
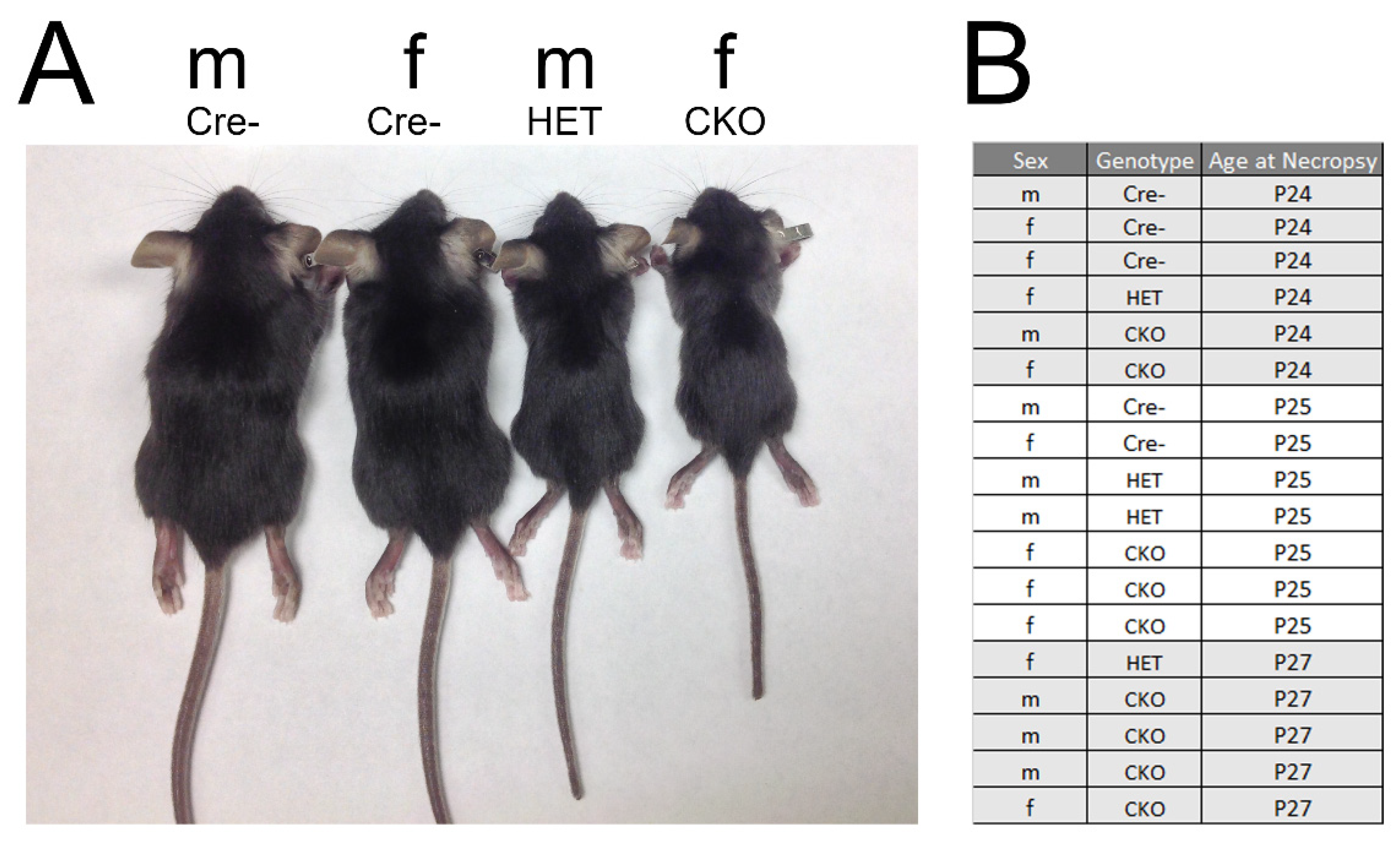
Skeletal Deformities in Osterix-Cre;Tgfbr2f/f Mice May Cause Postnatal Death


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Pathology
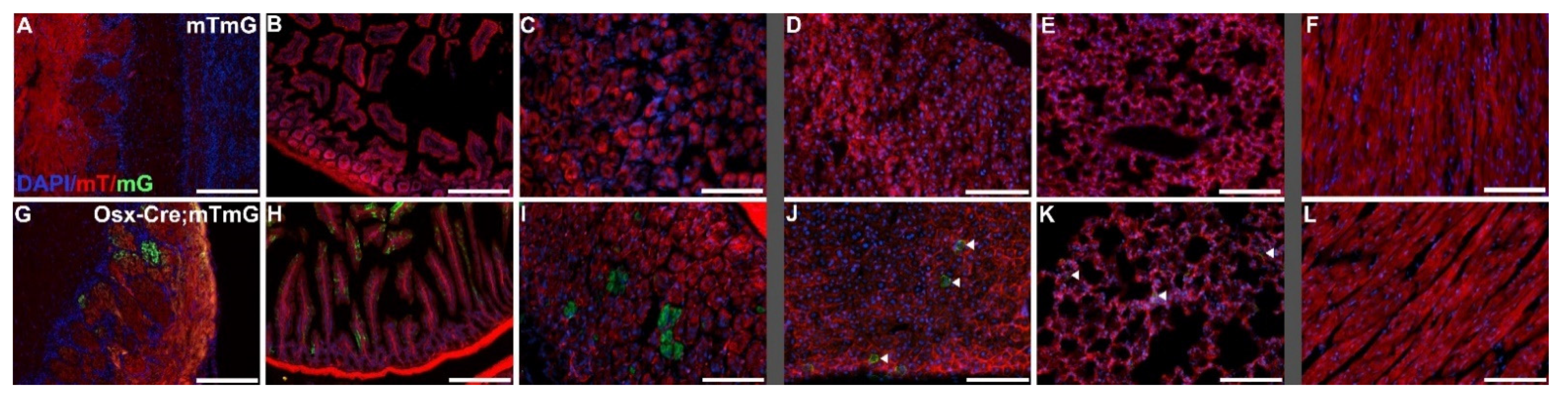
2.3. Imaging of Off-Target Cre Expression in Osterix-Cre:ROSA26mTmG Reporter Mice
3. Results
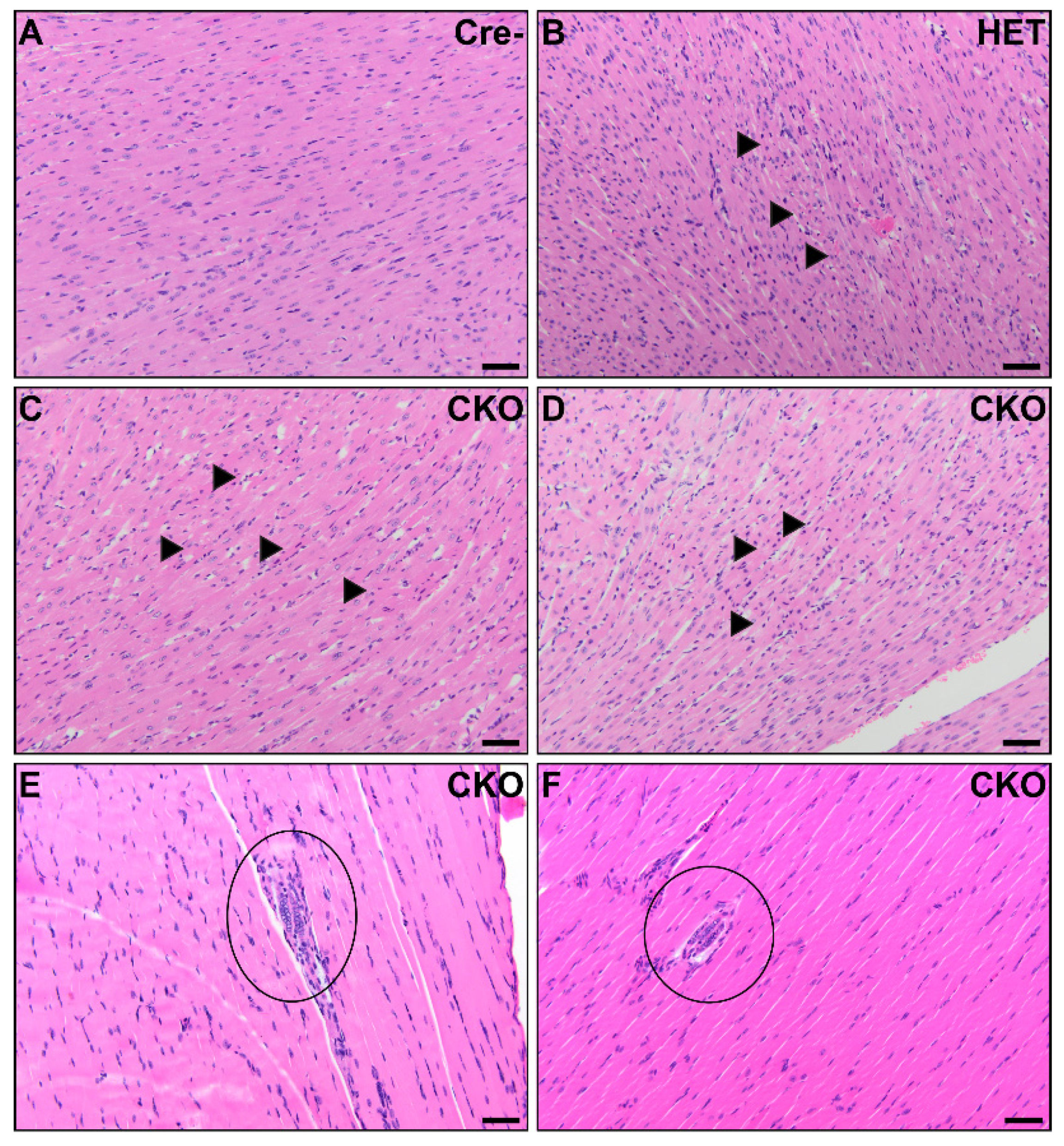
3.1. Visceral Muscle Injury Phenotypes Demonstrated in CKO Mice
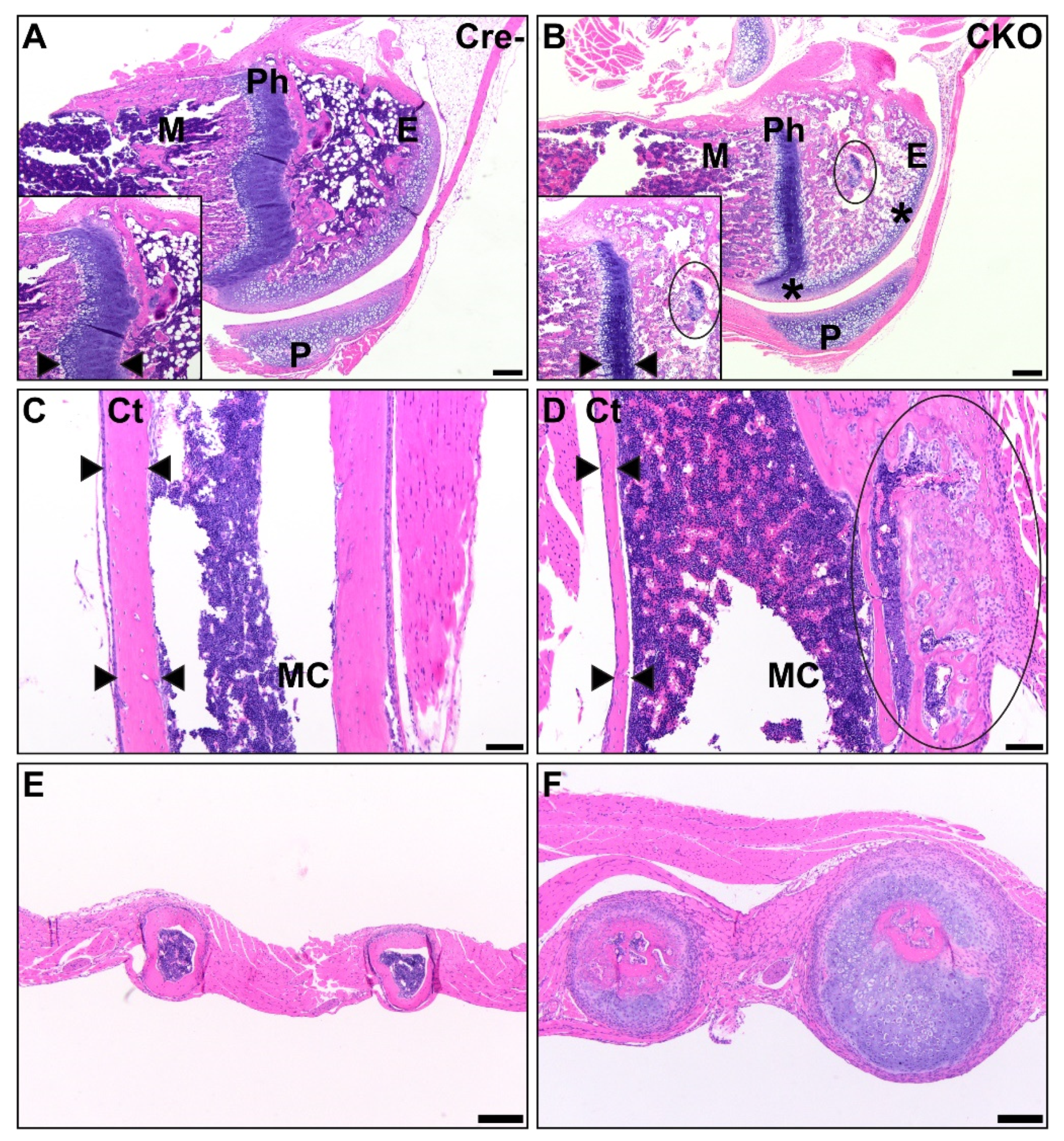
3.2. Osteochondral Dysplasia in Appendicular Skeletons of CKO Mice
3.3. Osteochondral Dysplasia in Axial Skeletons of CKO Mice
3.4. Facial Abnormalities in CKO Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Long, F. Building strong bones: Molecular regulation of the osteoblast lineage. Nat. Rev. Mol. Cell Biol. 2012, 13, 27–38. [Google Scholar] [CrossRef]
- Xing, W.; Godwin, C.; Pourteymoor, S.; Mohan, S. Conditional disruption of the osterix gene in chondrocytes during early postnatal growth impairs secondary ossification in the mouse tibial epiphysis. Bone Res. 2019, 7, 1–8. [Google Scholar] [CrossRef]
- Liu, Q.; Li, M.; Wang, S.; Xiao, Z.; Xiong, Y.; Wang, G. Recent Advances of Osterix Transcription Factor in Osteoblast Differentiation and Bone Formation. Front. Cell Dev. Biol. 2020, 8, 601224. [Google Scholar] [CrossRef]
- Kaback, L.A.; Soung, D.Y.; Naik, A.; Smith, N.; Schwarz, E.M.; O’Keefe, R.J.; Drissi, H. Osterix/Sp7 regulates mesenchymal stem cell mediated endochondral ossification. J. Cell. Physiol. 2008, 214, 173–182. [Google Scholar] [CrossRef]
- Nakashima, K.; Zhou, X.; Kunkel, G.; Zhang, Z.; Deng, J.M.; Behringer, R.R.; de Crombrugghe, B. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell 2002, 108, 17–29. [Google Scholar] [CrossRef] [Green Version]
- Peters, S.B.; Wang, Y.; Serra, R. Tgfbr2 is required in osterix expressing cells for postnatal skeletal development. Bone 2017, 97, 54–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Cox, M.K.; Coricor, G.; MacDougall, M.; Serra, R. Inactivation of Tgfbr2 in Osterix-Cre expressing dental mesenchyme disrupts molar root formation. Dev. Biol. 2013, 382, 27–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.M.; Arthur, A.; Paton, S.; Hemming, S.; Panagopoulos, R.; Codrington, J.; Walkley, C.R.; Zannettino, A.C.W.; Gronthos, S. Loss