
CLN8 Mutations Presenting with a Phenotypic Continuum of Neuronal Ceroid Lipofuscinosis—Literature Review and Case Report

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
3. Results
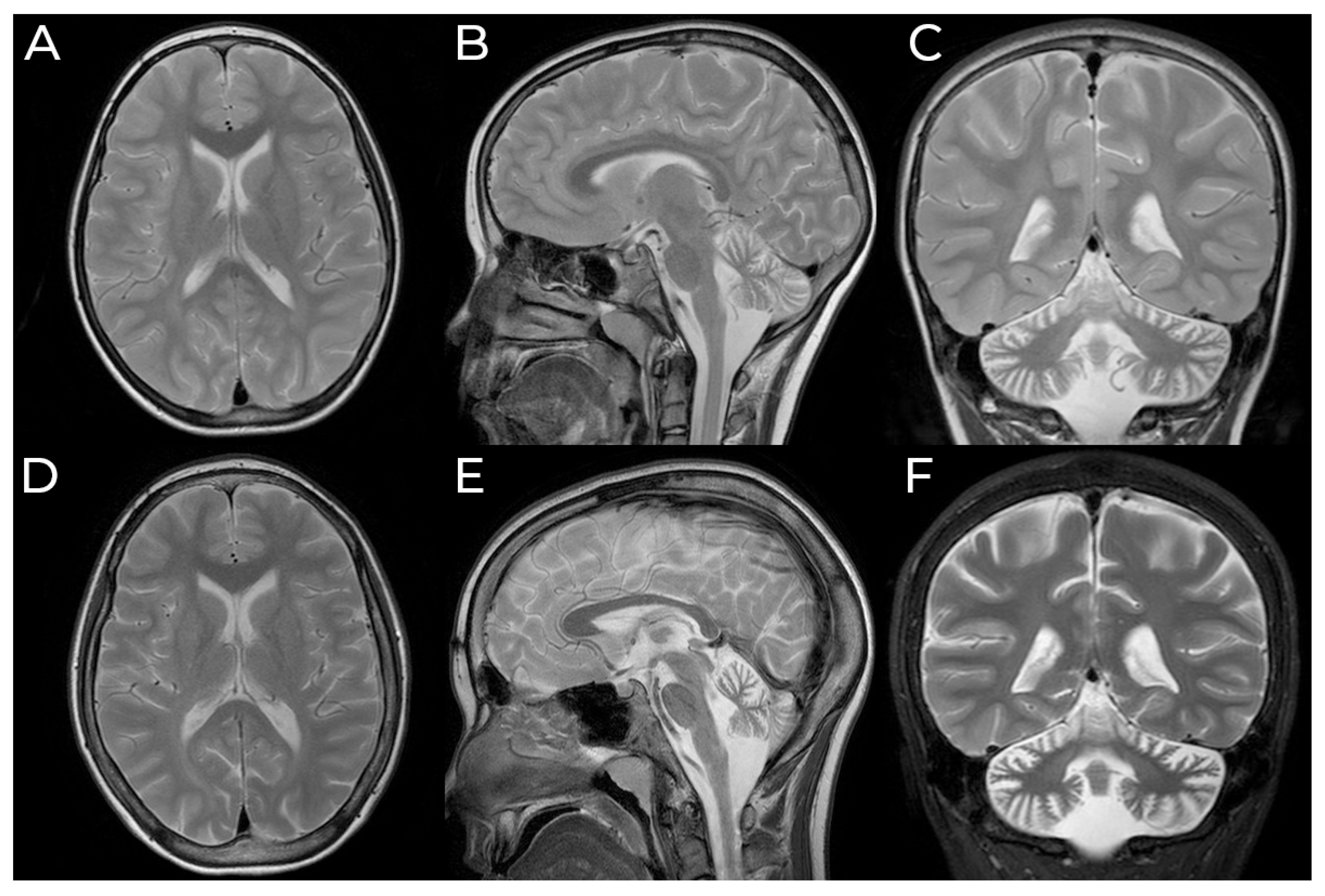
3.1. Case Presentation
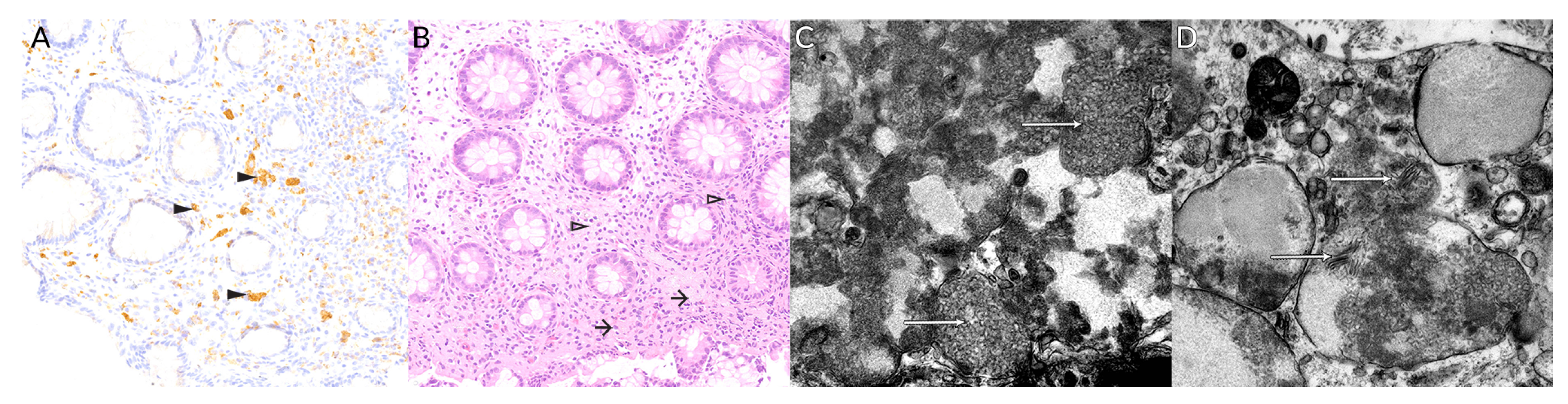
3.2. Molecular and Histological Testing
3.3. Follow-Up
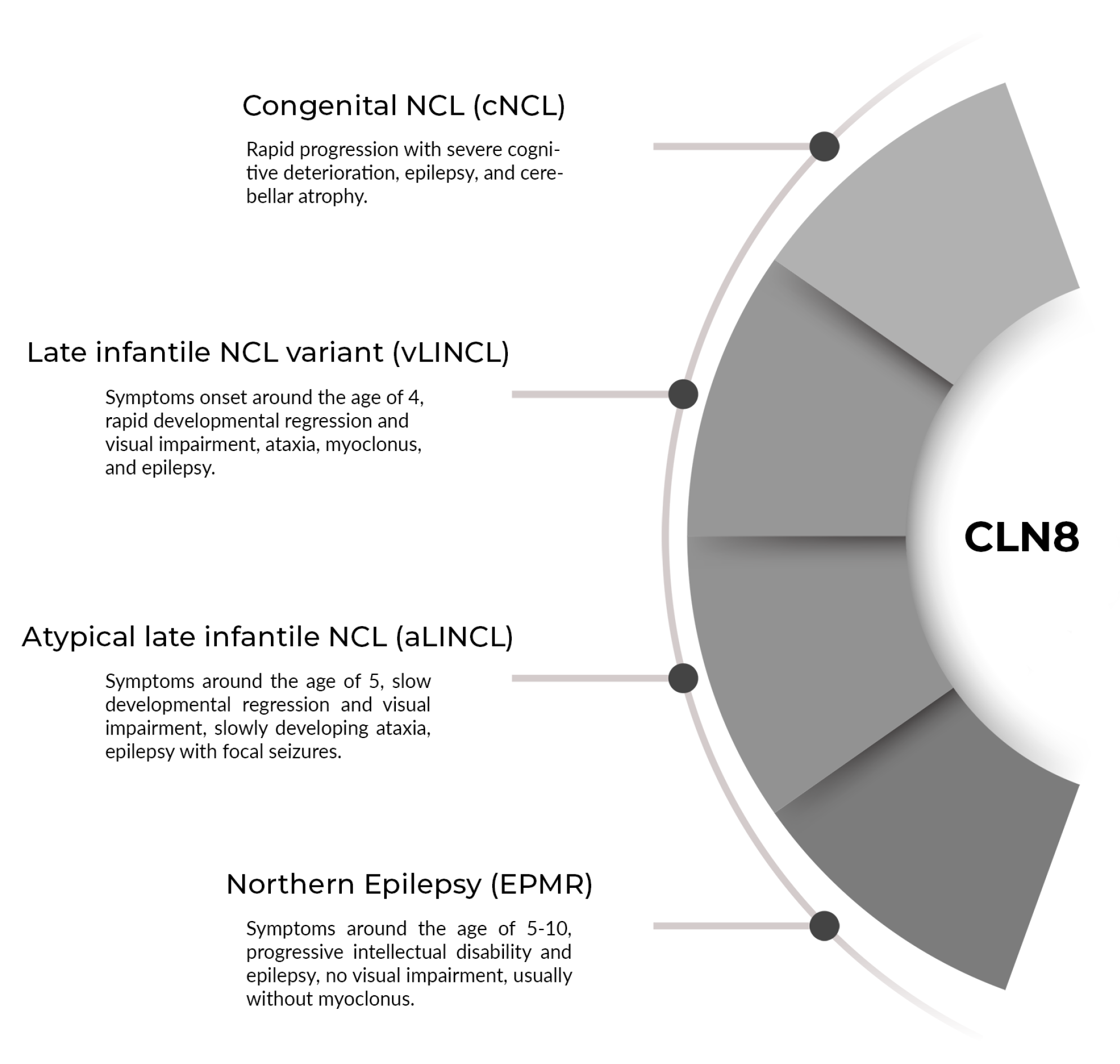
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nita, D.A.; Mole, S.E.; Minassian, B.A. Neuronal ceroid lipofuscinoses. Epileptic Disord. 2016, 18, 73–88. [Google Scholar] [CrossRef]
- Anderson, G.W.; Goebel, H.H.; Simonati, A. Human pathology in NCL. Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832, 1807–1826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeman, W.; Dyken, P. Neuronal ceroid-lipofuscinosis (Batten’s disease): Relationship to amaurotic family idiocy? Pediatrics 1969, 44, 570–583. [Google Scholar]
- Goebel, H.H.; Mole, S.E.; Sara, E.; Lake, B.D. The Neuronal Ceroid Lipofuscinoses (Batten Disease); IOS Press: Amsterdam, The Netherlands, 1999; p. 197. [Google Scholar]
- Stengel, O. Beretning om et maerkeligt Sygdomstilfaelde hos fire Sødskende I Nærheden af Roraas. Eyr Med. Tidskr. 1826, 1, 347–352. [Google Scholar]
- Haltia, M.; Rapola, J.; Santavuori, P. Infantile type of so-called neuronal ceroid-lipofuscinosis. Acta Neuropathol. 1973, 26, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Jansky, J. Dosud nepopsany pripad familiarni amauroticke idiotie komplikovane s hypoplasii mozeckovou. Sborn Lek. 1908, 13, 165–196. [Google Scholar]
- Bielschowsky, M. Über spatinfantile familiäre amaurotische Idiotie mit Kleinhirnsymptonen. Dtsch Z. Nerven-heilkd. 1913, 50, 7–29. [Google Scholar]
- Batten, F. Cerebral degeneration with symmetrical changes in the maculae in two members of a family. Trans. Ophthalmol. Soc. 1903, 23, 386–390. [Google Scholar]
- Spielmeyer, W. Über familiäre amaurotische Idiotien. Neurol. Cbl. 1905, 24, 612–620. [Google Scholar]
- Vogt, H. Über familiäre amaurotische Idiotie und verwandte Krankheitsbilder. Mschr. Psychiatr. Neurol. 1905, 18, 161–171. [Google Scholar] [CrossRef]
- Kufs, H. Über eine spatform der amaurotischen idiotie und ihre heredofamiliaren Grundlagen. Z. Ges. Neurol. Psych. 1925, 95, 168–188. [Google Scholar] [CrossRef]
- Boehme, D.; Cottrell, J.; Leonberg, S.; Zeman, W. A dominant form of neuronal ceroid-lipofuscinosis. Brain 1971, 94, 745–760. [Google Scholar] [CrossRef]
