaswami, G.; Gandal, M.J.; Hartl, C.; Leppa, V.; de la Torre Ubieta, L.; Huang, J. Genome-wide changes in lncRNA, splicing, and regional gene expression patterns in autism. Nature 2016, 540, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Velmeshev, D.; Schirmer, L.; Jung, D.; Haeussler, M.; Perez, Y.; Mayer, S.; Bhaduri, A.; Goyal, N.; Rowitch, D.H.; Kriegstein, A.R. Single-cell genomics identifies cell type–specific molecular changes in autism. Science 2019, 364, 685–689. [Google Scholar] [CrossRef]
- Willsey, A.J.; Sanders, S.J.; Li, M.; Dong, S.; Tebbenkamp, A.T.; Muhle, R.A.; Reilly, S.K.; Lin, L.; Fertuzinhos, S.; Miller, J.A. Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell 2013, 155, 997–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Sova, P.; Xu, Q.; Dombek, K.M.; Xu, E.Y.; Vu, H.; Tu, Z.; Brem, R.B.; Bumgarner, R.E.; Schadt, E.E. Stitching together multiple data dimensions reveals interacting metabolomic and transcriptomic networks that modulate cell regulation. PLoS Biol. 2012, 10, e1001301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scarpa, J.R.; Jiang, P.; Gao, V.D.; Fitzpatrick, K.; Millstein, J.; Olker, C.; Gotter, A.; Winrow, C.J.; Renger, J.J.; Kasarskis, A. Cross-species systems analysis identifies gene networks differentially altered by sleep loss and depression. Sci. Adv. 2018, 4, eaat1294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greene, C.S.; Krishnan, A.; Wong, A.K.; Ricciotti, E.; Zelaya, R.A.; Himmelstein, D.S.; Zhang, R.; Hartmann, B.M.; Zaslavsky, E.; Sealfon, S.C. Understanding multicellular function and disease with human tissue-specific networks. Nat. Genet. 2015, 47, 569–576. [Google Scholar] [CrossRef] [Green Version]
- Krishnan, A.; Zhang, R.; Yao, V.; Theesfeld, C.L.; Wong, A.K.; Tadych, A.; Volfovsky, N.; Packer, A.; Lash, A.; Troyanskaya, O.G. Genome-wide prediction and functional characterization of the genetic basis of autism spectrum disorder. Nat. Neurosci. 2016, 19, 1454–1462. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Gaiteri, C.; Bodea, L.-G.; Wang, Z.; McElwee, J.; Podtelezhnikov, A.A.; Zhang, C.; Xie, T.; Tran, L.; Dobrin, R. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell 2013, 153, 707–720. [Google Scholar] [CrossRef] [Green Version]
- Shu, L.; Chan, K.H.K.; Zhang, G.; Huan, T.; Kurt, Z.; Zhao, Y.; Codoni, V.; Trégouët, D.-A.; Yang, J.; Wilson, J.G. Shared genetic regulatory networks for cardiovascular disease and type 2 diabetes in multiple populations of diverse ethnicities in the United States. PLoS Genet. 2017, 13, e1007040. [Google Scholar] [CrossRef] [Green Version]
- Chai, L.E.; Loh, S.K.; Low, S.T.; Mohamad, M.S.; Deris, S.; Zakaria, Z. A review on the computational approaches for gene regulatory network construction. Comput. Biol. Med. 2014, 48, 55–65. [Google Scholar] [CrossRef]
- Pearl, J.R.; Colantuoni, C.; Bergey, D.E.; Funk, C.C.; Shannon, P.; Basu, B.; Casella, A.M.; Oshone, R.T.; Hood, L.; Price, N.D. Genome-Scale Transcriptional Regulatory Network Models of Psychiatric and Neurodegenerative Disorders. Cell Syst. 2019, 8, 122–135.e127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klengel, T.; Binder, E.B. Epigenetics of stress-related psychiatric disorders and gene × environment interactions. Neuron 2015, 86, 1343–1357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.E.; Parikshak, N.N.; Belgard, T.G.; Geschwind, D.H. Genome-wide, integrative analysis implicates microRNA dysregulation in autism spectrum disorder. Nat. Neurosci. 2016, 19, 1463–1476. [Google Scholar] [CrossRef] [PubMed]