of ephrinB1 in osteogenic progenitor cells impedes endochondral ossification and compromises bone strength integrity during skeletal development. Bone 2016, 93, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Liang, G.; Huang, Z.; Doty, S.B.; Boskey, A.L. Conditional inactivation of the CXCR4 receptor in osteoprecursors reduces postnatal bone formation due to impaired osteoblast development. J. Biol. Chem. 2011, 286, 26794–26805. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Ma, J.; Zhu, G.; Jules, J.; Wu, M.; McConnell, M.; Tian, F.; Paulson, C.; Zhou, X.; Wang, L.; et al. Cbfβ deletion in mice recapitulates cleidocranial dysplasia and reveals multiple functions of Cbfβ required for skeletal development. Proc. Natl. Acad. Sci. USA 2014, 111, 8482–8487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Snider, T.N.; Wimer, H.F.; Yamada, S.S.; Yang, T.; Holmbeck, K.; Foster, B.L. Multiple essential MT1-MMP functions in tooth root formation, dentinogenesis, and tooth eruption. Matrix Biol. 2016. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Strecker, S.; Wang, L.; Kronenberg, M.S.; Wang, W.; Rowe, D.W.; Maye, P. Osterix-Cre Labeled Progenitor Cells Contribute to the Formation and Maintenance of the Bone Marrow Stroma. PLoS ONE 2013, 8, e71318. [Google Scholar] [CrossRef] [Green Version]
- Abou-Ezzi, G.; Supakorndej, T.; Zhang, J.; Anthony, B.; Krambs, J.; Celik, H.; Karpova, D.; Craft, C.S.; Link, D.C. TGF-β Signaling Plays an Essential Role in the Lineage Specification of Mesenchymal Stem/Progenitor Cells in Fetal Bone Marrow. Stem Cell Rep. 2019, 13, 48–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodda, S.J.; McMahon, A.P. Distinct roles for Hedgehog and canonical Wnt signaling in specification, differentiation and maintenance of osteoblast progenitors. Development 2006, 133, 3231–3244. [Google Scholar] [CrossRef] [Green Version]
- Ono, N.; Ono, W.; Nagasawa, T.; Kronenberg, H.M. A subset of chondrogenic cells provides early mesenchymal progenitors in growing bones. Nat. Cell Biol. 2014, 16, 1157–1167. [Google Scholar] [CrossRef] [PubMed]