- Kozina, A.A.; Okuneva, E.G.; Baryshnikova, N.V.; Kondakova, O.B.; Nikolaeva, E.A.; Fedoniuk, I.D.; Mikhailova, S.V.; Krasnenko, A.Y.; Stetsenko, I.F.; Plotnikov, N.A.; et al. Neuronal ceroid lipofuscinosis in the Russian population: Two novel mutations and the prevalence of heterozygous carriers. Mol. Genet. Genomic. Med. 2020, 8, e1228. [Google Scholar] [CrossRef]
- Naseri, N.; Sharma, M.; Velinov, M. Autosomal dominant neuronal ceroid lipofuscinosis: Clinical features and molecular basis. Clin. Genet. 2021, 99, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Kousi, M.; Lehesjoki, A.E.; Mole, S.E. Update of the mutation spectrum and clinical correlations of over 360 mutations in eight genes that underlie the neuronal ceroid lipofuscinoses. Hum. Mutat. 2012, 33, 42–63. [Google Scholar] [CrossRef]
- Williams, R.E.; Mole, S.E. New nomenclature and classification scheme for the neuronal ceroid lipofuscinoses. Neurology 2012, 79, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Ranta, S.; Topcu, M.; Tegelberg, S.; Tan, H.; Üstübütün, A.; Saatci, I.; Dufke, A.; Enders, H.; Pohl, K.; Alembik, Y.; et al. Variant late infantile neuronal ceroid lipofuscinosis in a subset of Turkish patients is allelic to Northern epilepsy. Hum. Mutat. 2004, 23, 300–305. [Google Scholar] [CrossRef]
- Zemojtel, T.; Köhler, S.; Mackenroth, L.; Jäger, M.; Hecht, J.; Krawitz, P.; Graul-Neumann, L.; Doelken, S.; Ehmke, N.; Spielmann, M.; et al. Effective diagnosis of genetic disease by computational phenotype analysis of the disease-associated genome. Sci. Transl. Med. 2014, 6, 252ra123. [Google Scholar] [CrossRef] [Green Version]
- Hirvasniemi, A.; Lang, H.; Lehesjoki, A.E.; Leisti, J. Northern epilepsy syndrome: An inherited childhood onset epilepsy with associated mental deterioration. J. Med. Genet. 1994, 31, 177–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranta, S.; Lehesjoki, A.E. Northern epilepsy, a new member of the NCL family. Neurol. Sci. 2000, 21, S43–S47. [Google Scholar] [CrossRef]
- Mitchell, W.A.; Wheeler, R.B.; Sharp, J.D.; Bate, S.L.; Gardiner, R.M.; Ranta, U.S.; Lonka, L.; Williams, R.E.; Lehesjoki, A.E.; Mole, S.E. Turkish variant late infantile neuronal ceroid lipofuscinosis (CLN7) may be allelic to CLN8. Eur. J. Paediatr. Neurol. 2001, 5 (Suppl. A), 21–27. [Google Scholar] [CrossRef]