- Ravasi, T.; Suzuki, H.; Cannistraci, C.V.; Katayama, S.; Bajic, V.B.; Tan, K.; Akalin, A.; Schmeier, S.; Kanamori-Katayama, M.; Bertin, N. An atlas of combinatorial transcriptional regulation in mouse and man. Cell 2010, 140, 744–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Repunte-Canonigo, V.; Shin, W.; Vendruscolo, L.F.; Lefebvre, C.; van der Stap, L.; Kawamura, T.; Schlosburg, J.E.; Alvarez, M.; Koob, G.F.; Califano, A. Identifying candidate drivers of alcohol dependence-induced excessive drinking by assembly and interrogation of brain-specific regulatory networks. Genome Biol. 2015, 16, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Fletcher, M.N.; Castro, M.A.; Wang, X.; De Santiago, I.; O’Reilly, M.; Chin, S.-F.; Rueda, O.M.; Caldas, C.; Ponder, B.A.; Markowetz, F. Master regulators of FGFR2 signalling and breast cancer risk. Nat. Commun. 2013, 4, 1–12. [Google Scholar] [CrossRef]
- Pfaffenseller, B.; da Silva Magalhães, P.; De Bastiani, M.A.; Castro, M.A.A.; Gallitano, A.L.; Kapczinski, F.; Klamt, F. Differential expression of transcriptional regulatory units in the prefrontal cortex of patients with bipolar disorder: Potential role of early growth response gene 3. Transl. Psychiatry 2016, 6, e805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krämer, A.; Green, J.; Pollard, J., Jr.; Tugendreich, S. Causal analysis approaches in ingenuity pathway analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef]
- Bam, M.; Yang, X.; Zhou, J.; Ginsberg, J.P.; Leyden, Q.; Nagarkatti, P.S.; Nagarkatti, M. Evidence for epigenetic regulation of pro-inflammatory cytokines, interleukin-12 and interferon γ, in peripheral blood mononuclear cells from PTSD patients. J. Neuroimmune Pharmacol. 2016, 11, 168–181. [Google Scholar] [CrossRef] [Green Version]
- Rossin, E.J.; Lage, K.; Raychaudhuri, S.; Xavier, R.J.; Tatar, D.; Benita, Y.; Cotsapas, C.; Daly, M.J.; Constortium, I.I.B.D.G. Proteins encoded in genomic regions associated with immune-mediated disease physically interact and suggest underlying biology. PLoS Genet 2011, 7, e1001273. [Google Scholar] [CrossRef] [Green Version]
- Jia, P.; Zheng, S.; Long, J.; Zheng, W.; Zhao, Z. dmGWAS: Dense module searching for genome-wide association studies in protein–protein interaction networks. Bioinformatics 2011, 27, 95–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ideker, T.; Ozier, O.; Schwikowski, B.; Siegel, A.F. Discovering regulatory and signalling circuits in molecular interaction networks. Bioinformatics 2002, 18, S233–S240. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Cai, M.; Xing, X.; Ji, J.; Yang, E.; Wu, J. PINA 3.0: Mining cancer interactome. Nucleic Acids Res. 2021, 49, D1351–D1357. [Google Scholar] [CrossRef] [PubMed]
- Blizinsky, K.D.; Diaz-Castro, B.; Forrest, M.P.; Schürmann, B.; Bach, A.P.; Martin-de-Saavedra, M.D.; Wang, L.; Csernansky, J.G.; Duan, J.; Penzes, P. Reversal of dendritic phenotypes in 16p11. 2 microduplication mouse model neurons by pharmacological targeting of a network hub. Proc. Natl. Acad. Sci. USA 2016, 113, 8520–8525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gulsuner, S.; Walsh, T.; Watts, A.C.; Lee, M.K.; Thornton, A.M.; Casadei, S.; Rippey, C.; Shahin, H.; Braff, D.; Cadenhead, K.S. Spatial and temporal mapping of de novo mutations in schizophrenia to a fetal prefrontal cortical network. Cell 2013, 154, 518–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pomaznoy, M.; Ha, B.; Peters, B. GOnet: A Tool for Interactive Gene Ontology Analysis. BMC Bioinform. 2018, 19, 470. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Gilman, S.R.; Chiang, A.H.; Sanders, S.J.; Vitkup, D. Genotype to phenotype relationships in autism spectrum disorders. Nat. Neurosci. 2015, 18, 191–198. [Google Scholar] [CrossRef] [Green Version]
- Sandor, C.; Beer, N.L.; Webber, C. Diverse type 2 diabetes genetic risk factors functionally converge in a phenotype-focused gene network. PLoS Comput. Biol. 2017, 13, e1005816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gazestani, V.H.; Pramparo, T.; Nalabolu, S.; Kellman, B.P.; Murray, S.; Lopez, L.; Pierce, K.; Courchesne, E.; Lewis, N.E. A perturbed gene network containing PI3K–AKT, RAS–ERK and WNT–β-catenin pathways in leukocytes is linked to ASD genetics and symptom severity. Nat. Neurosci. 2019, 22, 1624–1634. [Google Scholar] [CrossRef]