- Canalis, E.; Parker, K.; Feng, J.Q.; Zanotti, S. Osteoblast lineage-specific effects of notch activation in the skeleton. Endocrinology 2013, 154, 623–634. [Google Scholar] [CrossRef] [Green Version]
- Razidlo, D.F.; Whitney, T.J.; Casper, M.E.; McGee-Lawrence, M.E.; Stensgard, B.A.; Li, X.; Secreto, F.J.; Knutson, S.K.; Hiebert, S.W.; Westendorf, J.J. Histone Deacetylase 3 Depletion in Osteo/Chondroprogenitor Cells Decreases Bone Density and Increases Marrow Fat. PLoS ONE 2010, 5, e11492. [Google Scholar] [CrossRef] [PubMed]
- Xian, L.; Wu, X.; Pang, L.; Lou, M.; Rosen, C.J.; Qiu, T.; Crane, J.; Frassica, F.; Zhang, L.; Rodriguez, J.P.; et al. Matrix IGF-1 maintains bone mass by activation of mTOR in mesenchymal stem cells. Nat. Med. 2012, 18, 1095–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joeng, K.S.; Long, F. Constitutive Activation of Gli2 Impairs Bone Formation in Postnatal Growing Mice. PLoS ONE 2013, 8, e55134. [Google Scholar] [CrossRef]
- Wang, L.; Mishina, Y.; Liu, F. Osterix-Cre Transgene Causes Craniofacial Bone Development Defect. Calcif. Tissue Int. 2015, 96, 129–137. [Google Scholar] [CrossRef] [Green Version]
- Davey, R.A.; Clarke, M.V.; Sastra, S.; Skinner, J.P.; Chiang, C.; Anderson, P.H.; Zajac, J.D. Decreased body weight in young Osterix-Cre transgenic mice results in delayed cortical bone expansion and accrual. Transgenic Res. 2012, 21, 885–893. [Google Scholar] [CrossRef]
- Huang, W.; Olsen, B.R. Skeletal defects in Osterix-Cre transgenic mice. Transgenic Res. 2015, 24, 167–172. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Shi, Y.; Regan, J.; Karuppaiah, K.; Ornitz, D.M.; Long, F. Osx-Cre targets multiple cell types besides osteoblast lineage in postnatal mice. PLoS ONE 2014, 9, e85161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, H.-S.; Serra, R. Deletion of Tgfbr2 in Prx1-cre expressing mesenchyme results in defects in development of the long bones and joints. Dev. Biol. 2007, 310, 304–316. [Google Scholar] [CrossRef] [Green Version]
