
Uncovering Signals from the Coronavirus Genome

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
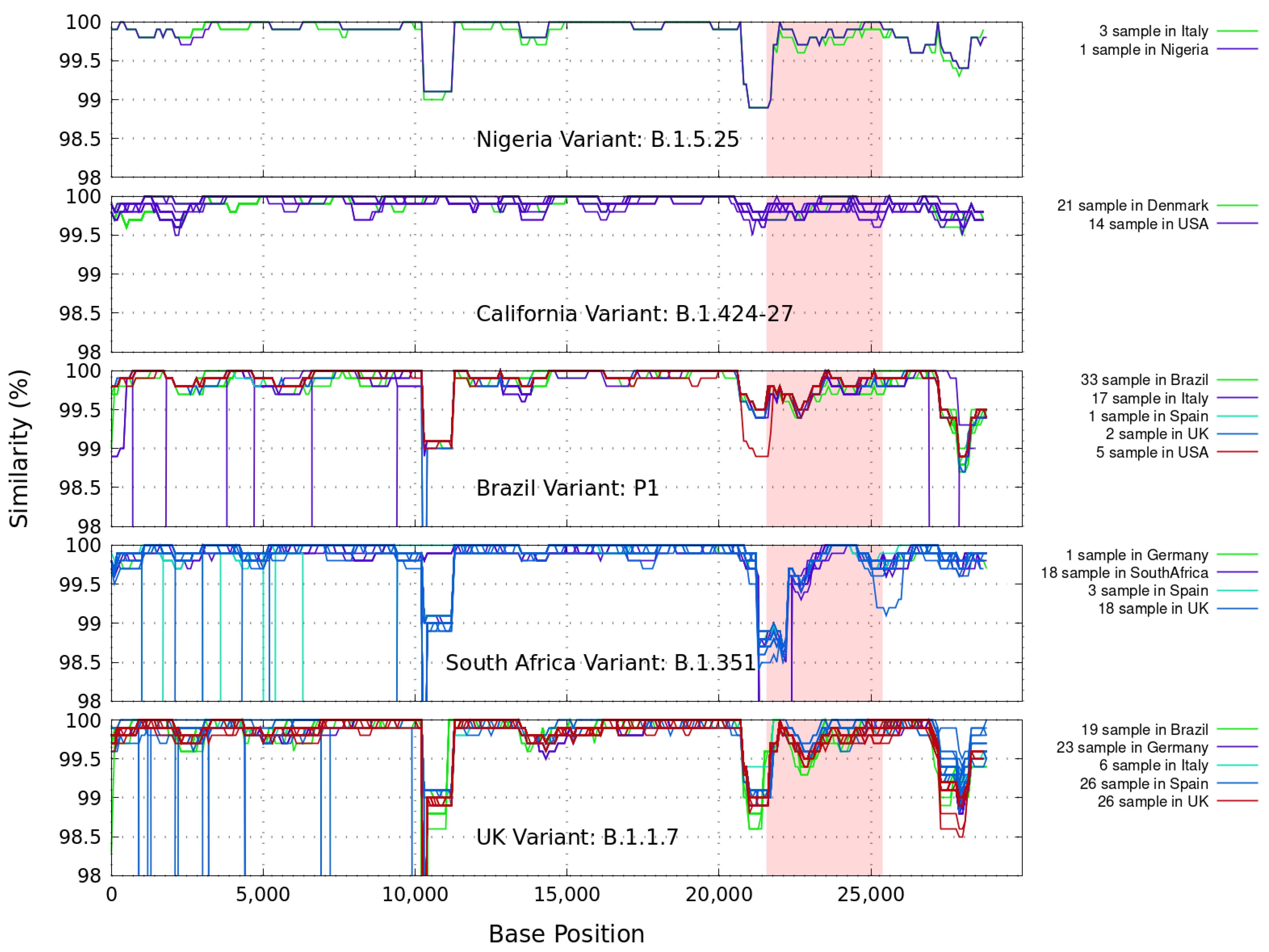
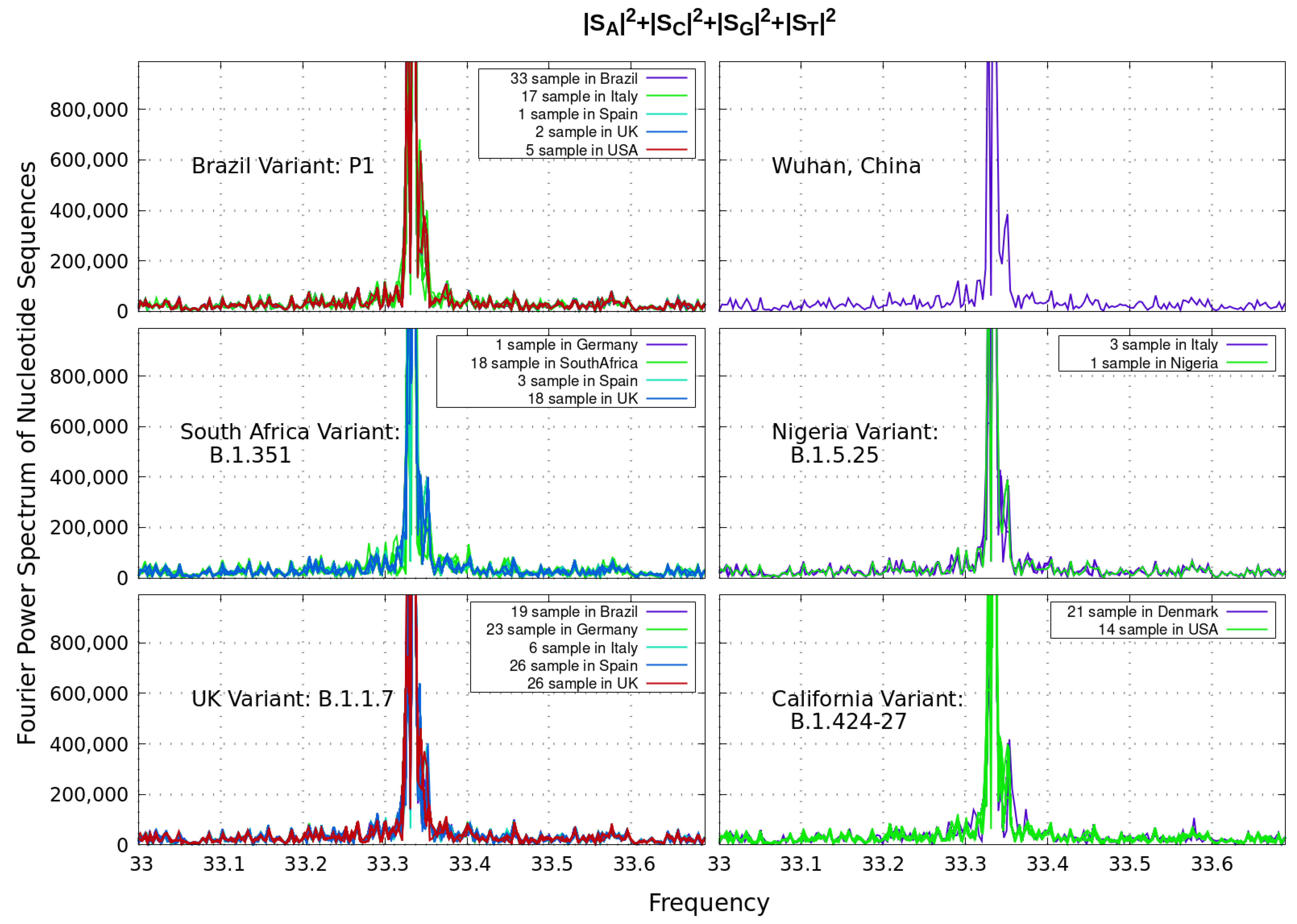
2. Similarity and Power Spectrum Analysis