- Gandal, M.J.; Haney, J.R.; Parikshak, N.N.; Leppa, V.; Ramaswami, G.; Hartl, C.; Schork, A.J.; Appadurai, V.; Buil, A.; Werge, T.M. Shared molecular neuropathology across major psychiatric disorders parallels polygenic overlap. Science 2018, 359, 693–697. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; Ripke, S.; Neale, B.M.; Faraone, S.V.; Purcell, S.M.; Perlis, R.H.; Mowry, B.J.; Thapar, A.; Goddard, M.E.; Witte, J.S. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat. Genet. 2013, 45, 984. [Google Scholar]
- The Network; Pathway Analysis Subgroup of the Psychiatric Genomics Consortium. Psychiatric genome-wide association study analyses implicate neuronal, immune and histone pathways. Nat. Neurosci. 2015, 18, 10. [Google Scholar] [CrossRef]
- Gandal, M.J.; Zhang, P.; Hadjimichael, E.; Walker, R.L.; Chen, C.; Liu, S.; Won, H.; Van Bakel, H.; Varghese, M.; Wang, Y. Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science 2018, 362, eaat8127. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Hwang, Y.; Webster, M.; Lee, D. Differential activation of immune/inflammatory response-related co-expression modules in the hippocampus across the major psychiatric disorders. Mol. Psychiatry 2016, 21, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Breen, M.; White, C.; Shekhtman, T.; Lin, K.; Looney, D.; Woelk, C.; Kelsoe, J. Lithium-responsive genes and gene networks in bipolar disorder patient-derived lymphoblastoid cell lines. Pharm. J. 2016, 16, 446–453. [Google Scholar] [CrossRef]
- Yoo, M.; Shin, J.; Kim, J.; Ryall, K.A.; Lee, K.; Lee, S.; Jeon, M.; Kang, J.; Tan, A.C. DSigDB: Drug signatures database for gene set analysis. Bioinformatics 2015, 31, 3069–3071. [Google Scholar] [CrossRef]
- Voineagu, I.; Wang, X.; Johnston, P.; Lowe, J.K.; Tian, Y.; Horvath, S.; Mill, J.; Cantor, R.M.; Blencowe, B.J.; Geschwind, D.H. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 2011, 474, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Sanders, S.J.; He, X.; Willsey, A.J.; Ercan-Sencicek, A.G.; Samocha, K.E.; Cicek, A.E.; Murtha, M.T.; Bal, V.H.; Bishop, S.L.; Dong, S. Insights into autism spectrum disorder genomic architecture and biology from 71 risk loci. Neuron 2015, 87, 1215–1233. [Google Scholar] [CrossRef] [Green Version]
- De Rubeis, S.; He, X.; Goldberg, A.P.; Poultney, C.S.; Samocha, K.; Cicek, A.E.; Kou, Y.; Liu, L.; Fromer, M.; Walker, S. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014, 515, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Lin, Y.; Ji, G. Cell Type-Specific Gene Network-Based Analysis Depicts the Heterogeneity of Autism Spectrum Disorder. Front. Cell. Neurosci. 2020, 14, 59. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Ellis, S.E.; Ashar, F.N.; Moes, A.; Bader, J.S.; Zhan, J.; West, A.B.; Arking, D.E. Transcriptome analysis reveals dysregulation of innate immune response genes and neuronal activity-dependent genes in autism. Nat. Commun. 2014, 5, 1–8. [Google Scholar] [CrossRef]
- Luo, Y.; Eran, A.; Palmer, N.; Avillach, P.; Levy-Moonshine, A.; Szolovits, P.; Kohane, I.S. A multidimensional precision medicine approach identifies an autism subtype characterized by dyslipidemia. Nat. Med. 2020, 26, 1375–1379. [Google Scholar] [CrossRef]
- Werling, D.M.; Parikshak, N.N.; Geschwind, D.H. Gene expression in human brain implicates sexually dimorphic pathways in autism spectrum disorders. Nat. Commun. 2016, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Mamdani, M.; Williamson, V.; McMichael, G.O.; Blevins, T.; Aliev, F.; Adkins, A.; Hack, L.; Bigdeli, T.; Van Der Vaart, A.D.; Web, B.T. Integrating mRNA and miRNA weighted gene co-expression networks with eQTLs in the nucleus accumbens of subjects with alcohol dependence. PLoS ONE 2015, 10, e0137671. [Google Scholar] [CrossRef]