- Seo, H.-S.; Serra, R. Tgfbr2 is required for development of the skull vault. Dev. Biol. 2009, 334, 481–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, Y.; Yeo, J.Y.; Chytil, A.; Han, J.; Bringas, P.; Nakajima, A.; Shuler, C.F.; Moses, H.L.; Chai, Y. Conditional inactivation of Tgfbr2 in cranial neural crest causes cleft palate and calvaria defects. Development 2003, 130, 5269–5280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwata, J.; Hacia, J.G.; Suzuki, A.; Sanchez-Lara, P.A.; Urata, M.; Chai, Y. Modulation of noncanonical TGF-β signaling prevents cleft palate in Tgfbr2 mutant mice. J. Clin. Investig. 2012, 122, 873–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oshima, M.; Oshima, H.; Taketo, M.M. TGF-beta receptor type II deficiency results in defects of yolk sac hematopoiesis and vasculogenesis. Dev. Biol. 1996, 179, 297–302. [Google Scholar] [CrossRef] [Green Version]
- Dünker, N.; Krieglstein, K. Tgfbeta2 -/- Tgfbeta3 -/- double knockout mice display severe midline fusion defects and early embryonic lethality. Anat. Embryol. 2002, 206, 73–83. [Google Scholar] [CrossRef]
- Choi, H.; Ahn, Y.-H.; Kim, T.-H.; Bae, C.-H.; Lee, J.-C.; You, H.-K.; Cho, E.-S. TGF-β Signaling Regulates Cementum Formation through Osterix Expression. Sci. Rep. 2016, 6, 26046. [Google Scholar] [CrossRef] [Green Version]
- Yang, G.; Zhou, J.; Teng, Y.; Xie, J.; Lin, J.; Guo, X.; Gao, Y.; He, M.; Yang, X.; Wang, S. Mesenchymal TGF-β Signaling Orchestrates Dental Epithelial Stem Cell Homeostasis Through Wnt Signaling. Stem Cells 2014, 32, 2939–2948. [Google Scholar] [CrossRef]
- Muzumdar, M.D.; Tasic, B.; Miyamichi, K.; Li, L.; Luo, L. A global double-fluorescent Cre reporter mouse. Genesis 2007, 45, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Li, S.; Han, D.; Kaartinen, V.; Chai, Y. Alk5-mediated transforming growth factor β signaling acts upstream of fibroblast growth factor 10 to regulate the proliferation and maintenance of dental epithelial stem cells. Mol. Cell. Biol. 2011, 31, 2079–2089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baffi, M.O.; Slattery, E.; Sohn, P.; Moses, H.L.; Chytil, A.; Serra, R. Conditional deletion of the TGF-β type II receptor in Col2a expressing cells results in defects in the axial skeleton without alterations in chondrocyte differentiation or embryonic development of long bones. Dev. Biol. 2004, 276, 124–142. [Google Scholar] [CrossRef] [PubMed]