- Talbot, J.; Singh, P.; Puvirajasinghe, C.; Sisodiya, S.M.; Rugg-Gunn, F. Moyamoya and progressive myoclonic epilepsy secondary to CLN6 bi-allelic mutations—A previously unreported association. Epilepsy Behav. Rep. 2020, 14, 100389. [Google Scholar] [CrossRef] [PubMed]
- Sahin, Y.; Güngör, O.; Gormez, Z.; Demirci, H.; Ergüner, B.; Güngör, G.; Dilber, C. Exome sequencing identifies a novel homozygous CLN8 mutation in a Turkish family with Northern epilepsy. Acta Neurol. Belg. 2017, 117, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Herva, R.; Tyynelä, J.; Hirvasniemi, A.; Syrjäkallio-Ylitalo, M.; Haltia, M. Northern Epilepsy: A Novel Form of Neuronal Ceroid-Lipofuscinosis. Brain Pathol. 2000, 10, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Kose, M.; Kose, E.; Ünalp, A.; Yılmaz, Ü.; Edizer, S.; Tekin, H.G.; Karaoğlu, P.; Özdemir, T.R.; Er, E.; Onay, H.; et al. Neuronal ceroid lipofuscinosis: Genetic and phenotypic spectrum of 14 patients from Turkey. Neurol. Sci. 2021, 42, 1103–1111. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.T.; Wang, X.H.; Ding, C.H.; Shen, X.; Zhang, H.; Zhang, W.H.; Li, J.W.; Ren, C.H.; Fang, F. Next-Generation Sequencing Analysis Reveals Novel Pathogenic Variants in Four Chinese Siblings With Late-Infantile Neuronal Ceroid Lipofuscinosis. Front. Genet. 2019, 10, 370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cannelli, N.; Cassandrini, D.; Bertini, E.; Striano, P.; Fusco, L.; Gaggero, R.; Specchio, N.; Biancheri, R.; Vigevano, F.; Bruno, C.; et al. Novel mutations in CLN8 in Italian variant late infantile neuronal ceroid lipofuscinosis: Another genetic hit in the Mediterranean. Neurogenetics 2006, 7, 111–117. [Google Scholar] [CrossRef]
- Reinhardt, K.; Grapp, M.; Schlachter, K.; Brück, W.; Gärtner, J.; Steinfeld, R. Novel CLN8 mutations confirm the clinical and ethnic diversity of late infantile neuronal ceroid lipofuscinosis. Clin. Genet. 2010, 77, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Mahajnah, M.; Zelnik, N. Phenotypic heterogeneity in consanguineous patients with a common cln8 mutation. Pediatr Neurol. 2012, 47, 303–305. [Google Scholar] [CrossRef]
- Allen, N.M.; O’hIci, B.; Anderson, G.; Nestor, T.; Lynch, S.A.; King, M.D. Variant late-infantile neuronal ceroid lipofuscinosis due to a novel heterozygous CLN8 mutation and de novo 8p23.3 deletion. Clin. Genet. 2012, 81, 602–604. [Google Scholar] [CrossRef]