- Tapocik, J.D.; Solomon, M.; Flanigan, M.; Meinhardt, M.; Barbier, E.; Schank, J.; Schwandt, M.; Sommer, W.H.; Heilig, M. Coordinated dysregulation of mRNAs and microRNAs in the rat medial prefrontal cortex following a history of alcohol dependence. Pharm. J. 2013, 13, 286–296. [Google Scholar] [CrossRef] [Green Version]
- Xiang, B.; Liu, K.; Yu, M.; Liang, X.; Zhang, J.; Lei, W.; Huang, C.; Chen, J.; Gu, X.; Li, N. Systematic genetic analyses of genome-wide association study data reveal an association between the key nucleosome remodeling and deacetylase complex and bipolar disorder development. Bipolar Disord. 2018, 20, 370–380. [Google Scholar] [CrossRef]
- Akula, N.; Wendland, J.R.; Choi, K.H.; McMahon, F.J. An integrative genomic study implicates the postsynaptic density in the pathogenesis of bipolar disorder. Neuropsychopharmacology 2016, 41, 886–895. [Google Scholar] [CrossRef]
- Toma, C.; Shaw, A.D.; Overs, B.J.; Mitchell, P.B.; Schofield, P.R.; Cooper, A.A.; Fullerton, J.M. De Novo Gene Variants and Familial Bipolar Disorder. JAMA Netw. Open 2020, 3, e203382. [Google Scholar] [CrossRef]
- Schubert, K.O.; Stacey, D.; Arentz, G.; Clark, S.R.; Air, T.; Hoffmann, P.; Baune, B.T. Targeted proteomic analysis of cognitive dysfunction in remitted major depressive disorder: Opportunities of multi-omics approaches towards predictive, preventive, and personalized psychiatry. J. Proteom. 2018, 188, 63–70. [Google Scholar] [CrossRef]
- Ciobanu, L.G.; Sachdev, P.S.; Trollor, J.N.; Reppermund, S.; Thalamuthu, A.; Mather, K.A.; Cohen-Woods, S.; Stacey, D.; Toben, C.; Schubert, K.O. Co-expression network analysis of peripheral blood transcriptome identifies dysregulated protein processing in endoplasmic reticulum and immune response in recurrent MDD in older adults. J. Psychiatr. Res. 2018, 107, 19–27. [Google Scholar] [CrossRef] [Green Version]
- Zeng, D.; He, S.; Ma, C.; Wen, Y.; Xie, Y.; Zhao, N.; Sun, X.; Wang, D.; Shen, Y.; Yu, Y. Co-expression network analysis revealed that the ATP5G1 gene is associated with major depressive disorder. Front. Genet. 2019, 10, 703. [Google Scholar] [CrossRef] [Green Version]
- Breen, M.S.; Tylee, D.S.; Maihofer, A.X.; Neylan, T.C.; Mehta, D.; Binder, E.B.; Chandler, S.D.; Hess, J.L.; Kremen, W.S.; Risbrough, V.B. PTSD blood transcriptome mega-analysis: Shared inflammatory pathways across biological sex and modes of trauma. Neuropsychopharmacology 2018, 43, 469–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torkamani, A.; Dean, B.; Schork, N.J.; Thomas, E.A. Coexpression network analysis of neural tissue reveals perturbations in developmental processes in schizophrenia. Genome Res. 2010, 20, 403–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacDonald, M.L.; Ding, Y.; Newman, J.; Hemby, S.; Penzes, P.; Lewis, D.A.; Yates, N.A.; Sweet, R.A. Altered glutamate protein co-expression network topology linked to spine loss in the auditory cortex of schizophrenia. Biol. Psychiatry 2015, 77, 959–968. [Google Scholar] [CrossRef]
- Kim, M.; Haney, J.R.; Zhang, P.; Hernandez, L.M.; Wang, L.-k.; Perez-Cano, L.; Gandal, M.J. Network signature of complement component 4 variation in the human brain identifies convergent molecular risk for schizophrenia. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Torshizi, A.D.; Armoskus, C.; Zhang, H.; Forrest, M.P.; Zhang, S.; Souaiaia, T.; Evgrafov, O.V.; Knowles, J.A.; Duan, J.; Wang, K. Deconvolution of transcriptional networks identifies TCF4 as a master regulator in schizophrenia. Sci. Adv. 2019, 5, eaau4139. [Google Scholar] [CrossRef] [Green Version]
- Gilman, S.R.; Chang, J.; Xu, B.; Bawa, T.S.; Gogos, J.A.; Karayiorgou, M.; Vitkup, D. Diverse types of genetic variation converge on functional gene networks involved in schizophrenia. Nat. Neurosci. 2012, 15, 1723. [Google Scholar] [CrossRef] [PubMed]
- Anttila, V.; Bulik-Sullivan, B.; Finucane, H.K.; Walters, R.K.; Bras, J.; Duncan, L.; Escott-Price, V.; Falcone, G.J.; Gormley, P.; Malik, R. Analysis of shared heritability in common disorders of the brain. Science 2018, 360, eeap8757. [Google Scholar]