- Sohn, P.; Cox, M.; Chen, D.; Serra, R. Molecular profiling of the developing mouse axial skeleton: A role for Tgfbr2 in the development of the intervertebral disc. BMC Dev. Biol. 2010, 10, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maes, C.; Kobayashi, T.; Selig, M.K.; Torrekens, S.; Roth, S.I.; Mackem, S.; Carmeliet, G.; Kronenberg, H.M. Osteoblast precursors, but not mature osteoblasts, move into developing and fractured bones along with invading blood vessels. Dev. Cell 2010, 19, 329–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, J.-H.; Park, S.-Y.; de Crombrugghe, B.; Kim, J.-E. Chondrocyte-specific ablation of Osterix leads to impaired endochondral ossification. Biochem. Biophys. Res. Commun. 2012, 418, 634–640. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, L.; Taiyab, A.; Melvin, V.S.; Jones, K.L.; Williams, T. Increased FGF8 signaling promotes chondrogenic rather than osteogenic development in the embryonic skull. Dis. Models Mech. 2018, 11. [Google Scholar] [CrossRef] [Green Version]
- Govindarajan, V.; Overbeek, P.A. FGF9 can induce endochondral ossification in cranial mesenchyme. BMC Dev. Biol. 2006, 6, 7. [Google Scholar] [CrossRef] [Green Version]
- Van Laer, L.; Dietz, H.; Loeys, B. Loeys-Dietz syndrome. Adv. Exp. Med. Biol. 2014, 802, 95–105. [Google Scholar] [CrossRef]
- Taya, Y.; O’Kane, S.; Ferguson, M. Pathogenesis of cleft palate in TGF-beta3 knockout mice. Development 1999, 126, 3869–3879. [Google Scholar] [CrossRef]
- Loeys, B.L.; Schwarze, U.; Holm, T.; Callewaert, B.L.; Thomas, G.H.; Pannu, H.; De Backer, J.F.; Oswald, G.L.; Symoens, S.; Manouvrier, S.; et al. Aneurysm Syndromes Caused by Mutations in the TGF-β Receptor. N. Engl. J. Med. 2006, 355, 788–798. [Google Scholar] [CrossRef] [PubMed]
- Iwata, J.; Parada, C.; Chai, Y. The mechanism of TGF-β signaling during palate development. Oral Dis. 2011, 17, 733–744. [Google Scholar] [CrossRef] [Green Version]
- Loeys, B.L.; Chen, J.; Neptune, E.R.; Judge, D.P.; Podowski, M.; Holm, T.; Meyers, J.; Leitch, C.C.; Katsanis, N.; Sharifi, N.; et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat. Genet. 2005, 37, 275–281. [Google Scholar] [CrossRef]