- Katata, Y.; Uematsu, M.; Sato, H.; Suzuki, S.; Nakayama, T.; Kubota, Y.; Kobayashi, T.; Hino-Fukuyo, N.; Saitsu, H.; Kure, S. Novel missense mutation in CLN8 in late infantile neuronal ceroid lipofuscinosis: The first report of a CLN8 mutation in Japan. Brain Dev. 2016, 38, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Xie, H.; Jiang, Q.; Wu, N.; Chen, X.; Chen, Q. Identification of two novel null variants in CLN8 by targeted next-generation sequencing: First report of a Chinese patient with neuronal ceroid lipofuscinosis due to CLN8 variants. BMC Med. Genet. 2018, 19, 4–9. [Google Scholar] [CrossRef] [Green Version]
- Alkhars, F.Z.; Bo Ali, A.Y.; Almohanna, M.A.; Almajhad, N.A. Neuronal ceroid lipofuscinoses type 8: Expanding genotype/phenotype diversity-first report from Saudi Arabia. Neurosciences 2020, 25, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Kohan, R.; Pesaola, F.; Guelbert, N.; Pons, P.; Oller-Ramírez, A.M.; Rautenberg, G.; Becerra, A.; Sims, K.; Xin, W.; Cismondi, I.A.; et al. The neuronal ceroid lipofuscinoses program: A translational research experience in Argentina. Biochim. Biophys. Acta Mol. Basis Dis. 2015, 1852, 2301–2311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez, R.L.; Yan, J.; Richards, S.; Mierau, G.; Wartchow, E.P.; Collins, C.D.; Shankar, S.P. Atypical presentation of neuronal ceroid lipofuscinosis type 8 in a sibling pair and review of the eye findings and neurological features. Am. J. Ophthalmol. Case Rep. 2016, 4, 50–53. [Google Scholar] [CrossRef] [Green Version]
- Avanzini, G.; Shibasaki, H.; Rubboli, G.; Canafoglia, L.; Panzica, F.; Franceschetti, S.; Hallett, M. Neurophysiology of myoclonus and progressive myoclonus epilepsies. Epileptic Disord. 2016, 18, 11–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butz, E.S.; Chandrachud, U.; Mole, S.E.; Cotman, S.L. Moving towards a new era of genomics in the neuronal ceroid lipofuscinoses. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165571. [Google Scholar] [CrossRef]
- Winter, E.; Ponting, C.P. TRAM, LAG1 and CLN8: Members of a novel family of lipid-sensing domains? Trends Biochem. Sci. 2002, 27, 381–383. [Google Scholar] [CrossRef]
- Lonka, L.; Salonen, T.; Siintola, E.; Kopra, O.; Lehesjoki, A.-E.; Jalanko, A. Localization of wild-type and mutant neuronal ceroid lipofuscinosis CLN8 proteins in non-neuronal and neuronal cells. J. Neurosci. Res. 2004, 76, 862–871. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Type | Eponym | Original Reports | Age of Onset | Distinctive Clinical Features |
|---|---|---|---|---|