- Fromer, M.; Roussos, P.; Sieberts, S.K.; Johnson, J.S.; Kavanagh, D.H.; Perumal, T.M.; Ruderfer, D.M.; Oh, E.C.; Topol, A.; Shah, H.R. Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nat. Neurosci. 2016, 19, 1442–1453. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.A.; Peñagarikano, O.; Belgard, T.G.; Swarup, V.; Geschwind, D.H. The emerging picture of autism spectrum disorder: Genetics and pathology. Annu. Rev. Pathol. Mech. Dis. 2015, 10, 111–144. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Pedrosa, E.; Hrabovsky, A.; Chen, J.; Puliafito, B.R.; Gilbert, S.R.; Zheng, D.; Lachman, H.M. Integrative transcriptome network analysis of iPSC-derived neurons from schizophrenia and schizoaffective disorder patients with 22q11. 2 deletion. BMC Syst. Biol. 2016, 10, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedroso, I.; Lourdusamy, A.; Rietschel, M.; Nöthen, M.M.; Cichon, S.; McGuffin, P.; Al-Chalabi, A.; Barnes, M.R.; Breen, G. Common genetic variants and gene-expression changes associated with bipolar disorder are over-represented in brain signaling pathway genes. Biol. Psychiatry 2012, 72, 311–317. [Google Scholar] [CrossRef] [Green Version]
- Katrinli, S.; Lori, A.; Kilaru, V.; Carter, S.; Powers, A.; Gillespie, C.F.; Wingo, A.P.; Michopoulos, V.; Jovanovic, T.; Ressler, K.J. Association of HLA locus alleles with posttraumatic stress disorder. Brain Behav. Immun. 2019, 81, 655–658. [Google Scholar] [CrossRef] [PubMed]
- Jia, P.; Kao, C.-F.; Kuo, P.-H.; Zhao, Z. A comprehensive network and pathway analysis of candidate genes in major depressive disorder. BMC Syst. Biol. 2011, 5, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagy, C.; Maitra, M.; Tanti, A.; Suderman, M.; Théroux, J.-F.; Davoli, M.A.; Perlman, K.; Yerko, V.; Wang, Y.C.; Tripathy, S.J. Single-nucleus transcriptomics of the prefrontal cortex in major depressive disorder implicates oligodendrocyte precursor cells and excitatory neurons. Nat. Neurosci. 2020, 23, 771–781. [Google Scholar] [CrossRef]
- Breen, M.S.; Maihofer, A.X.; Glatt, S.J.; Tylee, D.S.; Chandler, S.D.; Tsuang, M.T.; Risbrough, V.B.; Baker, D.G.; O’Connor, D.T.; Nievergelt, C.M. Gene networks specific for innate immunity define post-traumatic stress disorder. Mol. Psychiatry 2015, 20, 1538–1545. [Google Scholar] [CrossRef] [Green Version]
- Bam, M.; Yang, X.; Zumbrun, E.E.; Zhong, Y.; Zhou, J.; Ginsberg, J.P.; Leyden, Q.; Zhang, J.; Nagarkatti, P.S.; Nagarkatti, M. Dysregulated immune system networks in war veterans with PTSD is an outcome of altered miRNA expression and DNA methylation. Sci. Rep. 2016, 6, 1–13. [Google Scholar] [CrossRef]
- Mehta, D.; Voisey, J.; Bruenig, D.; Harvey, W.; Morris, C.P.; Lawford, B.; Young, R.M. Transcriptome analysis reveals novel genes and immune networks dysregulated in veterans with PTSD. Brain Behav. Immun. 2018, 74, 133–142. [Google Scholar] [CrossRef]
- Logue, M.W.; Smith, A.K.; Baldwin, C.; Wolf, E.J.; Guffanti, G.; Ratanatharathorn, A.; Stone, A.; Schichman, S.A.; Humphries, D.; Binder, E.B. An analysis of gene expression in PTSD implicates genes involved in the glucocorticoid receptor pathway and neural responses to stress. Psychoneuroendocrinology 2015, 57, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Bagot, R.C.; Cates, H.M.; Purushothaman, I.; Lorsch, Z.S.; Walker, D.M.; Wang, J.; Huang, X.; Schlüter, O.M.; Maze, I.; Peña, C.J. Circuit-wide transcriptional profiling reveals brain region-specific gene networks regulating depression susceptibility. Neuron 2016, 90, 969–983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunez, Y.O.; Truitt, J.M.; Gorini, G.; Ponomareva, O.N.; Blednov, Y.A.; Harris, R.A.; Mayfield, R.D. Positively correlated miRNA-mRNA regulatory networks in mouse frontal cortex during early stages of alcohol dependence. BMC Genom. 2013, 14, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Erickson, E.K.; Blednov, Y.A.; Harris, R.A.; Mayfield, R.D. Glial gene networks associated with alcohol dependence. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Blencowe, M.; Arneson, D.; Ding, J.; Chen, Y.-W.; Saleem, Z.; Yang, X. Network modeling of single-cell omics data: Challenges, opportunities, and progresses. Emerg. Top. Life Sci. 2019, 3, 379–398. [Google Scholar]