- Xu, X.; Han, J.; Ito, Y.; Bringas, P.; Urata, M.M.; Chai, Y. Cell autonomous requirement for Tgfbr2 in the disappearance of medial edge epithelium during palatal fusion. Dev. Biol. 2006, 297, 238–248. [Google Scholar] [CrossRef] [Green Version]
- Tarr, J.T.; Lambi, A.G.; Bradley, J.P.; Barbe, M.F.; Popoff, S.N. Development of normal and Cleft Palate: A central role for connective tissue growth factor (CTGF)/CCN2. J. Dev. Biol. 2018, 6, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Debbache, J.; Parfejevs, V.; Sommer, L. Cre-driver lines used for genetic fate mapping of neural crest cells in the mouse: An overview. Genesis 2018, 56, e23105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacCarrick, G.; Black, J.H.; Bowdin, S.; El-Hamamsy, I.; Frischmeyer-Guerrerio, P.A.; Guerrerio, A.L.; Sponseller, P.D.; Loeys, B.; Dietz, H.C. Loeys-Dietz syndrome: A primer for diagnosis and management. Genet. Med. 2014, 16, 576–587. [Google Scholar] [CrossRef] [PubMed]
- Jani, P.; Nguyen, Q.C.; Almpani, K.; Keyvanfar, C.; Mishra, R.; Liberton, D.; Orzechowski, P.; Frischmeyer-Guerrerio, P.A.; Duverger, O.; Lee, J.S. Severity of oro-dental anomalies in Loeys-Dietz syndrome segregates by gene mutation. J. Med. Genet. 2020, 57, 699–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loeys, B.L. Loeys-Dietz Syndrome. In Aneurysms-Osteoarthritis Syndrome: SMAD3 Gene Mutations; Elsevier Inc.: Amsterdam, The Netherlands, 2017; pp. 55–61. ISBN 9780128027110. [Google Scholar]
- Adès, L.C. Evolution of the face in Loeys-Dietz syndrome type II: Longitudinal observations from infancy in seven cases. Clin. Dysmorphol. 2008, 17, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.K.; Rommel, K.; Mishra, A.; Karck, M.; Haverich, A.; Schmidtke, J.; Arslan-Kirchner, M. TGFBR1 and TGFBR2 mutations in patients with features of Marfan syndrome and Loeys-Dietz syndrome. Hum. Mutat. 2006, 27, 770–777. [Google Scholar] [CrossRef] [PubMed]
- Riera, C.E.; Tsaousidou, E.; Halloran, J.; Follett, P.; Hahn, O.; Pereira, M.M.A.; Ruud, L.E.; Alber, J.; Tharp, K.; Anderson, C.M.; et al. The Sense of Smell Impacts Metabolic Health and Obesity. Cell Metab. 2017, 26, 198–211.e5. [Google Scholar] [CrossRef] [Green Version]