| Infantile | Haltia-Santavuori | Haltia and Santavuori [6] | 0–2 y | Rapid psychomotor deterioration, seizures, visual impairment, purposeless hand movements (Rett-like). Death around age 10 y [1]. |
| Late-infantile | Jansky-Bielschowsky | Jansky [7] and Bielschowsky [8] | 2–4 y | Initially epilepsy, followed by cognitive deterioration, myoclonus, ataxia, vision impairment. Death by age 6–15 y. |
| Juvenile | Batten, Batten-Spielmeyer-Vogt | Stengel [5], Batten [9], Spielmeyer [10] and Vogt [11] | 4–10 y | Initially rapidly progressive visual loss, followed by epilepsy. Death by age 30 y. |
| Adult | Kufs (AR), Parry (AD) | Kufs [12], Boehme [13] | 15–50 y | Cognitive decline, ataxia, myoclonus, epilepsy, behavioral changes, no visual impairment |
| Year/Phenotype | 1994/EPMR | 2000/EPMR | 2017/EPMR | 2001/vLINCL | 2006/vLINCL | 2010/vLINCL | 2010/vLINCL | 2010/vLINCL |
|---|---|---|---|---|---|---|---|---|
| Authors | Hirvasniemi et al. [20] | Ranta et al. [21] | Sahin et al. [24] | Mitchell et al. [22] | Cannelli et al. [28] | Reinhardt et al. [29] | Reinhardt et al. [29] | Reinhardt et al. [29] |
| Nationality | Finnish | Turkish | Turkish | Turkish | Italian | German | Pakistani | Turkish |
| No of cases, sex | 23, M(11), F(12) | 6 | 5, M(5) | 5 | 3, M(3) | 1, F(1) | 1, F(1) | 2, M(1), F(1) |
| Mutation | Homoz. c.70C>G, p.(Arg24Gly) | (1) Homoz. c.789G>C, p.(Trp263Cys); (2) Homoz. c.610C>T, p.(Arg204Cys); (3) Comp. heteroz. c.789G>C, p.(Trp263Cys)+c.610C>T, p.(Arg204Cys) | Homoz. c.677T>C, p. (Leu226Pro) | (1) Homoz. c.610C>T, p.(Arg204Cys); (2) Comp. heteroz. c.46C>A, p.(Leu16Met)+ c.509C>T, p.(Thr170Met); (3) Homoz. c.88delG, p.(Ala30Leufs20) | (1) Comp. heteroz c.581A>G, p.(Gln194Arg) + c.66delG, p.(Ala30Leufs*20); (2) Comp. heteroz c.473A>G, p.(Tyr158Cys) +c.66delG, p.(Ile23Serfs*5); (3) Homoz. c.88G>C, p.(Ala30Pro) | Homoz. c.611G>T, p.(Arg204Leu) | Homoz. c.709G>A, p.(Gly237Arg) | Homoz. c.544-2566_590del2613, p.0 |
| Infancy psychomotor development | Normal (20/23), Delayed (3/23) | N/A | Normal (5/5) | Delayed | Delayed | Normal | N/A | Delayed |
| Age of symptoms onset‡ | 6.7 y (5–10) | 5.7 (3,5–7) | 8.4 y (8–10) | 3.5 (3–4) | 4.5 y (3,5–6) | 3.5 y | 4 y | 3.25 y (3–3,5) |
| First symptoms‡ | Seizures | N/A | Seizures | Mixed | Seizures | Attention, sleep, speech disturbance | Ataxic gait | Seizures |
| Epilepsy | Yes, GTCS (23/23), complex focal (7/23) | Yes (6/6) | Yes, GTCS (5/5) | Yes (4/4) | Yes, myoclonic (2/3), tonic-clonic (1/3) | Yes, absence | Yes, drop attacks | Yes, myoclonic and drop attacks |