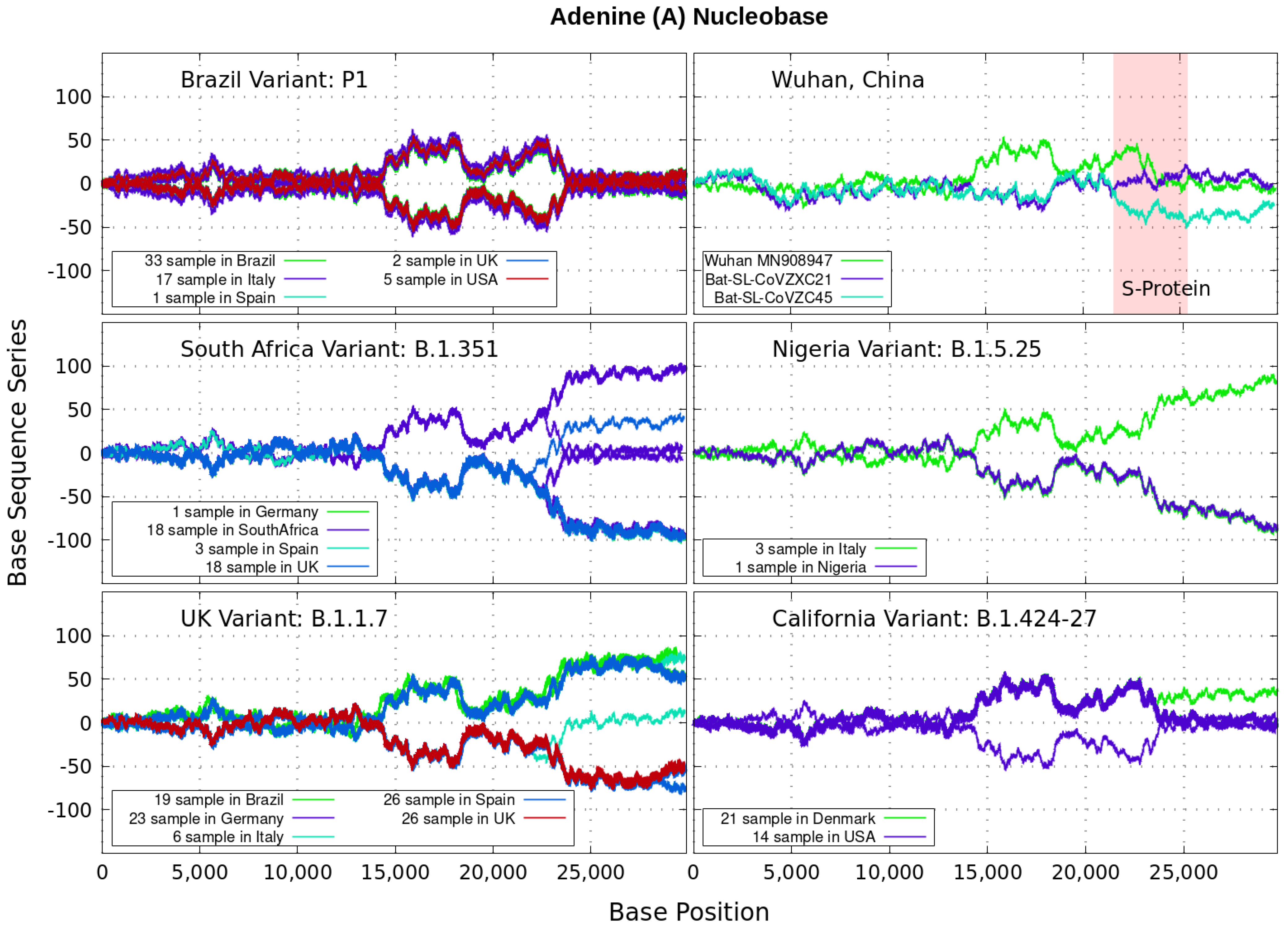
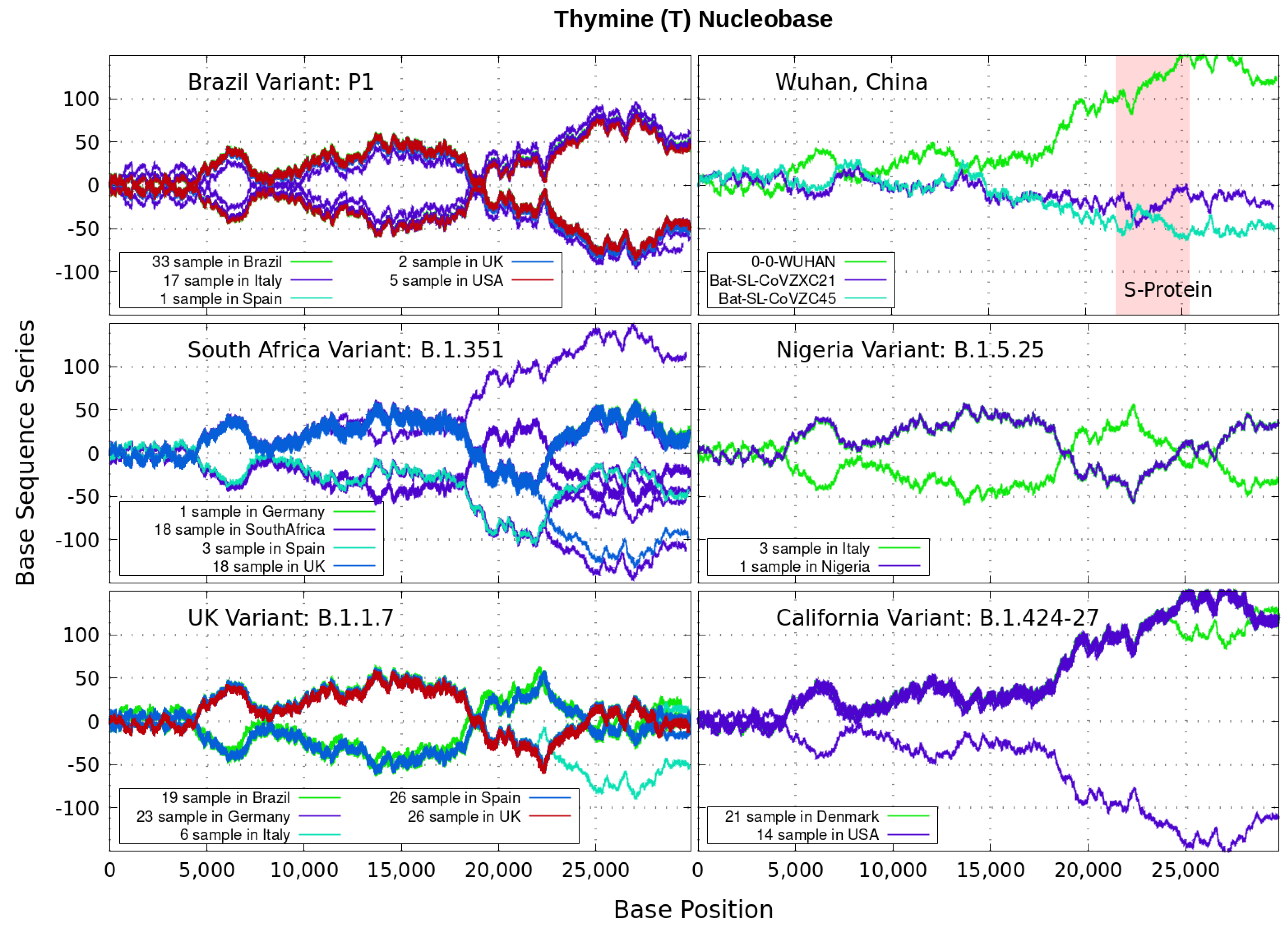
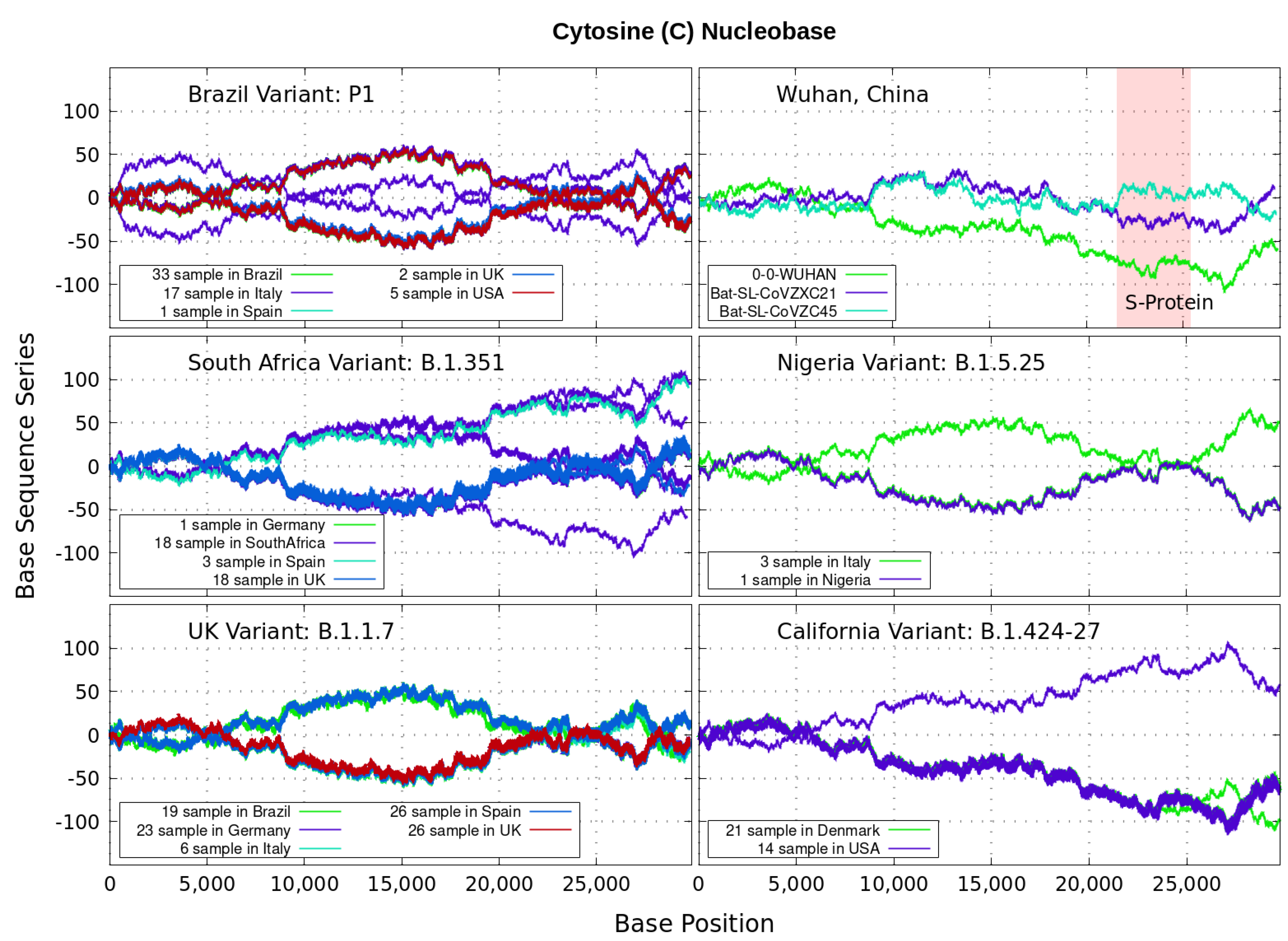
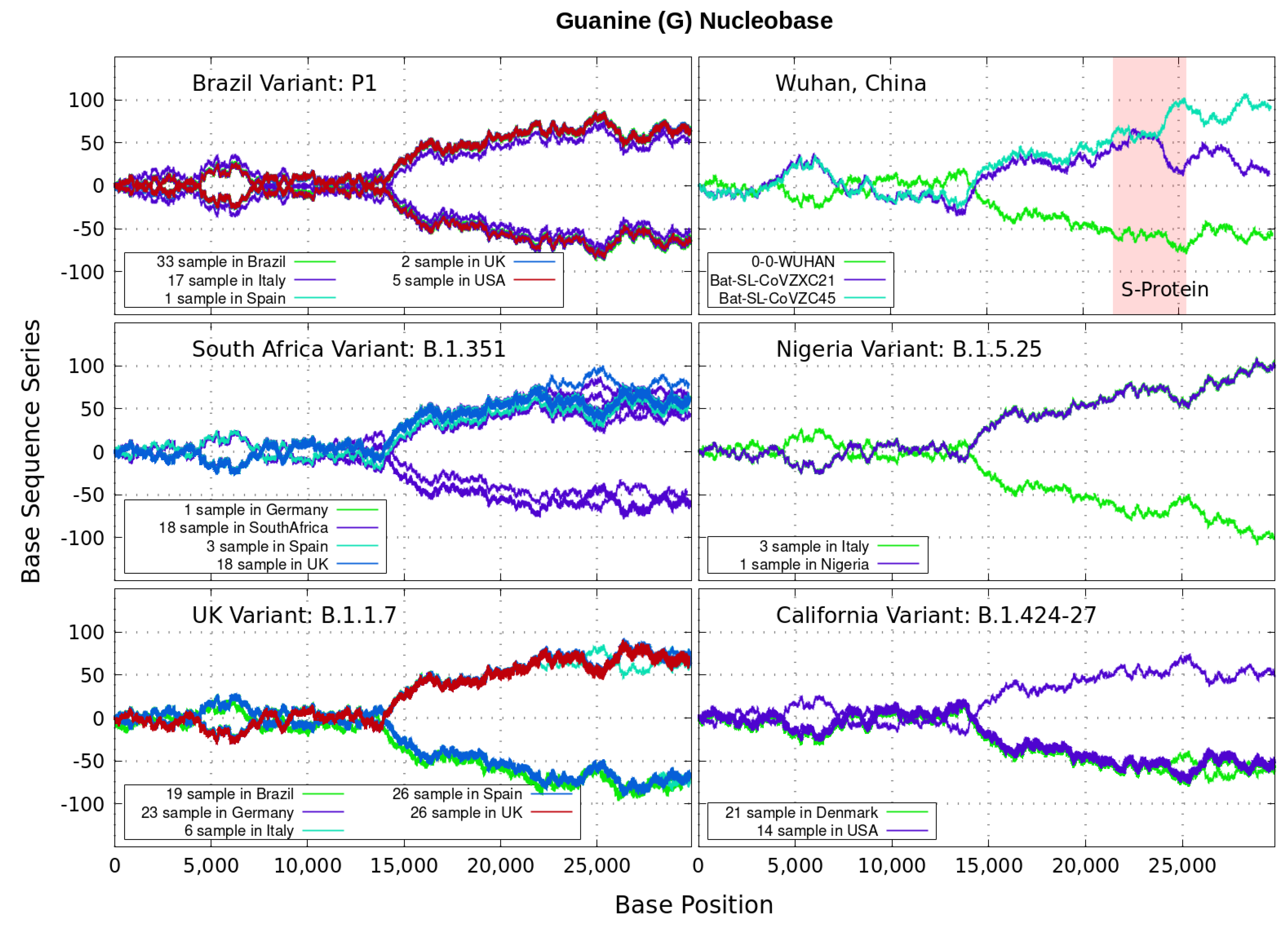
3. Finite and Alternating Sum Series of Virus Genome
4. Future Perspective
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
References
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Giorgi, E.E.; Marichannegowda, M.H.; Foley, B.; Xiao, C.; Kong, X.P.; Chen, Y.; Gnanakaran, S.; Korber, B.; Gao, F. Emergence of SARS-CoV-2 through recombination and strong purifying selection. Sci. Adv. 2020, 6, eabb9153. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; He, Y.; Zhou, Y.; Liu, S.; Zheng, B.-J.; Jiang, S. The spike protein of SARS-CoV—A target for vaccine and therapeutic development. Nat. Rev. Microbiol. 2009, 7, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Koch, L.; Poteski, C.; Trenkmann, M. (Eds.) Milestones in Genomic Sequencing. Nature Milestones. February 2021. Available online: www.nature.com/collections/genomic-sequencing-milestones (accessed on 22 June 2021).