- Efremova, M.; Vento-Tormo, M.; Teichmann, S.A.; Vento-Tormo, R. CellPhoneDB: Inferring cell–cell communication from combined expression of multi-subunit ligand–receptor complexes. Nat. Protoc. 2020, 15, 1484–1506. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, R.; Zhang, S.; Song, S.; Jiang, C.; Han, G.; Wang, M.; Ajani, J.; Futreal, A.; Wang, L. iTALK: An R package to characterize and illustrate intercellular communication. BioRxiv 2019, 507871. [Google Scholar]
- Aibar, S.; González-Blas, C.B.; Moerman, T.; Imrichova, H.; Hulselmans, G.; Rambow, F.; Marine, J.-C.; Geurts, P.; Aerts, J.; van den Oord, J. SCENIC: Single-cell regulatory network inference and clustering. Nat. Methods 2017, 14, 1083–1086. [Google Scholar] [CrossRef] [Green Version]
- Li, W.V.; Li, Y. scLink: Inferring Sparse Gene Co-expression Networks from Single-cell Expression Data. bioRxiv 2020. [Google Scholar] [CrossRef]
- Hu, Y.; Peng, T.; Gao, L.; Tan, K. CytoTalk: De novo construction of signal transduction networks using single-cell transcriptomic data. Sci. Adv. 2021, 7, eabf1356. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.; Wang, K.; Brunetti, T.; Xia, Y.; Jiao, C.; Dai, R.; Fitzgerald, D.; Thomas, A.; Jay, L.; Eckart, H. The DGCR5 long noncoding RNA may regulate expression of several schizophrenia-related genes. Sci. Transl. Med. 2018, 10, eeat6912. [Google Scholar] [CrossRef] [Green Version]
- Huckins, L.M.; Chatzinakos, C.; Breen, M.S.; Hartmann, J.; Klengel, T.; da Silva Almeida, A.C.; Dobbyn, A.; Girdhar, K.; Hoffman, G.E.; Klengel, C. Analysis of genetically regulated gene expression identifies a prefrontal PTSD gene, SNRNP35, specific to military cohorts. Cell Rep. 2020, 31, 107716. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Omics | Database | Description | URL | Usage in Network Applications |
|---|---|---|---|---|
| Genetics | GWAS catalog | Collections of the GWAS summary statistics files | https://www.ebi.ac.uk/gwas/ (accessed on 11 January 2021) | Find trait-related genes, pathways, and subnetworks |
| LD-hub | http://ldsc.broadinstitute.org/ (accessed on 11 January 2021) | |||
| PGC | https://www.med.unc.edu/pgc/ (accessed on 11 January 2021) | |||
| Genomics/Functional genomics/Transcriptomics | GTEx | Genotype, transcriptome, eQTLs, and sQTLs profiles across 13 brain regions from 948 donors and 2642 samples | https://www.gtexportal.org/home/ (accessed on 11 January 2021) | “Building bricks” for gene regulatory networks |
| GEO | A repository for various data types including genotypes, bulk tissue RNA-seq and single-cell RNA-seq datasets | https://www.ncbi.nlm.nih.gov/geo/ (accessed on 11 January 2021) | ||
| PsychENCODE | A repository specifically for neuropsychiatric disorders including RNA-seq datasets, SNP genotypes, epigenomic datasets and gene regulatory networks | http://resource.psychencode.org/ (accessed on 11 January 2021) https://www.synapse.org/#!Synapse:syn4921369/wiki/235539 (accessed on 11 January 2021) | ||
| BrainSpan | Transcriptional profiles of 16 cortical and subcortical regions with a temporal coverage across pre- and post-natal development in both males and females | http://www.brainspan.org/static/download.html (accessed on 11 January 2021) | ||
| Epigenomics | ENCODE | Transcriptional regulator and epigenomic factor profiles from 706 brain samples | https://www.encodeproject.org/ (accessed on 11 January 2021) | Provide regulator-target pair information |