| Epilepsy onset | 6.7 y (5–10) | 6.2 y (3.5–8) | 8.4 y (8–10) | 3.5 y (3–4) | 4.5 y (3.5–6) | 4 y | 6,5 y | 3.25 y (3–3.5) |
| Cognitive deterioration and onset | Yes (23/23), 2–5 y, after epilepsy onset | Yes (6/6) | Yes (5/5), 6.5 y (5–7y), after epilepsy onset | Yes | Yes | Yes, 3.5y | Yes, After ataxia onset | Yes, After epilepsy onset |
| Cognitive outcome | Borderline (4/19), Mild (4/19), Moderate (5/19), Severe (6/19) | N/A | Mild (3), Moderate (1), Severe (1) | N/A | N/A | Severe | Severe | Severe |
| Ataxia and onset | Yes (16/19), <30 y | yes (5/5), 8.2 y (4–12) | yes (5/5) | yes, 3.8 y (3.5–4) | yes | yes, 4 y | yes, 4 y | yes, 4 y |
| Abnormal speech onset | N/A | 8.6 y (7–9) | 16 y (15–18) | N/A | N/A | 4 y | 5.5 y | 4 y |
| Abnormal gait onset | <30 y | N/A | 14.6 y (14–16) | N/A | N/A | N/A | N/A | N/A |
| Myoclonus and onset | No | Yes (4/4), 7.1 y (3.5–9) | No | Yes, 3.9 y (3–4.5) | Yes | Yes, 5 y | Yes, 5 y | Yes, 4 y |
| Visual loss and onset | No or minor | Yes (6/6), 7.5 y (5–9) | No | Yes, 3.6 y (3–4) | Yes | Yes, 9 y | Yes, 7 y | Yes, 6 y |
| Optic pathology | None | N/A | None | N/A | N/A | Retinal degeneration | Optic nerve atrophy | Retinal degeneration |
| Progression rate | Slow | Rapid | Slow | Rapid | Rapid | Rapid | Rapid | Rapid |
| EEG | Slow background, disappearance of sleep patterns, scanty interictal epileptiform activity | N/A | Normal | N/A | N/A | N/A | N/A | Generalized polyspike slow wave |
| Cerebral MRI | Atrophy at later stages | N/A | Atrophy | N/A | Atrophy | Cortical atrophy | Atrophy, T2/FLAIR PV WM hyperintensivities | Atrophy, T2/FLAIR WM hyperintensivities |
| Cerebellar atrophy | Yes | N/A | Yes | N/A | Yes | Yes | Yes | Yes |
| Other | Behavioural difficulties in puberty (11/23) | N/A | N/A | N/A | Patients with deletions: earlier onset, myoclonus more prominent | N/A | Noise elicited myoclonus | N/A |
| Year/Phenotype | 2012/vLINCL | 2012/vLINCL | 2016/vLINCL | 2018/vLINCL | 2020/vLINCL | 2015/cNCL | 2016/aLINCL | 2021/aLINCL |
| Authors | Mahajnah et al. [30] | Allen et al. [31] | Katata et al. [32] | Gao et al. [33] | Alkhars et al. [34] | Kohan et al. [35] | Sanchez et al. [36] | Badura-Stronka et al. |
| Nationality | Israeli/Arabian | Irish | Japanese | Chinese | Arabian | Argentinian | Hispanic | Polish |
| No of cases, sex | 3, M(2), F(1) | 1, M(1) | 1, M(1) | 1, M(1) | 2, M(1), F(1) | 1, F(1) | 2, M(1), F(1) | 1, F(1) |