- Fischer, J.M.; Calabrese, P.P.; Miller, A.J.; Munoz, N.M.; Grady, W.M.; Shibata, D.; Liskay, R.M. Single cell lineage tracing reveals a role for TgfβR2 in intestinal stem cell dynamics and differentiation. Proc. Natl. Acad. Sci. USA 2016, 113, 12192–12197. [Google Scholar] [CrossRef] [Green Version]
- Batlle, E.; Massagué, J. Transforming Growth Factor-β Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef]
- Yang, L.; Pang, Y.; Moses, H.L. TGF-β and immune cells: An important regulatory axis in the tumor microenvironment and progression. Trends Immunol. 2010, 31, 220–227. [Google Scholar] [CrossRef] [Green Version]
- Kasagi, S.; Chen, W. TGF-beta1 on osteoimmunology and the bone component cells. Cell Biosci. 2013, 3, 4. [Google Scholar] [CrossRef] [Green Version]
- Masucci, M.T.; Minopoli, M.; Carriero, M.V. Tumor Associated Neutrophils. Their Role in Tumorigenesis, Metastasis, Prognosis and Therapy. Front. Oncol. 2019, 9, 1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerritsma, J.S.J.; Van Kooten, C.; Gerritsen, A.F.; Van Es, L.A.; Daha, M.R. Transforming growth factor-β1 regulates chemokine and complement production by human proximal tubular epithelial cells. Kidney Int. 1998, 53, 609–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bierie, B.; Chung, C.H.; Parker, J.S.; Stover, D.G.; Cheng, N.; Chytil, A.; Aakre, M.; Shyr, Y.; Moses, H.L. Abrogation of TGF-β signaling enhances chemokine production and correlates with prognosis in human breast cancer. J. Clin. Investig. 2009, 119, 1571–1582. [Google Scholar] [CrossRef] [Green Version]
- Spaulding-Barclay, M.A.; Stern, J.; Mehler, P.S. Cardiac changes in anorexia nervosa. Cardiol. Young 2016, 26, 623–628. [Google Scholar] [CrossRef]
- Casiero, D.; Frishman, W.H. Cardiovascular complications of eating disorders. Cardiol. Rev. 2006, 14, 227–231. [Google Scholar] [CrossRef]
- Fayssoil, A.; Melchior, J.C.; Hanachi, M. Heart and anorexia nervosa. Heart Fail. Rev. 2021, 26, 65–70. [Google Scholar] [CrossRef]
- Akdeniz, O.; Yılmaz, E.; Çelik, M.; Özgün, N. Cardiac evaluation in children with malnutrition. Turk. Arch. Pediatrics 2019, 54, 157–165. [Google Scholar] [CrossRef]