| FANTOM | Atlases of transcriptional regulatory networks, promoters, enhancers, lncRNAs, and miRNAs | https://fantom.gsc.riken.jp/ (accessed on 11 January 2021) | ||
| Proteomics | STRING DB | Curated protein interactions including 24.6 million proteins from 5090 organisms | https://string-db.org/ (accessed on 11 January 2021) | Provide protein-protein interaction information |
| Networks | Relationship Captured | Disadvantages | Example Construction Methods | |
|---|---|---|---|---|
| Gene regulatory network | Co-expression network | Covariation and co-regulation among gene clusters |
| WGCNA [42]; MEGENA [43] |
| Bayesian network | Causality of regulation between gene pairs |
| RIMBANet [44] | |
| Regulator-target pair network | Specific regulation of certain transcriptional factors/non-coding RNAs |
| From database (FANTOM) [38]; ARACNe [45]; TargetScan [46] | |
| Protein-protein interaction network | Physical interaction affinity between pairs of proteins |
| From database (STRINGDB) [40] | |
| Literature-based network | Background likelihood network | Possibility of gene pairs participating in a similar genetic phenotype |
| Gilman et al., 2011 [47] |
| Phenotypic-linkage network | Gene clusters related with disease-related phenotypes curated from the literature | Ward et al., 2020 [48] | ||
| Hybrid network | General gene-gene interactions, PPIs, and literature co-occurrence | Use premade networks (e.g., PCNet) [49]; Custom script | ||
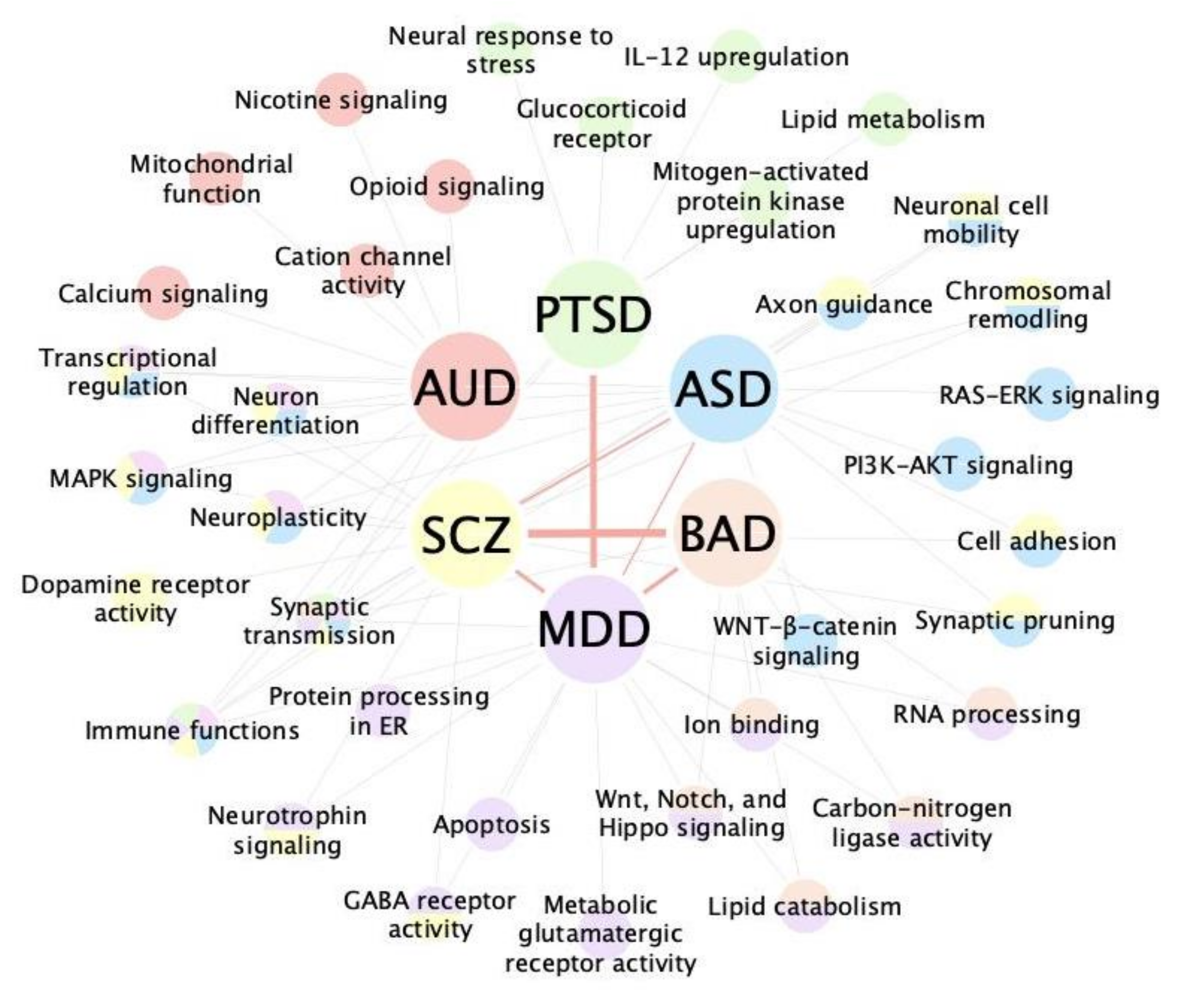
| Disorder | Networks | Key Findings | Ref. |
|---|---|---|---|
| ASD | Co-expression network | Synapse and immune response-related modules are affected in frontal and temporal cortex from ASD subjects; ASD rare variants affects early transcriptional regulation and synaptic development pathways and are enriched in superficial cortical layers and glutamatergic projection neurons in developing and adult human cortex. | Voineagu et al. [91]; Parikshak et al. [53] |
| Protein-protein interaction network | ASD rare variant related protein interactions are enriched in synaptic transmission, cell junction, TGFβ pathway, neurodegeneration, and transcriptional regulation. | de Rubies et al. [93]; Sanders et al. [92] | |
| Bayesian network | Synaptic transmission, MAPK signaling, histone modification, and immune response are the top affected functions in predicted ASD risk genes using a brain-specific network. | Krishnan et al. [61] | |
| Literature-based network | ASD rare variant genes form a network related to synapse development, axon targeting, and neuron motility; Genes in ASD rare variant and single nucleotide variants informed network are expressed at the highest level in cortical interneurons, pyramidal neurons, and the medium spiny neurons of the striatum. | Gilman et al. [47]; Chang et al. [81] | |
| Hybrid network | The ASD network constructed with the peripheral blood transcriptome in children with ASD was enriched for ASD rare mutation genes, as well as their regulatory targets and regulators. RAS–ERK, PI3K–AKT, and WNT–β-catenin signaling pathways are enriched in ASD-specific networks. | Gazestani et al. [83] | |
| AUD | Co-expression network | In prefrontal cortex samples from human AUD subjects, a module functioning in calcium signaling, nicotine response and opioid signaling are down-regulated in AUD, while another module functioning in immune signaling are up-regulated in AUD; In nucleus accumbens samples from human AUD subjects, two neuronal modules enriched for genes in oxidative phosphorylation, mitochondrial dysfunction, and MAPK signaling pathways are down-regulated in AUD, while four immune-related modules enriched for astrocyte and microglia markers are up-regulated in AUD. | Kapoor et al. [52]; Mamdani et al. [98] |
| Transcription factor/miRNA regulons | Pathways related to synaptic processes and neuroplasticity are disrupted in a rat AUD model; Nr3c1 acts as a master regulator in multiple brain regions in alcohol-dependent rats. | Tapocik et al., 2013 [99]; Repunte-Canonigo et al., 2015 [69] | |