| Mutation | Homoz. c.763C>G, p.(Gln255*) | Comp. heteroz. c.562_563delCT, p.(Leu188Valfs*58) + 8p23.3 terminal deletion | Homoz. c.620T>G, p.(Leu207Arg) | Comp. heteroz. c.298C>T, p.(Gln100*) + c.551G>A, p.(Trp184*) | (1) Homoz. c.699_700delGT, p.Phe234Profs12; (2) Homoz. c.(?_-1)_(5431_544-1)del | (rs143730802) + (rs587779411) | Comp. heteroz. c.200C>T, p.(Ala67Val) + CLN8 deletion | Homoz. c.531C>T, p.(Trp177Cys) |
| Infancy psychomotor development | Normal | Delayed | Delayed | Normal | Normal (1/2), Delayed (1/2) | Delayed | Normal | Delayed |
| Age of symptoms onset‡ | 5 y (4–6) | 4 y | 3 y | 4 y | 2 y | Congenital | 11.5 (7–16) | 5.5 y |
| First symptoms‡ | Seizures (2/3), Mixed (1/3) | Ataxia | Seizures | Seizures | Seizures (1/2), Ataxia (1/2) | Seizures | Seizures (1/2), Visual (1/2) | Seizures |
| Epilepsy | Yes, tonic-clonic | Yes | Yes, drop attacks, myoclonic | Yes | Yes, GTCS, myoclonic | Yes, GTCS | Yes, GTCS | Yes, GTCS, complex focal |
| Epilepsy onset | 6.7 y (5–9) | 4.5 y | 3 y | 4 y | 1.5 y (1–2) | 3 y | 11.5 y (7–16) | 5.5 y |
| Cognitive deterioration and onset | Yes, After epilepsy onset (2/3) | Yes, 4 y | Yes, N/A | Yes, 3 y, after epilepsy onset | Yes, 3 y, after epilepsy onset (1/2), Birth (1/2) | Yes, Birth | No, N/A | Yes, <2 y, after epilepsy onset |
| Cognitive outcome | Profound (1/3), Moderate (1/3), Mild (1/3) | N/A | N/A | N/A | Severe | Severe | Excellent | Moderate |
| Ataxia and onset | yes (1/3), 5 y, No (2/3) | yes, 4 y | yes, 4 y | yes, 7 y | yes, 2.5 y (1–4) | No | yes | yes, 6.6 y |
| Abnormal speech onset | N/A | 4 y | N/A | 7 y | 3 y (1/2) | N/A | N/A | 3 y |
| Abnormal gait onset | N/A | 4 y | 4 y | 7 y | 3 y (2-4) | Never achieved | N/A | 9.5 y |
| Myoclonus and onset | Yes (1/3) | N/A | Yes, 5 y | N/A | Yes (2/2), 1.5 y (1–2) | Yes, 6 y | No | No |
| Visual loss and onset | Yes, 6.5 y (6–7.5) | Yes, 5.5 y | Yes, 5 y | Yes, 7 y | Yes (2/2), 2.5 y (2–3) | N/A | Yes, 9 y (5–13) | Yes, 9.5 y |
| Optic pathology | Retinal degeneration, Optic nerve atrophy (2/3) | Abnormal ERG | Retinal and macular | N/A | Optic nerve atrophy | N/A | Complex macular (1/2), Retinal and optic nerve atrophy (1/2) | Macular dystrophy, optic nerve atrophy |
| Progression rate | Rapid (1/3), Slow (2/3) | Rapid | Rapid | Rapid | Rapid | Rapid | Slow | Slow |