- King, B.R.; Aburdene, M.; Thompson, A.; Warres, Z. Application of discrete Fourier inter-coefficient difference for assessing genetic sequence similarity. EURASIP J. Bioinform. Syst. Biol. 2014, 2014, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, C.; Chen, Y.; Yau, S.S.-T. A measure of DNA sequence similarity by Fourier Transform with applications on hierarchical clustering. J. Theor. Biol. 2014, 359, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Chechetkin, V.; Turygin, A. Size-dependence of three-periodicity and long-range correlations in DNA sequences. Phys. Lett. A 1995, 199, 75–80. [Google Scholar] [CrossRef]
- Hoang, T.; Yin, C.; Zheng, H.; Yu, C.; He, R.L.; Yau, S.S.-T. A new method to cluster DNA sequences using Fourier power spectrum. J. Theor. Biol. 2015, 372, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Pal, J.; Ghosh, S.; Maji, B.; Bhattacharya, D.K. Use of FFT in Protein Sequence Comparison under Their Binary Representations. Comput. Mol. Biosci. 2016, 6, 33–40. [Google Scholar] [CrossRef] [Green Version]
- Touati, R.; Haddad-Boubaker, S.; Ferchichi, I.; Messaoudi, I.; Ouesleti, A.E.; Triki, H.; Lachiri, Z.; Kharrat, M. Comparative genomic signature representations of the emerging COVID-19 coronavirus and other coronaviruses: High identity and possible recombination between Bat and Pangolin coronaviruses. Genomics 2020, 112, 4189–4202. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.-L.; Hu, B.; Wang, B.; Wang, M.-N.; Zhang, Q.; Zhang, W.; Wu, L.-J.; Ge, X.-Y.; Zhang, Y.-Z.; Daszak, P.; et al. Isolation and Characterization of a Novel Bat Coronavirus Closely Related to the Direct Progenitor of Severe Acute Respiratory Syndrome Coronavirus. J. Virol. 2016, 90, 3253–3256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voss, R.F. Evolution of long-range fractal correlations and 1/fnoise in DNA base sequences. Phys. Rev. Lett. 1992, 68, 3805–3808. [Google Scholar] [CrossRef] [PubMed]
- Naqvi, A.A.T.; Fatima, K.; Mohammad, T.; Fatima, U.; Singh, I.K.; Singh, A.; Atif, S.M.; Hariprasad, G.; Hasan, G.M.; Hassan, I. Insights into SARS-CoV-2 genome, structure, evolution, pathogenesis and therapies: Structural genomics approach. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2020, 1866, 165878. [Google Scholar] [CrossRef] [PubMed]
- Canessa, E. Modeling of body mass index by Newton’s second law. J. Theor. Biol. 2007, 248, 646–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canessa, E. Multifractality in time series. J. Phys. A Math. Gen. 2000, 33, 3637–3651. [Google Scholar] [CrossRef] [Green Version]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Canessa, E. Uncovering Signals from the Coronavirus Genome. Genes 2021, 12, 973. https://doi.org/10.3390/genes12070973
Canessa E. Uncovering Signals from the Coronavirus Genome. Genes. 2021; 12(7):973. https://doi.org/10.3390/genes12070973
Chicago/Turabian StyleCanessa, Enrique. 2021. "Uncovering Signals from the Coronavirus Genome" Genes 12, no. 7: 973. https://doi.org/10.3390/genes12070973
APA StyleCanessa, E. (2021). Uncovering Signals from the Coronavirus Genome. Genes, 12(7), 973. https://doi.org/10.3390/genes12070973