- Liepinsh, E.; Makrecka, M.; Kuka, J.; Makarova, E.; Vilskersts, R.; Cirule, H.; Sevostjanovs, E.; Grinberga, S.; Pugovics, O.; Dambrova, M. The heart is better protected against myocardial infarction in the fed state compared to the fasted state. Metabolism 2014, 63, 127–136. [Google Scholar] [CrossRef]
- Agarwal, S.K.; Heiss, G.; Barr, R.G.; Chang, P.P.; Loehr, L.R.; Chambless, L.E.; Shahar, E.; Kitzman, D.W.; Rosamond, W.D. Airflow obstruction, lung function, and risk of incident heart failure: The Atherosclerosis Risk in Communities (ARIC) study. Eur. J. Heart Fail. 2012, 14, 414–422. [Google Scholar] [CrossRef] [PubMed]
- Engström, G.; Melander, O.; Hedblad, B. Population-based study of lung function and incidence of heart failure hospitalisations. Thorax 2010, 65, 633–638. [Google Scholar] [CrossRef] [Green Version]
- Wannamethee, S.G.; Shaper, A.G.; Papacosta, O.; Lennon, L.; Welsh, P.; Whincup, P.H. Lung function and airway obstruction: Associations with circulating markers of cardiac function and incident heart failure in older men—The British Regional Heart Study. Thorax 2016, 71, 526–534. [Google Scholar] [CrossRef] [Green Version]
- Faa, A.; Iacovidou, N.; Xanthos, T.; Locci, A.; Pampaloni, P.; Aroni, F.; Papalois, A.; Faa, G.; Fanos, V. Hypoxia/reoxygenation-induced myocardial lesions in newborn piglets are related to interindividual variability and not to oxygen concentration. Clinics 2012. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Corps, K.; Stanwick, M.; Rectenwald, J.; Kruggel, A.; Peters, S.B. Skeletal Deformities in Osterix-Cre;Tgfbr2f/f Mice May Cause Postnatal Death. Genes 2021, 12, 975. https://doi.org/10.3390/genes12070975
Corps K, Stanwick M, Rectenwald J, Kruggel A, Peters SB. Skeletal Deformities in Osterix-Cre;Tgfbr2f/f Mice May Cause Postnatal Death. Genes. 2021; 12(7):975. https://doi.org/10.3390/genes12070975
Chicago/Turabian StyleCorps, Kara, Monica Stanwick, Juliann Rectenwald, Andrew Kruggel, and Sarah B. Peters. 2021. "Skeletal Deformities in Osterix-Cre;Tgfbr2f/f Mice May Cause Postnatal Death" Genes 12, no. 7: 975. https://doi.org/10.3390/genes12070975
APA StyleCorps, K., Stanwick, M., Rectenwald, J., Kruggel, A., & Peters, S. B. (2021). Skeletal Deformities in Osterix-Cre;Tgfbr2f/f Mice May Cause Postnatal Death. Genes, 12(7), 975. https://doi.org/10.3390/genes12070975