| EEG | Slow background (1/3), Multiple spikes and bursts of generalized spike-slow wave (1/3) | Slow background, complex focal seizures | Diffuse spikes and slow waves | Slow background, abundant generalized atypical spike-slow wave | Generalized slow background with polyspike epileptiform discharges | N/A | N/A | Progressive slowing, paroxysmal slow waves; generalized sharp-slow wave after 12 y |
| Cerebral MRI | Diffuse atrophy (1/3), Mild atrophy (1/3), T2/FLAIR WM hyperintensivities (2/3) | T2/FLAIR posterior deep WM hyperintensivities | T2/FLAIR diffuse PV WM hyperintensivities | Diffuse atrophy | Diffuse atrophy (1/2) | N/A | N/A | T2/FLAIR WM hyperintensivities |
| Cerebellar atrophy | Yes | Yes | Yes | Yes | Yes | Yes | N/A | Yes |
| Other | Severily affected, rapid deterioration (1/3) | N/A | N/A | N/A | N/A | N/A | Polidactyly, ADHD (1/2) | Myoclonus on lamotrigine; hypersensitive to noise |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Badura-Stronka, M.; Winczewska-Wiktor, A.; Pietrzak, A.; Hirschfeld, A.S.; Zemojtel, T.; Wołyńska, K.; Bednarek-Rajewska, K.; Seget-Dubaniewicz, M.; Matheisel, A.; Latos-Bielenska, A.; et al. CLN8 Mutations Presenting with a Phenotypic Continuum of Neuronal Ceroid Lipofuscinosis—Literature Review and Case Report. Genes 2021, 12, 956. https://doi.org/10.3390/genes12070956
Badura-Stronka M, Winczewska-Wiktor A, Pietrzak A, Hirschfeld AS, Zemojtel T, Wołyńska K, Bednarek-Rajewska K, Seget-Dubaniewicz M, Matheisel A, Latos-Bielenska A, et al. CLN8 Mutations Presenting with a Phenotypic Continuum of Neuronal Ceroid Lipofuscinosis—Literature Review and Case Report. Genes. 2021; 12(7):956. https://doi.org/10.3390/genes12070956
Chicago/Turabian StyleBadura-Stronka, Magdalena, Anna Winczewska-Wiktor, Anna Pietrzak, Adam Sebastian Hirschfeld, Tomasz Zemojtel, Katarzyna Wołyńska, Katarzyna Bednarek-Rajewska, Monika Seget-Dubaniewicz, Agnieszka Matheisel, Anna Latos-Bielenska, and et al. 2021. "CLN8 Mutations Presenting with a Phenotypic Continuum of Neuronal Ceroid Lipofuscinosis—Literature Review and Case Report" Genes 12, no. 7: 956. https://doi.org/10.3390/genes12070956
APA StyleBadura-Stronka, M., Winczewska-Wiktor, A., Pietrzak, A., Hirschfeld, A. S., Zemojtel, T., Wołyńska, K., Bednarek-Rajewska, K., Seget-Dubaniewicz, M., Matheisel, A., Latos-Bielenska, A., & Steinborn, B. (2021). CLN8 Mutations Presenting with a Phenotypic Continuum of Neuronal Ceroid Lipofuscinosis—Literature Review and Case Report. Genes, 12(7), 956. https://doi.org/10.3390/genes12070956