| BAD | Co-expression network | BAD common variants are enriched in the hippocampus and amygdala across developmental stages. In dorsolateral frontal cortex samples from human BAD subjects, modules enriched for genes related to postsynaptic density, RNA processing, and carbon-nitrogen ligase activity are downregulated, while modules enriched for genes related to ion binding and lipid catabolism are upregulated. | Xiang et al. [100]; Akula et al. [101] |
| Transcription factor regulons | EGR3, TSC22D4, ILF2, YBX1 and MADD are predicted as master regulators in human prefrontal cortex with BAD. | Pfaffenseller et al. [71] | |
| Protein-protein interaction network | CDH4, MTA2, RBBP4, and HDAC2 are the core genes predicted by PPI analysis, involved in early brain development regulation. HP and PC are related to BAD de novo mutations; MAP4, WDHD1, EIF4E and STRN are related to the BAD common variant loci. | Xiang et al. [100]; Toma et al. [102] | |
| MDD | Co-expression network | CCND3, TXND5, TRI26 are the driver genes for cognitive dysfunction in MDD, validated by plasma protein level in MDD subjects; Immune response and protein processing in the ER are disrupted in older adults with recurrent MDD | Schubert et. al. [103]; Ciobanu et al. [104] |
| Protein-protein interaction network | The ATP5G1 gene is associated with the pathogenesis of MDD | Zeng et al. [105] | |
| PTSD | Co-expression network | Differential responses to PTSD are observed in correlated modules constructed from the peripheral blood transcriptome of PTSD subjects. In men, an IL-12 signaling module is upregulated; In women, a module related to lipid metabolism and mitogen-activated protein kinase is upregulated. Cytokine, innate immune, and type I interferon-related modules are shared between sexes. | Breen et. al. [106] |
| miRNA regulons | Downregulated miRNAs in peripheral blood transcriptome of PTSD subjects are predicted to target IFNG and IL-12. | Bam et al. [73] | |
| SCZ | Co-expression network | Genes related to central nervous system development failed to attenuate with age in SCZ subjects; Synaptic protein co-expression was significantly decreased in the auditory cortex of SCZ subjects; SCZ common variants are enriched in negative co-expression genes of C4A | Torkamani et al. [107]; MacDonald et. al. [108]; Kim et. al. [109] |
| Transcription factor regulons | TCF4 is a master regulator identified from postmortem dorsolateral prefrontal cortex of SCZ subjects and cultured olfactory neuroepithelium | Torshizi et. al. [110] | |
| Protein-protein interaction network | MAPK3/ERK1 is the top hub for the 16p11.2 microduplication network | Blizinsky et. al. [78] | |
| Literature-based network | SCZ rare variant-derived network genes function mainly in axon guidance, neuronal cell mobility, synaptic function, and chromosomal remodeling, and are highly expressed in the brain during prenatal development. | Gilman et. al. [111] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zuo, Y.; Wei, D.; Zhu, C.; Naveed, O.; Hong, W.; Yang, X. Unveiling the Pathogenesis of Psychiatric Disorders Using Network Models. Genes 2021, 12, 1101. https://doi.org/10.3390/genes12071101
Zuo Y, Wei D, Zhu C, Naveed O, Hong W, Yang X. Unveiling the Pathogenesis of Psychiatric Disorders Using Network Models. Genes. 2021; 12(7):1101. https://doi.org/10.3390/genes12071101
Chicago/Turabian StyleZuo, Yanning, Don Wei, Carissa Zhu, Ormina Naveed, Weizhe Hong, and Xia Yang. 2021. "Unveiling the Pathogenesis of Psychiatric Disorders Using Network Models" Genes 12, no. 7: 1101. https://doi.org/10.3390/genes12071101
APA StyleZuo, Y., Wei, D., Zhu, C., Naveed, O., Hong, W., & Yang, X. (2021). Unveiling the Pathogenesis of Psychiatric Disorders Using Network Models. Genes, 12(7), 1101. https://doi.org/10.3390/genes12071101